
Abstract
Ficus species (Moraceae) play pivotal roles in tropical and subtropical ecosystems. Thriving across diverse habitats, from rainforests to deserts, they harbor a multitude of mutualistic and antagonistic interactions with insects, nematodes, and pathogens. Despite their ecological significance, knowledge about the genomic background of Ficus remains limited. In this study, we report a chromosome-level reference genome of F. hirta, with a total size of 297.27 Mb, containing 28,625 protein-coding genes and 44.67% repeat sequences. These findings illuminate the genetic basis of Ficus responses to environmental challenges, offering valuable genomic resources for understanding genome size, adaptive evolution, and co-evolution with natural enemies and mutualists within the genus.
Similar content being viewed by others
Background & Summary
Ficus is a highly species rich genus of mainly pantropical woody plants with a diverse range of growth forms. Fig trees occupy a broad range of habitats1,2 and are among the most ecologically important plant groups in tropical forests3,4. The genus is characterized by its enclosed inflorescences (figs, also called syconia) that vary in size and location, but have remained unchanged in fundamental structure since the genus first appeared around 45 mya5,6,7. The evolutionary history of the genus has therefore combined extensive radiation and ecological diversification with a reproductive conservatism that is linked to their unique interaction with the trees’ only pollinators (fig wasps, Hymenoptera Agaonidae). Perhaps the most significant innovation involving fig anatomy has involved the modification of breeding systems, with some Ficus species monoecious, others gynodioecious (but functionally dioecious), that involves associated changes in floral anatomy8. Ficus belongs to the Eudicot family Moraceae, placed by recent phylogenies within the ‘urticalean’ clade of Rosales. Dioecy is believed to be the ancestral state within Moraceae as a whole5 but the ancestral breeding system in Ficus remains uncertain8. Most Ficus species are diploid with 2n = 26, irrespective of their phylogenetic relations within the genus9, but tetraploid species are known from Africa10. The significance of hybridization in Ficus diversification has been debated, but Gardner et al. have shown that while introgression has taken place, it has not had a major impact on evolution in the genus7.
In addition to pollinating fig wasps, Ficus also has symbiotic non-pollinating fig wasps, beetles, flies, moths, nematodes and pathogens that are likely to have a negative impact on the host. More than 300 leaf-chewing and more than 400 sap-sucking insect species were recorded from just 15 Ficus species from Papua New Guinea11,12,13,14. Ficus species possess diversified direct defense strategies, including physical structures and differing chemical defenses15,16. They are known to contain hundreds of different secondary metabolites17,18, but we know little of the underlying genetics.
Here, we assembled a high-quality chromosome-level genome of F. hirta using a combination of PacBio HiFi sequencing and Hi-C techniques and compared this with previously published genomes of four congeners. The assembled F. hirta genome had a combined length of 297.27 Mb, featuring a contig N50 of 19.71 Mb and achieving a complete BUSCO score of 98.50%. A substantial 282.12 Mb (94.90%) of the sequences were successfully anchored to the 13 pseudochromosomes. The genome annotation predicted 28,625 protein-coding genes. This high-quality F. hirta genome provides novel genomic resources for future researchers on genome and adaptive evolution within fig trees, as well as Ficus-natural enemy and mutualist co-evolution.
Methods
Sample collection and sequencing
F. hirta material came from a natural population growing in the South China Botanical Garden (23.18°N, 113.36°E), Guangzhou, China. Fresh young leaves of F. hirta were collected for genome sequencing. Organs (leaves, stems, inflorescences and roots) were collected from three individual trees to provide biological replicates of the F. hirta sampled for its transcriptome. All samples were immediately flash-frozen using liquid nitrogen and stored at −80 °C for subsequent nucleic acid extraction. High-quality genomic DNA was isolated from young leaves of F. hirta using the CTAB method19. The genomic DNA was then fragmented into random fragments, and short-read libraries of F. hirta were constructed according to Illumina’s standard protocol, and paired-end reads (150 bp) were sequenced on an Illumina NovaSeq platform. Additionally, a 15 kb HiFi library was constructed following the protocol for the PacBio Sequel2 platform, and circular consensus sequencing (CCS) was performed. A Hi-C library20 was also sequenced on an Illumina NovaSeq platform with paired-end reads of 150 bp. Total RNA was extracted using CTAB and RNA-seq libraries were constructed and sequenced on an Illumina NovaSeq platform with a read length of 150 bp on both sides. All Illumina sequencing data were filtered to obtain clean data using the fastp v0.23.1 software21 for subsequent analysis. All analyses were performed on a laboratory server with 60 TB storage and 100 threads, operating on Linux.
Genome assembly
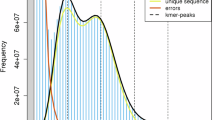
Before assembly, we first estimated the genome size and heterozygosity of F. hirta by calculating the 17-mer frequency distribution using Jellyfish v2.3.0 and GenomeScope v2.0 software22,23. Next, Pacbio HiFi reads were assembled into contigs using hifiasm v0.15.4 with the default parameters24. To obtain clean Hi-C data, we used HiC-Pro v3.1.0 to filter the raw Hi-C data25. After that, the clean Hi-C data were aligned to the final assembled contigs by the juicer pipeline v1.6 to obtain the interaction matrix26. The contigs were then ordered and anchored using 3D de novo assembly (3D-DNA) v18041927. Finally, the Hi-C contact maps of the final assembly result were reviewed manually with Juicebox v1.11.0826.
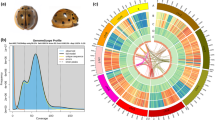
The genome of F. hirta was estimated to be 283.52 Mb in size, with a heterozygosity of 1.26% (Figure S1). We performed de novo assembly of the F. hirta genome at the chromosome-level based on PacBio reads generated in CCS mode (HiFi reads), with 31.76 Gb (106-fold coverage), 65-fold coverage of clean Illumina short reads amounting to 19.49 Gb, and 124-fold coverage of high-throughput chromatin conformation capture (Hi-C) data amounting to 37.05 Gb (Table S1). The assembled genome size was 297.27 Mb, with 282.12 Mb anchored onto 13 pseudochromosomes (anchor rate: 94.90%) (Fig. 1A; Figure S2; Table 1). The contig N50 was 19.71 Mb, which has higher integrity and continuity (contigs N50: 0.18 to 2.29 Mb) (Table S2), compared to F. carica (8.23 Mb)28, F. microcarpa (1.77 Mb)29, F. hispida (2.16 Mb)29, and F. religiosa (5.53 Mb)30.

The genomic features of Ficus hirta. (A) The 13 pseudochromosomes; (B) gene density; (C) histogram of GC content; (D–F) the density of total repeat sequences, Gypsy LTR-REs, and Copia LTR-REs; (G–J) tRNA, snRNA, miRNA, and rRNA density; (K) intragenomic collinearity. (B–J) were drawn in 100 kb overlapping sliding windows.
Genome annotation
For repeated elements identification and masking, we used homology-based and de novo approaches to identification. Briefly, a de novo repeat library was constructed using RepeatModeler v2.0.231. Then the obtained library was combined with the Repbase database v21.1232 to identify repetitive sequences in the F. hirta genome using RepeatMasker v4.1.233. For noncoding RNA prediction, the tRNA genes were predicted using tRNAscan-SE v2.0.634. Others, including miRNA, rRNA and snRNA genes, were detected by comparison with the Rfam database using CMsearch v1.1.3 with the default parameters35,36. Protein-coding gene annotation was conducted using homology-based, transcriptome-based, and ab initio prediction methods. First, we used homologies from 11 different species (Table S3) as protein-based evidence for predicting gene sets using GeneWise v2.4.137. Transcriptome data, including leaf, stem, inflorescence, and root RNA-seq reads were mapped using HISAT2 v2.1.038. Ab initio prediction using packages AUGUSTUS v3.4.039, trained by the transcriptome data. To generate a comprehensive protein-coding gene set, we used the GETA pipeline (https://github.com/chenlianfu/geta) to integrate annotations from all homology-based, transcriptome-based, and ab initio predictions. To functionally annotate the predicted gene models, we searched several different databases, including the NCBI nr40, Swiss-Port41, KOG42, eggNOG43, Pfam44, GO45, and KEGG46.
In total, 28,625 protein-coding genes were predicted using a combination of de novo homolog-based searches and RNA-seq data, of which 92.39% could be functionally annotated (Fig. 1B,C; Table 1; Table S4). The predicted proteome contained 98.50% complete and 0.80% fragmented BUSCO genes (Table S5). A total of 132.79 Mb repeat elements were identified, which accounted for 44.67% of the F. hirta genome (Fig. 1D; Table 2). The most abundant repetitive elements were LTR retrotransposon (LTR-RE) elements (59.31 Mb; LTR-RE/Copia: 13.59 Mb; LTR-RE/Gypsy: 41.60 Mb), followed by DNA transposons (11.58 Mb), with an additional 46.13 Mb of unclassified repetitive sequences (Fig. 1E,F; Table 2). Furthermore, our analysis revealed the presence of 9,830 noncoding RNAs, which included 133 miRNAs, 574 transfer RNAs (tRNA), 8,717 ribosomal RNAs (rRNA), and 406 small nuclear RNAs (snRNA) (Fig. 1G–J; Table S6).
Data Records
The National Genomics Data Center (NGDC) database BioProject accession number for the sequence reported in this paper is PRJCA019243. The raw sequencing data for HiFi, Hi-C, and RNA-seq were submitted to NGDC GSA with accession numbers CRR857341-CRR85735647. The chromosomal-level genome assembly file was deposited in the NCBI GenBank with accession number GCA_038430175.148. Moreover, the gene structure annotation, gene function annotation and TE annatition files have been deposited at the Figshare49 database.
Technical Validation
To assess genome assembly quality, the Illumina genomic and RNA-seq reads were mapped to the genome using BWA v0.7.1750 and HISAT2 v2.1.038, respectively. To evaluate the completeness and accuracy of the genome, we used the LTR assembly index (LAI)51 and BUSCO v4.1.252 evaluation with the embryophyta_odb10 database to examine. Finally, the mapping rates of Illumina and HiFi reads to the genome were 98.52% and 99.13%, respectively (Table S7). The LAI had a score of 19.98 (Table 1), which is similar to the scores for Oryza sativa and Arabidopsis thaliana51. Benchmarking Universal Single-Copy Orthologs (BUSCO) analyses showed the assembled genome contained 1,590 (98.50% of 1,614) complete sets of the core orthologous genes in the Embryophyta_odb10 database, which is higher than that of the seven previously reported Ficus genomes (89.7%–96.4%) (Table S5). All these values suggest a high quality of F. hirta genome sequence.
Code availability
No custom code was used for this study. All sofware and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and code/parameters of sofware have been detailed described in Methods.
References
Harrison, R. D. Figs and the diversity of tropical rainforests. Bioscience 55, 1053–1064 (2005).
Pierantoni, M. et al. Mineral deposits in Ficus leaves: morphologies and locations in relation to function. Plant Physiol. 176, 1751–1763 (2018).
Shanahan, M., So, S., Compton, S. G. & Corlett, R. Fig-eating by vertebrate frugivores: a global review. Biol. Rev. 76, 529–572 (2001).
Cottee-Jones, H. E. W., Bajpai, O., Chaudhary, L. B. & Whittaker, R. J. The importance of Ficus (Moraceae) trees for tropical forest restoration. Biotropica 48, 413–419 (2016).
Datwyler, S. L. & Weiblen, G. D. On the origin of the fig: phylogenetic relationships of Moraceae from ndhF sequences. Am. J. Bot. 91, 767–777 (2004).
Compton, S. G. et al. Ancient fig wasps indicate at least 34 Myr of stasis in their mutualism with fig trees. Biol. lett. 6, 838–842 (2010).
Gardner, E. M. et al. Echoes of ancient introgression punctuate stable genomic lineages in the evolution of figs. Proc. Natl. Acad. Sci. USA 120, e2222035120 (2023).
Zhang, Q., Onstein, R. E., Little, S. A. & Sauquet, H. Estimating divergence times and ancestral breeding systems in Ficus and Moraceae. Ann. Bot. 123, 191–204 (2019).
Condit, I. J. Cytological studies in the genus Ficus. III. Chromosome numbers in sixty-two species. Madrono. 17, 153–155 (1964).
Hans, A. S. Cytomorphology of arborescent Moraceae. J. Arnold. Arbor. 53, 216–225 (1972).
Basset, Y. & Novotny, V. Species richness of insect herbivore communities on Ficus in Papua New Guinea. Biol. J. Linn. Soc. Lond. 67, 477–499 (1999).
Elbeaino, T., Digiaro, M. & Martelli, G. P. Complete sequence of fig fleck-associated virus, a novel member of the family Tymoviridae. Virus. Res. 161, 198–202 (2011).
Hosomi, A., Miwa, Y., Furukawa, M. & Kawaradani, M. Growth of fig varieties resistant to ceratocystis canker following infection with Ceratocystis fimbriata. J. Jpn. Soc. Hortic.Sci. 81, 159–165 (2012).
Zhao, C. et al. Ficophagus giblindavisi n. sp (Nematoda: Aphelenchoididae), an associate of Ficus variegata in China. Nematology. 24, 901–914 (2022).
Borges, R. M., Bessière, J. M. & Ranganathan, Y. Diel variation in fig volatiles across syconium development: making sense of scents. J. Chem. Ecol. 39, 630–642 (2013).
Villard, C., Larbat, R., Munakata, R. & Hehn, A. Defence mechanisms of Ficus: pyramiding strategies to cope with pests and pathogens. Planta 249, 617–633 (2019).
Sirisha, N., Sreenivasulu, M., Sangeeta, K. & Chetty, C. M. Antioxidant properties of Ficus species-a review. Int. J. Pharmtech. Res. 2, 2174–2182 (2010).
Volf, M. et al. Community structure of insect herbivores is driven by conservatism, escalation and divergence of defensive traits in Ficus. Ecol. Lett. 21, 83–92 (2018).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15 (1997).
Xie, T. et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol. Plant 8, 489–492 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Feng, X., Cheng, H., Portik, D. & Li, H. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat. Methods 19, 671–674 (2022).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. de novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Usai, G. et al. Epigenetic patterns within the haplotype phased fig (Ficus carica L.) genome. Plant J. 102, 600–614 (2020).
Zhang, X. et al. Genomes of the banyan tree and pollinator wasp provide insights into fig-wasp coevolution. Cell 183, 875–889 (2020).
Chakraborty, A., Mahajan, S., Bisht, M. S. & Sharma, V. K. Genome sequencing and comparative analysis of Ficus benghalensis and Ficus religiosa species reveal evolutionary mechanisms of longevity. Iscience 25, 105100 (2022).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 24, 4.10.11–14.10.14 (2009).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Cui, X. et al. CMsearch: simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 32, i332–i340 (2016).
Gardner, P. P. et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. methods 12, 357–360 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Wheeler, D. L. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 35, D5–D12 (2007).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54 (1999).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 4, 41 (2003).
Hernandez-Plaza, A. et al. eggNOG 6.0: enabling comparative genomics across 12 535 organisms. Nucleic Acids Res. 51, D389–D394 (2023).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA012347 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_038430175.1 (2024).
Huang, W. C. A high-quality chromosome-level genome assembly of Ficus hirta. figshare https://doi.org/10.6084/m9.figshare.25246813 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126–e126 (2018).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work was supported by National Key R & D Program of China (2023YFE0107400), Science and Technology Projects in Guangzhou (E33309), Guangzhou Ecological Landscape Technology Collaborative Innovation Center (202206010058) and the Chinese Academy of Sciences PIFI Fellowship for Visiting Scientists (2022VBA0002).
Author information
Authors and Affiliations
Contributions
H.Y. conceived the project and supervised this study. W.H., S.F. and H.C. collected samples. W.H. Y.D. and W.L. performed genome analysis. H.Y., W.H., and S.G.C. wrote the manuscript. All authors read and approved the final manuscript and all authors commented on the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, W., Ding, Y., Fan, S. et al. A high-quality chromosome-level genome assembly of Ficus hirta. Sci Data 11, 526 (2024). https://doi.org/10.1038/s41597-024-03376-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03376-z
- Springer Nature Limited