
Abstract
Helwingia, a shrub of the monotypic cosmopolitan family Helwingiaceae, is distinguished by its inflorescence, in which flowers are borne on the midrib of the leaf—a trait not commonly observed in related plant families. Previous studies have investigated the development of this unusual structure using comparative anatomical methods. However, the scarcity of genomic data has hindered our understanding of the origins and evolutionary history of this uncommon trait at the molecular level. Here, we report the first high-quality genome of the family Helwingiaceae. Assembled using HiFi sequencing and Hi-C technologies, the genome of H. omeiensis is anchored to 19 chromosomes, with a total length of 2.75 Gb and a contig N50 length of 6.78 Mb. The BUSCO completeness score of the assembled genome was 98.2%. 53,951 genes were identified, of which 99.7% were annotated in at least one protein database. The high-quality reference genome of H. omeiensis provides an essential genetic resource and sheds light on the phylogeny and evolution of specific traits in the family Helwingiaceae.
Similar content being viewed by others

Background & Summary
Helwingiaceae is a monotypic family in the order Aquifoliales, comprising a single genus Helwingia. The innovative structure of this genus is that the flowers are borne on the midrib of the leaf, which is known as an “epiphyllous inflorescence”, setting them apart from other plants. In addition, the pith, leaves, and fruits of plants in this genus are traditionally used in herbal medicine to treat dysentery and as diuretic and anti-inflammatory remedies1. The genus includes four species, H. chinensis, H. himalaica, H. japonica, and H. omeiensis, which are all dioecious shrubs mainly found in eastern Asia2,3. Specifically, H. omeiensis is indigenous to Southwest China, and thrives in moist woodlands and on mountain slopes2.
Previous comparative anatomical studies suggested that changes in the position of flower primordium initiation and intercalary growth may contribute to the formation of this distinct structure4,5,6. With the development of high-throughput sequencing technologies, the genomes of three closely related species in the genus Ilex of the family Aquifoliaceae have been published7,8. However, despite the fact that RNA-seq data and the complete chloroplast genomes of three Helwingia species have been released4,9,10, a lack of genomic data remains a barrier to studying the evolutionary origin of the family.
In this study, we leveraged a combination of short reads, high-fidelity (HiFi) reads, and chromosome conformation capture (Hi-C) sequencing data to construct a chromosome-level genome assembly for H. omeiensis, providing the first genome resource for the family Helwingiaceae. The length of the genome assembly was 2.75 Gb, with a scaffold N50 of 127.8 Mb and a contig N50 of 6.78 Mb. We identified 1.98 Gb of repetitive elements, accounting for 72.21% of the assembled genome, as well as 53,951 protein-coding genes. The genome assembly and annotation of H. omeiensis will provide a critical foundation for exploring the genetic basis underpinning of this unique inflorescence structure and the phylogenetic relationships within the family Helwingiaceae.
Methods
Plant materials
All of the fresh materials were collected from a female adult plant of Helwingia omeiensis cultivated in Mount Emei Botanical Garden, Sichuan Province, China (N29°35′40, E103°22′40), and the specimens were kept at the Museum of Sichuan University. The genomic DNA was extracted from young leaves, whereas RNA was extracted from mature leaves and terminal buds.
Library construction and sequencing
For short-read sequencing, the sample was randomly fragmented by an ultrasonic processor (Covaris S220; Woburn, MA, USA) to generate DNA fragments approximately 350 bp in length. The DNA fragments were subsequently constructed through end repair, the addition of a 3′ A tail and the ligation of adapters. Next, the library was sequenced with a DNBSEQ-G400 (BGI, Wuhan, China). The raw short reads were filtered by SOAPnuke v1.5.610 to remove adapters and low-quality reads. A total of 87.36 Gb of clean data were obtained for H. omeiensis (Table 1).
For HiFi (high-fidelity) sequencing, high-quality genomic DNA was sheared using Megaruptor® 3 (Diagenode), and subreads with a length of 20 kb were further selected using Sage ELF to prepare the PacBio HiFi libraries in CCS mode on the Pacific Biosciences Sequel II System (Supplementary Figure S1). Finally, 50.32 Gb of long clean reads were generated (Table 1), with mean lengths of 13.0 kb and 14.5 kb, respectively.
Hi-C technology captures sequence interactions between all DNA segments within chromosomes to obtain information on interactions between segments of the genome for assisted genome assembly11. Fresh leaves of the same individual were used to construct Hi-C libraries, and the MboI restriction enzyme was used for DNA ligation. After tailing, pulldown, and adapter ligation, the DNA library was sequenced on an Illumina HiSeq X Ten System (BGI, Wuhan, China) with a strategy of 2 × 150 bp. After filtering low-quality reads, 221.52 Gb of clean Hi-C data were obtained (Table 1).
RNA sequencing
Mature leaves and young terminal buds of the same individual were collected for RNA extraction. The RNA-seq library was constructed using the Illumina standard protocol (San Diego, CA, United States) and sequenced on the Illumina HiSeq X Ten platform (BGI, Wuhan, China). The raw data were filtered by Cutadapt v1.1612 to remove adapters and low-quality reads. After quality control by FastQC v0.11.8 (https://github.com/s-andrews/FastQC), 10.46 Gb of paired-end short clean reads were generated from the RNA-seq library (Table 1).
Genome survey and de novo assembly
Jellyfish v2.1.413 was used to quickly count K-mer frequencies ranging from 17 to 31, and then GenomeScope14 predicted genomic features using a K-mer-based statistical approach (Supplementary Table S1). The H. omeiensis genome was estimated to be 2.54 Gb in size, with a heterozygosity rate of 1.19% and repetitive sequences accounting for 54.85% of the total length of the genome (Fig. 1). Using 50.32 Gb of clean HiFi reads with hifiasm v0.19.6-r59515, we generated a genome assembly of 2.92 Gb in size with a contig N50 of 6.21 Mb. Following that, Chromap v0.2.5-r47316 was utilized to align Hi-C clean reads to the contig assembly, and according to the strength of interactions between pairs of reciprocal sequences, YaHS v1.2a.117 was used to anchor contigs onto 1,584 scaffolds. Next, using Juicebox v1.11.0818, we visualized the Hi-C contact maps of the scaffold assembly and made final refinements to the genome assembly. With reference to chromosome counts indexed in the Chromosome Counts Database (CCDB)19 (https://ccdb.tau.ac.il/) and the whole-genome Hi-C interaction heatmap, we identified the 19 longest scaffolds as pseudo-chromosomes (Fig. 2). TGS-GapCloser v1.2.120 filled 75 of the 1,011 gaps in the scaffold assembly based on HiFi reads. The final assembly had a total length of 2.75 Gb, with a contig N50 of 6.78 Mb. The length of 19 pseudochromosomes was 2.38 Gb, with a maximum chromosome length of 153.79 Mb (Table 2). Since there is no reference genome for this species, we numbered the chromosomes in order from largest to smallest (Fig. 3 and Table 3).

Distribution profiles of 27-mer analysis of short reads.

The Hi-C interactive heatmap of 19 pseudo-chromosomes of H. omeiensis.

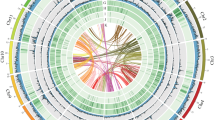
Circos plot of genomic characteristics and annotation of the H. omeiensis genome assembly (1 Mb window size). The data from the outer to the inner circles are as follows: (a) length of 19 pseudochromosomes, (b) GC density, (c) gene density, (d) Gypsy retrotransposon density, (e) Copia retrotransposon density, and (f) DNA transposon density.
Gene annotation
To perform a comprehensive prediction of protein-coding genes, the GETA v2.5.6 pipeline (https://github.com/chenlianfu/geta) was used for automatic genome-wide annotation. First, RepeatModeler v2.0.321 and DeepTE22 were used for self-training and to construct a repeat library. On this basis, RepeatMasker v4.1.2-p123 was employed to predict and combine repetitive elements for homology-based methods. The analysis revealed that 72.21% of the genome was composed of repetitive sequences, including 46.39% long-terminal repeat (LTR) retrotransposons and 19.43% DNA transposons (Table 4).
After masking repetitive sequences in the genome, three strategies (homology-based, RNA-seq-guided, and ab initio methods) were used for the annotation process. For the RNA-seq-guided method, the RNA sequencing data were provided to HISAT2 v2.1.024 and SAMtools v1.1125 to map the data to the repeat-masked genome. Then, TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder) was used to predict the open reading frame (ORF), and filter out the gene models with identities greater than 80% at the amino acid level between pairs to obtain nonredundant results. Protein sequences from Vitis vinifera, Arabidopsis thaliana, Solanum lycopersicum, Daucus carota, and Ilex latifolia were aligned to the query genome as homologous proteins using GeneWise v2.4.126 to estimate protein-coding genes (Supplementary Table S2). Ab initio prediction was carried out with AUGUSTUS v3.4.027, which guided by previous prediction results. Based on the GETA pipeline, all the outputs were validated using HMMER v3.3.228 and NCBI-BLAST + v2.13.0 + before being integrated into a complete and nonredundant set of gene annotations.
Following the alignments by DIAMOND v2.0.1529, gene functions were indicated using the Nonredundant Protein Sequence Database (NR)30, InterPro31, UniProt32, and EggNOG33 with an e-value of 1e-5. In addition, GO annotation was performed by KOBAS34 (http://kobas.cbi.pku.edu.cnwas) aligned with the Arabidopsis thaliana database.
Data Records
All the raw sequencing reads of H. omeiensis were uploaded to the NCBI database under accession number SRP43521335. The genome assembly had been submitted to Genome Warehouse in China National Center for Bioinformation under accession number GWHEQHK0000000036 and European Nucleotide Archive (ENA) with accession number GCA_964187755.237. The annotation files of the genome are available in the figshare database: https://doi.org/10.6084/m9.figshare.22817414.v338.
Technical Validation
Evaluation of the genome assembly and annotation
To assess the integrity of the assembly, short reads were mapped to the genomes using minimap239, giving a mapping rate of 96.59% and a genome coverage of 99.85%. The alignment rate of RNA sequencing reads was 96.95% and 94.10% for two H. omeiensis samples by HISAT2 v2.1.0 (Supplementary Table S3)24. The completeness and accuracy of the final genome assembly were checked by Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.240 with eudicots_odb10. The results showed that 98.2% of orthologs of eudicots could be identified in the assembly (Supplementary Figure S2). Moreover, the values evaluated by Merqury v1.341 based on short reads also showed high consensus quality (accuracy > 99.99%, QV > 58) and low base-level error rates (1.37 × 10−6). In addition, the LTR Assembly Index (LAI) score of the whole-genome assembly was calculated to be 24.52, exceeding that of rice (MSUV7) and Arabidopsis (TAIR10), reaching the ‘gold quality’42. These results demonstrated that the assembly is reliable and has high base-level accuracy, high completeness, and high contiguity.
Via multiple annotation approaches, we identified 53,951 protein-coding genes in the H. omeiensis genome (Table 5). BUSCO analysis showed the completeness of predicted genes was 94.5% (Supplementary Figure S2). The functional analysis revealed that 99.7% of the protein-encoding genes could be annotated in at least one of five public databases (Fig. 4).

Venn diagram displaying the matches of genes of H. omeiensis in five public protein databases.
Code availability
(1) SOAPnuke v1.5.6: parameters: -n 0.01 -l 20 -q 0.1 -i -Q 2 -G -M 2 -A 0.5 -d
(2) Cutadapt v1.16: parameters: -a AGATCGGAAG -q 20
All the other software and pipelines not listed or described in the methods section used the default parameters.
References
Chen Lin, L. W.-j. et al. Overview of Pharmaceutical Research on Helwingia Willd. Journal of Liaoning University of Traditional Chinese Medicine 14, 116–118 (2012).
Wu, R. H. W. Z., Raven, P. H., Hong, D. Y. Flora of China (Apiaceae through Ericaceae). Vol. 14 (Science Press, 2005).
Miller, C. The World Flora Online – Research Infrastructure for Plant Conservation. Biodiversity Information Science and Standards (2019).
Sun, C., Yu, G., Bao, M., Zheng, B. & Ning, G. Biological pattern and transcriptomic exploration and phylogenetic analysis in the odd floral architecture tree: Helwingia willd. BMC Res Notes 7, 402 (2014).
Ao, C. & Tobe, H. Floral morphology and embryology of Helwingia (Helwingiaceae, Aquifoliales): systematic and evolutionary implications. J Plant Res 128, 161–175 (2015).
Dickinson, T. A. & Sattler, R. Development of the epiphyllous inflorescence of helwingia japonica (helwingiaceae). American Journal of Botany 62, 962–973 (1975).
Yao, X., Lu, Z., Song, Y., Hu, X. & Corlett, R. T. A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in Southwest China. Hortic Res 9 (2022).
Kong, B. L. et al. Chromosomal level genome of Ilex asprella and insight into antiviral triterpenoid pathway. Genomics 114, 110366 (2022).
Zhang, C. et al. Asterid Phylogenomics/Phylotranscriptomics Uncover Morphological Evolutionary Histories and Support Phylogenetic Placement for Numerous Whole-Genome Duplications. Mol Biol Evol 37, 3188–3210 (2020).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Louwers, M., Splinter, E., van Driel, R., de Laat, W. & Stam, M. Studying physical chromatin interactions in plants using Chromosome Conformation Capture (3C). Nat Protoc 4, 1216–1229 (2009).
Martin, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal 17, 10–12.
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nature Communications 12 (2021).
Zhou, C., McCarthy, S. A., Durbin, R. & Alkan, C. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39 (2023).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101 (2016).
Rice, A. et al. The Chromosome Counts Database (CCDB) – a community resource of plant chromosome numbers. New Phytologist 206, 19–26 (2014).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Yan, H., Bombarely, A., Li, S. & Valencia, A. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4 10 11–14 10 14 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439 (2006).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Research 51, D29–D38 (2023).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43, D213–221 (2015).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
Bu, D. et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Research 49, W317–W325 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP435213 (2023).
National Genomics Data Center https://ngdc.cncb.ac.cn/gwh/Assembly/83104/show (2023).
European Nucleotide Archive http://identifiers.org/insdc.gca:GCA_964187755.2 (2024).
Chen, Y. The annotation of Helwingia omeiensis genome assembly. figshare https://doi.org/10.6084/m9.figshare.22817414.v3 (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46, e126–e126 (2018).
Acknowledgements
This research was supported by the Natural Science Foundation of China (32171606, 41771055).
Author information
Authors and Affiliations
Contributions
Y.C., L.F. and H.L. collected the materials and performed the genome sequencing and assembly. Y.C. performed the data validation and analyses. Y.C., J.L. and Q.H. wrote the manuscript. All the authors approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Feng, L., Lin, H. et al. Chromosome-level genome assembly of Helwingia omeiensis: the first genome in the family Helwingiaceae. Sci Data 11, 719 (2024). https://doi.org/10.1038/s41597-024-03568-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03568-7
- Springer Nature Limited