
Abstract
Osteoarthritis (OA) is a multifactorial and increasingly prevalent degenerative disease that affects the whole joint. The pathogenesis of OA is poorly understood and there is a lack of therapeutic interventions to reverse the pathological process of this disease. Accumulating studies have shown that the overproduction of reactive oxygen species (ROS) and ROS-induced lipid peroxidation are involved in the pathogenesis of OA. 4-Hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) have received considerable attention for their role in cartilage degeneration and subchondral bone remodeling during OA development. Ferroptosis is a form of cell death characterized by a lack of control of membrane lipid peroxidation and recent studies have suggested that chondrocyte ferroptosis contributes to OA progression. In this review, we aim to discuss lipid peroxidation-derived 4-HNE and MDA in the progression of OA. In addition, the therapeutic potential for OA by controlling the accumulation of lipid peroxidation and inhibiting chondrocyte ferroptosis are discussed.
Similar content being viewed by others


Facts
-
Lipid peroxidation contributes to OA progression.
-
The lipid peroxidation products 4-HNE and MDA are closely associated with OA pathogenesis.
-
Inflammatory mediators, ferroptosis inducers, mechanical overloading, and iron overload increase the accumulation of ROS and lipid peroxidation in chondrocytes, which can lead to chondrocytes ferroptosis and might contribute to the pathogenesis.
-
Inhibiting lipid peroxidation and ferroptosis by antioxidants might provide a novel therapy for OA.
Open questions
-
What are the underlying mechanisms that lipid peroxidation is involved in the progression of OA?
-
What is the specific role of 4-HNE, MDA, and ferroptosis in OA?
-
What are the links between GPX4, iron homeostasis, and IL-β in chondrocyte ferroptosis?
-
Are there any inducers or inhibitors that can target ferroptosis for the treatment of OA?
Introduction
Osteoarthritis (OA) is the most common joint disease; more than 303 million people worldwide suffer from OA [1]. OA is becoming more prevalent and burdensome, especially in the elderly population [2]. The current treatment of OA involves disease education, guidance for sports activities, medication, and surgical treatment [3]. Although pharmacotherapy can alleviate or control pain and inflammation of OA, it fails to reverse the disease progression and may produce side effects during chronic administration. Joint replacement surgery has been a routine treatment for end-stage OA, but the timing of surgery is crucial because joint replacements will not survive forever [4]. There are many known risk factors for OA such as aging, gender, obesity, genetics, injury, and abnormal loading [5]. The pathogenic mechanisms caused by these risk factors are poorly understood. However, oxidative stress plays a critical role and has been highlighted in these pathologic processes of OA [6, 7].
Oxidative stress represents an abnormal condition that reactive oxygen species (ROS) production and ROS elimination are unbalanced due to an increase in ROS [8]. Excessive ROS can attack proteins, DNA, and lipids such as polyunsaturated fatty acids, which is an important component of cellular membranes [9].
Lipid peroxidation is a specific process of lipid oxidation, which received more attention for its involvement in various and numerous pathological states [10]. Lipid peroxidation includes three different mechanisms: (1) enzymatic oxidation, (2) nonenzymatic free radical-mediated oxidation, and (3) nonenzymatic free radical-independent oxidation [11]. Free radicals can attack lipids that contain carbon-carbon double bond(s), such as polyunsaturated fatty acids (PUFAs), and generate lipid peroxidation products [12]. Lipid hydroperoxides (LOOH) are primary products, which can be degenerated into secondary aldehydes such as MDA, propanal, hexanal, and 4-HNE [12]. In this review, we focused on the role of free radical-mediated peroxidation of lipids and its products in OA.
Ferroptosis is a form of recently discovered cell death, which occurs with iron and ROS dependence, characterized by lipid peroxidation [13, 14]. A growing amount of studies have demonstrated that ferroptosis participates in the progression of many different diseases such as cancer [15], Alzheimer’s disease [16], Parkinson’s disease [17], traumatic brain injury [18], rheumatoid arthritis (RA) [19], ischemia-reperfusion injury [20] and intervertebral disc degeneration [21]. Recently, our group first reported the role of chondrocyte ferroptosis in the progression of OA [22]. In addition, several studies reconfirmed chondrocyte ferroptosis existing in OA and provided some potential therapeutic strategies [23,24,25,26].
Therefore, we will review the role of ROS-induced lipid peroxidation in chondrocytes and its decomposition end products 4-HNE and MDA in OA. Moreover, we will discuss the therapeutic potential of regulating lipid peroxidation and chondrocyte ferroptosis for OA.
Oxidative stress and lipid peroxidation
Oxidative stress is a concept for research in redox biology and medicine, and it results from the increased levels of ROS and reactive nitrogen species changing the normal redox status [27, 28]. ROS is a term that describes chemically reactive chemicals that contain oxygen [29]. Generally, ROS includes free radicals and non-radicals. Free radicals usually include superoxide radicals (O•2–), hydroxyl radicals(•OH), hydroperoxyl radicals (HOO•), alkoxy radicals (RO•), and peroxyl radicals (ROO•) [28]. The non-radicals include hydrogen peroxide (H2O2), ozone (O3), and nitric oxide (NO) [30]. Free radicals are very unstable (contain at least one unpaired electron in its outer orbital) and can attack biomolecules such as DNA, proteins, and lipids and cause cellular injury [30]. Among all the free radicals, O•2− is the most abundant, and •OH is the most harmful [30]. The mitochondria often act as the most source of ROS for their containing of the respiratory chain and utilizing most of the intracellular oxygen [31]. Besides, the other sources of endogenous ROS include the protoplasm (NADPH oxidases, hemoglobin, riboflavin), the endoplasmic reticulum (Cytochromes P450 and b5), the peroxisomes (oxidases, flavoproteins), and lysosomes (myeloperoxidase, metal ions) [32].
Lipid peroxidation is a typical biologically relevant free radical chain reaction [33]. Like other chain reactions, the phases of lipid peroxidation include initiation, propagation, and termination. In the initiation step, free radicals such as •OH abstract hydrogen from lipids and form lipid radicals (L•) [12]. In the propagation phase, a lipid radical reacts with oxygen, producing a lipid peroxyl radical (LOO•) that may interact with another lipid (L•), making a LOOH and another lipid radical (L•) that can repeat the cycle [34]. In the termination phase, the propagation stops by antioxidants or lipid peroxyl radicals interacting with other radicals to produce a stable nonradical.
Lipid peroxidation can generate a wide array of peroxidation products, including primary products and secondary products. LOOH are the most abundant primary products [12] and the decomposition of LOOH can generate reactive carbonyl species (RCS), such as short-chain carbonyl derivatives and oxidized truncated phospholipids [35]. RCS are more stable and toxic than free radicals because they can attack biomolecule targets (proteins, DNA, and aminophospholipids) far from their site of formation [36]. The short-chain carbonyl derivatives include a variety of lipid aldehydes, such as α, β-unsaturated aldehydes, di-aldehydes, and ketoaldehydes [35, 36]. Among these, the α, β-unsaturated aldehyde 4-hydroxy-2-nonenal (4-HNE) and di-aldehyde malondialdehyde (MDA) are most studied (Fig. 1).

Free radical-mediated oxidation lipid peroxidation includes three steps: initiation, propagation, and termination. In the initiation phase, ROS/ RNS free radicals react with PUFAs and abstract an allylic hydrogen thus forming lipid radicals. In the propagation phase, the lipid radicals react with oxygen and form lipid peroxyl radicals, and then lipid peroxyl radicals react with PUFAs to form new radicals and lipid hydroperoxides. In the termination phase, the propagation can be blocked by antioxidants or lipid radicals by donating a hydrogen atom to lipid peroxyl radicals resulting in the formation of nonradical products. Due to the instability of lipid hydroperoxides, they may degrade into secondary products. 4-HNE and MDA are the main products among the many reactive aldehydes.
The reaction between RCS and biomolecules, called lipoxidation, can generate a variety of adducts collectively called advanced lipoxidation end products (ALEs) [37]. In addition to acting as biomarkers of oxidative damage, ALEs also have a role in the pathological processes of some oxidative-based diseases [38]. Among them, 4-HNE-modified proteins [39] and MDA-modified proteins [40] are the most commonly studied.
Many studies have demonstrated that lipid peroxidation is involved in different diseases and disorders such as cardiovascular diseases [35], cancer [41], neurodegenerative diseases [42], and diabetic retinopathy [28]. In addition, lipid peroxidation products such as 4-HNE [43], MDA [44], and isoprostanes [45] are widely used as biomarkers of lipid peroxidation.
Pathological aspects of lipid peroxidation in OA
During the past two decades, mounting studies have demonstrated that lipid peroxidation has a role in the onset and progression of OA. Clinical studies have shown that lipid peroxidation levels are increased in OA patients. A previous study reported significantly increased lipid peroxidation (measured by 4-HNE and MDA) in the synovial cells of OA than in RA patients and controls [46]. Moreover, MDA and 4-HNE protein adducts were also identified in OA tissue sections by using immunohistochemical methods [47]. As is well known, isoprostanes can serve as reliable markers of lipid peroxidation [48]. Basu et al. [49] reported that the levels of 8-isoprostaglandin F2α (one of the major isoprostanes) in serum and synovial fluid were higher in OA patients compared to control subjects. Moreover, Franz and his colleagues [50] also found increased levels of 8-isoprostane F2α in both synovial fluid and serum from the OA group. The studies suggest that lipid peroxidation is increased in OA.
Lipid peroxidation damage in OA
Until now, the specific mechanisms of how lipid peroxidation is involved in the development of OA have not been completely clarified. Cartilage degeneration is one of the most prominent features of OA. A previous study from Tiku et al. [51] showed that chondrocyte lipid peroxidation may contribute to cartilage aging and OA. Furthermore, they demonstrated that chondrocyte lipid peroxidation contributed to the oxidation and degradation of collagen, which in turn may aggravate OA by increasing the sensibility of cartilage to mechanical fatigue [51]. Recently a study found that lipid peroxidation could regulate chondrocyte mitochondrial dynamics and cartilage injury response [52]. Animal experiments also demonstrated that lipid peroxidation has a role in the pathogenesis of the early stages of OA. Zubavlenko et al. [53] have observed the activation of the lipid peroxidation process in posttraumatic osteoarthrosis (PTOA) rat models. Moreover, Gladkova et al. [54] also found that increased production of primary and intermediate lipid peroxidation products (hydroxyl radicals, MDA) in PTOA rats. Previous studies have reported that lipid peroxidation has a role in osteoporotic bone loss [55] and synovial inflammation [56]. However, there is not much research directly examining the role of lipid peroxidation in the subchondral bone remodeling, synovial inflammation of OA. In order to introduce the role of lipid peroxidation in OA, we reviewed existing research focused on lipid peroxidation-derived products and lipid peroxidation-induced ferroptosis.
Lipid peroxidation-derived aldehydes in OA
As the most abundant and toxic lipid peroxidation-derived aldehyde, 4-HNE has been shown to be involved in the development of many disorders such as cardiovascular diseases [57], neurological diseases [58], inflammatory bowel disease [59], diabetes [60], RA [61], and different types of cancer [62,63,64]. Furthermore, 4-HNE can react with macromolecules (proteins, lipids, and nucleic acids) and regulate various signaling pathways such as Nrf2, NF-κB, PKC and ERK [65]. In addition, 4-HNE has a role in numerous cell processes, including but not limited to oxidative stress [66], inflammation [67], cell proliferation [68] and cell death [69].
MDA is usually used as a biomarker for lipid peroxidation assessment [44]. Moreover, MDA can react with amino acids and yield different adducts such as MDA-lysine [40], and MDA-glycine [70]. Moreover, the MDA-acetaldehyde protein adduct termed malondialdehyde–acetaldehyde (MAA) has been reported to have a role in several diseases, like pancreatitis [71], atherosclerosis [72], RA [73], and inflammatory bowel disease [74]. Following, we will discuss the role of lipid peroxidation-derived 4-HNE and MDA in the development of OA.
Effects of 4-HNE on cartilage homeostasis and subchondral bone
4-HNE is one of the secondary metabolites of oxidation of PUFAs, particularly omega-6 fatty acids such as arachidonic acid and linoleic acid [75]. As the most cytotoxic aldehyde [76], 4-HNE is an α, β-unsaturated aldehyde and a highly reactive lipid-derived electrophile [77]. Studies have shown that 4-HNE and 4-HNE-modified proteins are closely associated with cartilage homeostasis by affecting the anabolic and catabolic processes of the chondrocytes. Morquette et al. [78] reported that the level of 4-HNE was increased in OA cartilage. 4-HNE could induce transcriptional and post-translational modifications of collagen II and matrix metalloproteases 13 (MMP13) [78]. At mRNA levels, 4-HNE inhibited collagen II expression but induced MMP13 expression [78]. At the post-translational level, 4-HNE accelerated collagen II degradation and activated MMP13, possibly by post-translational modification [78]. Results from another study indicated that 4-HNE-binding to collagen II could lead to multiple abnormalities of chondrocyte phenotype and function, suggesting it may contribute to interrupting the extracellular matrix (ECM) homeostasis [79].
Moreover, studies reported that 4-HNE plays a role in inflammatory responses and apoptosis of osteoarthritic chondrocytes. Vaillancourt et al. [80] reported that 4-HNE could upregulate cyclooxygenase-2 (COX-2) via activating ATF/CRE and inhibit inducible nitric oxide synthase (iNOS) via inactivating NF-κB in OA chondrocytes. They also found that 4-HNE production in OA articular tissues contributed to inflammatory responses by upregulating COX-2 expression and limiting the magnitude of transcriptional expression of iNOS [80]. Apoptosis is a form of classical programmed cell death, and chondrocyte apoptosis has been proven to have a promoting role in OA [81]. ROS-mediated oxidative damage, inflammatory stimulus, and apoptosis are all involved in cartilage degeneration and the progression of arthritis [82]. Lipid peroxidation is a process of the oxidative degradation of lipids caused by free radicals, characterized by formatting lipid peroxides, and participates in different types of cell death [83]. In their later study, Vaillancourt and his colleagues demonstrated that 4-HNE was cytotoxic and could induce apoptosis in human osteoarthritic chondrocytes [84]. Moreover, 4-HNE could suppress pro-survival Akt kinase activity yet induce Fas/CD95 and p53 expression in chondrocytes [84]. However, GSH-S-transferase A4-4 knockdown could augment 4-HNE cytotoxicity, and its overexpression could obviously inhibit 4-HNE-induced death of chondrocytes [84]. In an animal experiment, Shi et al. [85] demonstrated that carnosine could abolish 4-HNE production and attenuate cartilage degeneration in the dog model of OA. Aldehyde dehydrogenase 2 (ALDH2) is an important enzyme in the detoxification of 4-HNE [86]. Ausra et al. [87] showed that ALDH2 may play a protective role in human OA chondrocytes. Moreover, a recent study demonstrated that ALDH2 could alleviate MIA‑induced oxidative stress, inflammation and apoptosis in chondrocytes via inhibiting aquaporin 4 expression [88].
In addition, 4-HNE binding to proteins was reported to disturb the phenotype and function of chondrocytes [89]. In a recent study, Geib et al. [89] identified 4-HNE-modified proteins such as 4-HNE-modified histones, H2A and H2B, and histone deacetylase in chondrocytes of OA patients via anti-4-HNE antibodies. Moreover, Niu et al. [90] recently found that 4-HNE has a role in dampening chondrogenesis. Their results demonstrated that 4-HNE could form adducts with SOX9 and lead to its ubiquitin-mediated degradation [90].
Subchondral bone is an essential component of the joint, and the subchondral bone remodeling dominated by osteoclasts and osteoblasts can reflect the OA process [91, 92]. Data from a study showed that 4-HNE/protein adduct levels were higher in OA osteoblasts than in normal osteoblasts and when OA osteoblasts were treated with hydrogen peroxide [93]. The results showed that 4-HNE could increase osteocalcin and type I collagen synthesis while inhibiting alkaline phosphatase activity [93]. In addition, 4-HNE could change the COX-2 and interleukin-6 (IL-6) expression by affecting their production signaling pathways in osteoblasts [93]. 4-HNE inhibited IL-6 expression while inducing prostaglandin E2 (PGE2) release and COX-2 expression [93]. In human OA osteoblasts, 4-HNE could activate p38 MAPK, ATF-2/CREB and JNK1/2, but inhibit NF-κB signal [93]. Furthermore, 4-HNE modulated IL-6 and PGE2 expression via IKKα and p38 MAPK signaling pathways, respectively [93]. Thus, 4-HNE may contribute to OA development by changing the phenotypic and function of osteoblasts in OA subchondral bone.
The results mentioned above confirm that lipid peroxidation exists in OA; in addition, 4-HNE/4-HNE-modified proteins may have a valuable and attractive role in exploring the pathophysiology of OA. More studies are warranted to explore the impact of 4-HNE on cartilage homeostasis and the therapeutic potential by scavenging 4-HNE in OA.
Effects of MDA on cartilage collagen oxidation
MDA is another main second product of lipid peroxidation, with the formula of CH2(CHO)2, usually used as a marker of oxidative stress [94]. Tiku et al. [95] demonstrated that MDA mediates the oxidation of cartilage collagens. A previous study found that serum MDA concentration and serum C2C concentration (a marker of collagen type II degradation) were significantly increased in ACLT-induced OA dogs [96]. Moreover, the levels of MDA and C2C continuously increased during the whole process of the experiment, and MDA levels had a high positive correlation with C2C [96]. Watari et al. [97] observed that the serum levels of MDA, c-telopeptide degradation products of type II collagen (CTX-II), carboxy propeptide of type II collagen (CPII) were higher in the STR/Ort (Str) mice than in CBA mice. They also found that the level of MDA was correlated with CTX-II, but not with CPII [97].
Several studies reported that the levels of MDA in plasma were markedly higher in OA patients than in healthy controls [98,99,100]. Furthermore, results showed that the MDA concentrations in synovial fluid were higher compared to paired plasma samples [98]. Contrarily, a more recent study reported that there was no difference in MDA concentrations in erythrocytes and blood plasma between healthy subjects and OA patients [101]. Reasons for the different results may include the limited sample sizes, the assays for detection, different stages of the disease, and the difference between local and systemic states of oxidative stress. The association between MDA concentrations in plasma and synovial fluid in OA patients warrants further investigation. More clinical trials with larger sample sizes and combinations with other lipid peroxidation biomarkers would help to draw more solid conclusions.
As is a highly reactive aldehyde, MDA can combine acetaldehyde (AcA) to form MAA or the MAA adduct [102]. AcA is derived from exogenous sources or the breakdown of MDA [103]. MAA-adducts that are derived from the reaction of MAA and proteins have been proven to be immunogenic and possess proinflammatory and profibrogenic properties [104]. Moreover, studies have demonstrated that MAA-adducts may play a role in several diseases, especially alcoholic liver injury and atherosclerosis [104, 105]. In addition, MDA modifications and anti-MDA-modified protein autoreactivity have been reported to play a role in RA [106]. In RA, MDA-related antigens and MDA reactive antibodies may have a role in the synovial pathogenesis of RA [106]. Moreover, hypermutated anti-MDA IgG clones could enhance osteoclastogenesis and may lead to joint destruction [106]. In a more recent study, Sakuraba et al. [107] also found that anti-MDA antibodies have a role in accelerating bone erosion in RA via inducing glycolysis and lipid biosynthesis of osteoclasts. Furthermore, methotrexate (MTX), the immunosuppressant commonly used for the treatment of RA, has been demonstrated that could reduce inflammation and subsequent tissue damage by inhibiting MAA-adducts formation and scavenging superoxide in RA [108].
However, studies focused on the role of MAA-adducts and anti-MDA antibodies in OA are lacking. In a previous study by Mikuls et al. [102], the serum levels of IgA anti-MAA and IgG anti-MAA were higher than health controls. Up to now, the research focus of MAA and MAA-adducts has been mainly on inflammatory diseases or autoimmune diseases. Nevertheless, with more and more in-depth research on lipid peroxidation in OA, the roles of MDA, MAA, or MAA-adducts may become increasingly important.
Lipid peroxidation-driven chondrocyte ferroptosis in OA
Ferroptosis was first reported by Dixon et al. [13] in 2012, and they identified it as a unique form of cell death caused by iron overload and lipid peroxidation, different from apoptosis and autophagy. The inactivation of antioxidant systems such as glutathione peroxidase 4 (GPX4), glutathione (GSH), and coenzyme Q10 (CoQ10) system promotes ferroptosis [109]. Ferroptosis is characterized by ROS generation, GPX4 depletion, LOOH accumulation, and iron overload [14]. Recent studies have demonstrated that these features, which can initiate ferroptosis, are accompanied by the pathogenesis of OA [22, 23, 110].
Our group first demonstrated that chondrocyte ferroptosis have an important role in the progression of OA. We found that both interleukin-1 beta (IL-1β) and ferric ammonium citrate (FAC) could induce ROS, lipid ROS accumulation, and ferroptosis-related proteins (GPX4, ACSL4, P53, and SLC7A11) expression changes in chondrocytes [22]. Following our study, many investigations have shown the involvement of chondrocyte ferroptosis in the development of OA and provided potential therapeutic strategies targeting lipid peroxidation and ferroptosis [23, 24, 111]. These studies are summarized in Table 1.
GPX4-dependent chondrocyte ferroptosis in OA
GPX4 has recently attracted more attention in OA due to its ability to eliminate lipid peroxides and inhibit ferroptosis [23]. System Xc−/GSH/GPX4 pathway is the most studied pathway in OA. Miao et al. [23] found that GPX4 could regulate ferroptosis in chondrocytes; on the other hand, GPX4 could regulate ECM degradation via MAPK/NF-κB signaling pathway [23]. The results enrich research on GPX4 and chondrocyte ferroptosis and provide a new idea for OA treatment.
Moreover, recent studies have demonstrated that IL-1β treatment could promote ferroptosis and decrease the level of GPX4 protein in chondrocytes [22, 112]. Lv and his colleagues reported that the RNA-binding protein SND1 promoted ferroptosis in IL-1β-treated chondrocytes by affecting the HSPA5-GPX4 axis [112]. The results provide new insights into the mechanisms by which IL-1β induces chondrocyte ferroptosis.
In another study, researchers found that transient receptor potential vanilloid 1 (TRPV1) could prevent chondrocyte ferroptosis by upregulating the expression of GPX4 [113]. In the primary chondrocytes, Trpv1 activation could restore TBHP-induced Gpx4 decrease, while Trpv1 activation substantially restored.
As is well known, exposure of chondrocytes to excessive mechanical loading accelerates the development of OA [114]. Piezo-type mechanosensitive ion channel component 1 (Piezo1) is one of the piezo channels present on the cell membrane and works as a mechanotransducer [115]. A recent study reported that mechanical overloading could induce GPX4-regulated chondrocyte ferroptosis in OA Piezo1 channel facilitated calcium influx [111]. Ferroptosis suppressor protein (FSP-1) and coenzyme Q10 (CoQ10) treatment could attenuate the progression of OA in GPX4-deficient mice [111]. Taken together, all results observed in mentioned studies indicate a new underlying mechanism of mechanical overloading to the progression of OA, that is, the GPX4-associated ferroptosis pathway (Fig. 2).

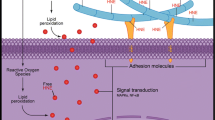
OA osteoarthritis, 4-HNE 4-hydroxy-2-noneal, MDA malondialdehyde, GPX4 glutathione peroxidase 4. Association of lipid peroxidation and its secondary products 4-HNE and MDA with OA, cartilage catabolism and inflammation, subchondral bone metabolism, chondrocyte apoptosis, chondrocyte ferroptosis.
GPX4-independent chondrocyte ferroptosis in OA
Iron homeostasis is essential for joint health, and iron overload in joints has proved to be strongly associated with the pathogenesis of OA [110]. Several studies have demonstrated that iron overload can cause oxidative stress damage and induce apoptosis [116] in chondrocytes. Jing et al. [117] demonstrated that iron overload could cause cartilage degeneration by inducing oxidative stress and mitochondrial dysfunction [117]. In addition, FAC treatment could greatly promote the expression apoptosis marker of cleaved caspase‑3 in chondrocytes [118]. In another study, Karim et al. [116] also found that high doses of FAC treatment induced the generation of ROS, inhibited collagen II syntheses, and facilitated the apoptosis of chondrocytes.
Excessive iron ions can induce membrane lipid peroxidation via Fenton reaction and cell damage. FAC has been shown to have a role in inducing ferroptosis in chondrocytes in our previous study [22]. We found that FAC could significantly increase the level of ROS and lipid ROS in chondrocytes [22]. Moreover, FAC treatment promoted the expression of P53 and ACSL4 while inhibiting GPX4 and SLC7A11 in chondrocytes [22]. In addition, Pan et al. [119] found that FAC and IL-1β treatment elevated levels of ROS and LPO in chondrocytes. Not only that, the levels of LPO biomarker MDA were greatly increased in chondrocytes treated with FAC and IL-1β [119].
IL-1β is a commonly used proinflammatory cytokine to establish in vitro model of OA [120]. The ability of IL-1β to induce chondrocyte catabolism [121] has been well known for a long time, but it is only recently that IL-1β has been shown to disrupt chondrocyte iron homeostasis [117] and trigger chondrocyte ferroptosis [22, 122]. Jing et al. [117] found IL- 1β could promote iron influx and cause chondrocytes iron overload. Meanwhile, excess iron could increase matrix metalloproteinases (MMPs) expression induced by IL- 1β [117]. Moreover, several studies recently reported the role of IL- 1β in inducing ferroptosis in chondrocytes [26, 119, 122]. Although IL-1β-mediated chondrocyte ferroptosis was observed in the aforementioned studies, the specific mechanisms are unclear. In addition to increased ROS and lipid ROS [122], the levels of intracellular iron can also be augmented after IL- 1β treatment in chondrocytes. Mo et al. [26] found that the concentration of Fe2+ increased in ATDC5 cells after stimulation with IL‐1β. In addition, Wang et al. [122] observed that IL-1β could increase the intracellular iron and mitochondrial iron levels in chondrocytes. Recently, we have demonstrated nuclear receptor coactivator 4 (NCOA4) could aggravate OA by promoting chondrocyte ferroptosis and ferritinophagy [123]. We found IL-1β could disturb chondrocyte iron homeostasis via JNK-CJUN-NCOA4 [123]. The role of NCOA4-mediated ferritinophagy in chondrocytes gives us further insight into the connection between IL-1β and ferroptosis in OA pathogenesis. Thus, IL-1β induced iron metabolism disorders may be responsible for chondrocyte ferroptosis and cartilage degradation in OA cartilage.
As an effective iron chelator, deferoxamine (DFO) can chelate free iron and is used to relieve the damage of iron overload. Previous research demonstrated that DFO could strongly prevent the formation of free radicals caused by IL-1α treatment in synovial fluid of temporomandibular joint arthritis in rats [124]. Another study showed that DFO could suppress excessive collagenase-mediated type II collagen cleavage and protease, production of cytokine, and COL10A1 expression while upregulating adenosine monophosphate-activated protein kinase and Krebs cycle genes in human osteoarthritic cartilage [125]. In more recent studies, researchers found that DFO could promote recovery of traumatic spinal cord injury [126] and slow the process of disc degeneration [21] by inhibiting ferroptosis in animal models. Moreover, Lin et al. [127] also found that DFO could alleviate iron overload-induced ECM degradation in FVIII-deficient hemophilic mice. In addition, our group and other researchers have recently demonstrated that DFO could alleviate OA by inhibiting chondrocyte ferroptosis [23, 128]. More details about DFO are summarized in Table 1.
Lipid peroxidation and ferroptosis promote bone loss in subchondral bone
The modification of subchondral bone, including early-stage bone loss, late-stage bone sclerosis and histopathological alterations, plays an important role in the pathogenesis of OA [129]. Recently studies have reported the role of iron accumulation and ferroptosis in bone loss. He et al. [130] recently find that subchondral bone loss in the iron-overload-induced model of knee OA. The mechanisms of iron-overload-induced bone loss mainly include the increased osteoclastogenesis and bone resorption capacity of osteoclasts [131], lipid peroxidation accumulation and ferroptosis in osteoblasts [132]. Moreover, studies have demonstrated that iron chelator DFO [133] and antioxidant N-acetylcysteine (NAC) [130] could save bone loss induced by iron accumulation. These results suggest that ferroptosis have a role in OA subchondral bone homeostasis and structural integrity. Therefore, targeting iron metabolism disorders, lipid peroxidation accumulation, and osteoblast ferroptosis in subchondral bone may provide new therapeutic strategies for OA.
The role of ferroptosis in osteoarthritis synovial membrane
As a degenerative disease of the whole joint, OA affects articular and periarticular tissues such as synovium. Moreover, synovial inflammation is a critical OA-related pathological change. Previous studies have reported that iron overload could promote synovial inflammation and synovial hyperplasia in joint diseases such as hemophilic arthropathy [110]. Although increasing attention has been given to the role of ferroptosis in OA to date, most researchers pay more attention to chondrocyte ferroptosis and cartilage degeneration rather than synovitis. Until recently, efforts in synoviocyte ferroptosis research have focused mainly on RA [134]. However, some investigators started exploring the role of ferroptosis in the synovial tissue of OA [135, 136]. By using bioinformatics analysis Xia et al. identified and verified ferroptosis-related genes (ATF3, IL-6, IL-1B and EGR1) in the synovial tissue of OA, which were possibly associated with synovial hyperplasia [135]. Notably, in addition to synoviocytes, synovial macrophages also have an important role in synovial inflammation of OA [137]. Therefore, future studies could focus on the role of synoviocytes and macrophages ferroptosis in OA synovial membrane, which may provide a new direction for future studies on OA.
Antioxidants
Antioxidants are reducing substances that inhibit oxidation and reduce the occurrence of multiple disorders [138]. The above studies have demonstrated the critical role of excessive ROS and lipid ROS in OA progression. Therefore, antioxidants may be valuable therapeutics for OA.
Enzymatic antioxidants
The antioxidative defense grid of the human body includes an array of enzymatic and nonenzymatic antioxidants [139]. The enzymatic antioxidative defense system commonly includes superoxide dismutases (SODs), catalase (CAT), and GPX. In addition, the aldehyde dehydrogenases and GSH-S-transferase are also important aldehydes detoxifying enzymes [86].
SODs are metalloenzymes that require a metal cofactor for their activity. By catalyzing the conversion of superoxide anion to hydrogen peroxide and oxygen, SODs can eliminate the damage of ROS. A previous study reported the levels of SOD family members (SOD1, SOD2, and SOD3) were drastically lower in OA cartilage than in macroscopically normal cartilage, especially SOD2 [140]. Depletion of SOD2 in chondrocytes gave rise to increased ROS levels yet decreased collagenase expression [140]. Besides, the decrease in the SOD activity in end-stage osteoarthritic synovium and cartilage was also reported in a recent study [141]. These studies showed that the total SOD activity was decreased and SOD down-regulation in cartilage may be involved in the development of OA. However, whether SOD can be a useful biomarker or a therapeutic target for OA are unknown.
Catalases can protect cells from oxidative damage by degrading hydrogen peroxide to water and oxygen (2H2O2 --> 2 H2O + O2) [142]. CAT is one of the antioxidant enzymes which can protect against free radical attack [139]. Researchers found that expression of mitochondrially targeted CAT promoted Akt phosphorylation and attenuated phosphorylation of proapoptotic signaling proteins under conditions of oxidative stress in human articular chondrocytes [143]. In addition, transgenic mice that express human catalase targeted to the mitochondria exhibited less severity of age-related OA compared to WT mice [143]. A study reported that CAT activity was markedly higher in OA cartilage compared to the healthy control group [144]. As an important antioxidant enzyme, CAT has a role in protecting chondrocytes by reducing TNF-α-induced apoptosis [145]. The CAT activity increase in OA patients may attribute to the compensating protective role of CAT.
GPXs can catalyze the reduction of hydrogen peroxide or organic hydroperoxides to water or corresponding alcohols with the help of GSH [146]. Mammalians possess four major selenium-dependent GPX isozymes (GPX1, GPX2, GPX3, and GPX4) that can protect against oxidative stress [147]. A previous study reported that activities of antioxidant enzymes such as SOD, CAT, GPX, GSH reductase and glutathione-S-transferase were significantly increased in knee OA synovial fluid [148]. GPX1 has a role in various cellular processes, such as anti-oxidation, anti-apoptosis, and regulation of cell differentiation [149]. Results showed that the expression of GPX1 considerably decreased in OA tissues and cells [149]. Meanwhile, GPX1 overexpression constrained oxidative stress and apoptosis in OA chondrocytes by regulating CREB/HO-1 [149]. GPX4 is an important enzyme that can reduce esterified phospholipid hydroperoxides and regulate ferroptosis [150]. Zhang et al. [151] demonstrated that homocysteine could enhance GPX4 methylation, thus inducing ferroptosis in the nucleus pulposus. Moreover, they found that folic acid could repress ferroptosis in nucleus pulposus cells by downregulating the methylation level of GPX4 [151]. Moreover, as a key regulator of ferroptosis, GPX4 and its role in OA have been demonstrated in three recently published studies [22, 23, 111]. First of all, GPX4 expression in the damaged cartilage of OA patients was markedly decreased compared to in undamaged cartilage [23]. Secondly, the key inflammatory cytokine in OA IL-1β could suppress the expression of GPX4 in chondrocytes [22]. Thirdly, GPX4 levels decreased in cartilage from the OA animal models, and knockout of GPX4 exacerbated mice experimental OA process [111]. Taken together, the protective role of GPX4 in OA is worth an in-depth study and may provide a novel insight into the molecular mechanism and potential treatment of OA.
Nonenzymatic antioxidants
The nonenzymatic antioxidant system consists of GSH, vitamin C, vitamin, and CoQ10. As a natural tripeptide, GSH consists of glutamic acid, cysteine and glycine, and it mainly exists in the forms of reduced GSH and oxidized glutathione (glutathione disulfide (GSSG)), and glutathione-protein mixed disulfides [152, 153]. GSH can prevent lipid peroxidation caused by toxic oxygen radicals by reducing the endogenously produced hydrogen peroxide with the help of GPX. In this process, the oxidation and reduction process between GSH and GSSG forms a redox cycle [154]. Moreover, GSH can detoxify 4-HNE by forming GS-4-HNE adducts or serving as a cofactor for alpha-class glutathione transferases (GSTAs) [155].
In OA, GSH was reported to enhance the antioxidative capacity of hyaluronic acid and modulate the expression of proinflammatory cytokines in human fibroblast-like synoviocytes [156]. Furthermore, GSH has a role in augmenting the effect caused by hyaluronic acid on OA patients [156]. Moreover, Zhu et al. [157] reported that GSH can act as a mediator of cartilage oxidative stress resistance and resilience during aging and OA. They found different stressors such as aging, inflammation, biomechanical loading, and pro-oxidant stimuli could affect GSH content and redox balance in experimental models of OA-related stress [157]. In addition, a recently published review introduced the protective role of GSH and its precursor NAC in OA [158]. Beyond that, as the reducing substrate of GPX4 activity, GSH plays an essential in inhibiting ferroptosis [159]. Therefore, regulation of GSH levels may be an effective approach to inhibit chondrocyte ferroptosis and may be one of the therapeutic strategies for OA.
Vitamin C and Vitamin E are both important nonenzymatic antioxidants that help reduce free radical damage, and act as nutritional supplements for OA treatment. A previous study reported that vitamin C could prevent monosodium iodoacetate (MIA) induced changes such as cell growth inhibiting and oxidative stress increasing, apoptosis, MMPs increasing, and proteoglycan loss in a chondrosarcoma cell line (SW1353) [160]. In addition, supplement with vitamin C could mitigate MIA-induced OA in rats [160]. Results from another study demonstrated that vitamin C could attenuate senescence of human osteoarthritic osteoblasts [161].
Vitamin E is a lipid-soluble essential micronutrient with a powerful antioxidant effect. A study reported the synovial fluid vitamin E concentration considerably decreased in OA patients [162]. Another research reported that Vitamin E could protect the cartilage matrix by preventing chondrocyte lipid peroxidation [51]. Furthermore, Vitamin E protected rat mesenchymal stem cells (MSCs) against hydrogen peroxide-induced oxidative stress in vitro and improved the therapeutic potential of MSCs in the surgically-induced rat model of OA [163]. Another study reported that Vitamin E ameliorated alterations to the cartilage of knee joints induced by monoiodoacetate and diabetes mellitus in rats, which manifested as inhibition of biomarkers of inflammation [164]. A clinical trial also reported a positive effect of Vitamin E in improving clinical symptoms and reducing oxidative stress conditions in patients with late-stage knee OA [165]. Interestingly, vitamin E could also prevent lipid peroxidation and inhibit ferroptosis [166, 167].
Although much research has reported the therapeutic value of Vitamin C or Vitamin E in preventing OA, negative results [168, 169] were also reported. However, more randomized placebo-controlled trials are desperately needed to verify the therapeutic efficiency of Vitamin C or Vitamin E for OA treatment.
CoQ10 is a crucial component of the electron transport chain in mitochondria and participates in cellular energy metabolism [170]. CoQ10 is a powerful antioxidant that inhibits lipid peroxidation and scavenges free radicals [171,172,173], and it also shows anti-inflammatory effects [174]. It has been reported that combined administration of CoQ10 and MTX suppressed adjuvant-induced arthritis progression in rats more effectively than did MTX alone [175]. CoQ10 could potentiate both the antiarthritic and the antioxidative effect of MTX, evident by the decreased levels of protein carbonyls in plasma and levels of 4-HNE adducts and MDA adducts to plasma proteins [175]. Moreover, results from a study showed that CoQ10 could improve pain and cartilage degradation in MIA-induced OA rats [176]. In a more recent study, researchers encapsulated CoQ10 into micelles and found that CoQ10-micelles had a better chondroprotective effect than CoQ10 in OA rats [177]. Moreover, CoQ10-micelles could decrease the expression levels of catabolic and necroptotic factors in human OA chondrocytes [177]. Another study also reported the anticatabolic and cartilage protective potential in rat chondrocytes [178]. In addition, CoQ10 is essential for the health of nearly all human tissues, especially for organs with high energy demands, such as skeletal muscle [179]. A clinical study showed that CoQ10 level was positively associated with antioxidant capacity, muscle mass, muscle strength and muscle endurance in OA patients [180]. Raising the level of CoQ10 in OA patients may improve their antioxidant capacity and muscle function [180]. CoQ10 is not only an essential scavenger of ROS but is also involved in the FSP1-CoQ10-NAD(P)H pathway that has recently been reported to inhibit ferroptosis [181, 182]. Thus, based on the above evidence, CoQ10 might represent a new therapeutic modality for OA (Fig. 3).

Extracellular stimuli such as inflammatory mediators, ferroptosis inducers, oxidative stress, mechanical overloading, or iron overload can elevate ROS levels and lipid peroxidation in chondrocytes and can ultimately contribute to OA progression. Inflammatory mediator IL-1β can increase the levels of ROS, lipid ROS, and the lipid peroxidation marker MDA in chondrocytes. Classical ferroptosis inducer erastin can induce chondrocyte ferroptosis by inhibiting systemic Xc-. Moreover, mechanical overloading can promote ferroptosis through Piezo1 activation and subsequent calcium influx in chondrocytes. Accumulated iron in chondrocytes can catalyze the formation of ROS, which further forms the lipid radical and leads to lipid peroxidation. Chondrocyte ferroptosis and lipid peroxidation end product 4-HNE can cause ECM degradation by breaking the equilibrium between synthesis and breakdown of extracellular matrix. Antioxidants including vitamin C, vitamin E, NAC, and CoQ10 can reduce lipid peroxidation and the formation of ROS. Iron chelator DFO can inhibit erastin-induced articular chondrocyte death, and delay articular cartilage degradation and OA progression. And the ferroptosis inhibitor Fer-1 can rescue the IL-1β–induced decrease in collagen II and increase in MMP13 expression. Antioxidant enzyme GPX4 can prevent chondrocyte ferroptosis by combating lipid peroxidation.
Conclusions and perspectives
Lipid peroxidation plays an important role in the development of OA. 4-HNE and MDA not only act as the main biomarkers for lipid peroxidation assessment and contribute to multiple pathological processes during OA development. The role of 4-4-HNE in cartilage catabolism, chondrocyte apoptosis, inflammation, and osteoclasts activity alteration suggests that 4-HNE may be a potential target in OA. Moreover, recently identified 4-HNE-modified proteins in OA chondrocytes may provide a unique opportunity to investigate how chondrocytes adapt to oxidative stress and lipid peroxidation. Not only that, 4-HNE detoxification pathways such as GSTA4-4, ALDH2, GSTs, and GSH may open new avenues to explore novel therapy for OA. MDA may be a good marker in OA but more research will be needed to confirm the levels of it in joint fluid and reveal the relationship between articular fluid MDA concentration and serum MDA concentration. The pathophysiological role of MAA and MAA-adducts in OA may be an intriguing line of investigation for future studies. In addition, growing evidence suggests that lipid peroxidation-induced ferroptosis has an important role in cartilage degradation, subchondral bone remodeling and synovitis of OA. Therefore, inhibiting lipid peroxidation and ferroptosis in chondrocytes might provide a novel therapy for OA.
References
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–858. https://doi.org/10.1016/s0140-6736(18)32279-7.
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–59. https://doi.org/10.1016/S0140-6736(19)30417-9.
Martinez-Armenta C, Camacho-Rea MC, Martínez-Nava GA, Espinosa-Velázquez R, Pineda C, Gomez-Quiroz LE, et al. Therapeutic potential of bioactive compounds in honey for treating osteoarthritis. Front Pharm. 2021;12:642836. https://doi.org/10.3389/fphar.2021.642836.
Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr Cartil. 2020;28:242–8. https://doi.org/10.1016/j.joca.2020.01.002.
Palazzo C, Nguyen C, Lefevre-Colau MM, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. 2016;59:134–8. https://doi.org/10.1016/j.rehab.2016.01.006.
Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73–82. https://doi.org/10.1016/j.freeradbiomed.2018.08.038.
Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: role of polyphenols. Biomed Pharmacother. 2020;129:110452. https://doi.org/10.1016/j.biopha.2020.110452.
Sies H. Oxidative stress: concept and some practical aspects. Antioxidants (Basel). 2020;9:852. https://doi.org/10.3390/antiox9090852.
Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111:5944–72. https://doi.org/10.1021/cr200084z.
Guéraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, et al. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010;44:1098–124. https://doi.org/10.3109/10715762.2010.498477.
Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochem Biophys Res Commun. 2005;338:668–76. https://doi.org/10.1016/j.bbrc.2005.08.072.
Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. https://doi.org/10.1155/2014/360438.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–900. https://doi.org/10.1016/j.bbagen.2017.05.019.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:e1904197. https://doi.org/10.1002/adma.201904197.
Ashraf A, Jeandriens J, Parkes HG, So PW. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: evidence of ferroptosis. Redox Biol. 2020;32:101494. https://doi.org/10.1016/j.redox.2020.101494.
Mahoney-Sanchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog Neurobiol. 2021;196:101890. https://doi.org/10.1016/j.pneurobio.2020.101890.
Bao Z, Liu Y, Chen B, Miao Z, Tu Y, Li C, et al. Prokineticin-2 prevents neuronal cell deaths in a model of traumatic brain injury. Nat Commun. 2021;12:4220. https://doi.org/10.1038/s41467-021-24469-y.
Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation. 2021;44:35–47. https://doi.org/10.1007/s10753-020-01338-2.
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–99. https://doi.org/10.1038/s41418-019-0299-4.
Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J Cell Physiol. 2021;236:2725–39. https://doi.org/10.1002/jcp.30039.
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33–43. https://doi.org/10.1016/j.jot.2020.09.006.
Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine. 2022;76:103847. https://doi.org/10.1016/j.ebiom.2022.103847.
Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, et al. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif. 2021;54:e13134. https://doi.org/10.1111/cpr.13134.
Yan J, Feng G, Ma L, Chen Z, Jin Q. Metformin alleviates osteoarthritis in mice by inhibiting chondrocyte ferroptosis and improving subchondral osteosclerosis and angiogenesis. J Orthop Surg Res. 2022;17:333. https://doi.org/10.1186/s13018-022-03225-y.
Mo Z, Xu P, Li H. Stigmasterol alleviates interleukin-1beta-induced chondrocyte injury by down-regulatingsterol regulatory element binding transcription factor 2 to regulateferroptosis. Bioengineered. 2021;12:9332–40. https://doi.org/10.1080/21655979.2021.2000742.
Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–3. https://doi.org/10.1016/j.redox.2015.01.002.
Augustine J, Troendle EP, Barabas P, McAleese CA, Friedel T, Stitt AW, et al. The role of lipoxidation in the pathogenesis of diabetic retinopathy. Front Endocrinol. 2020;11:621938. https://doi.org/10.3389/fendo.2020.621938.
D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–24. https://doi.org/10.1038/nrm2256.
Pinazo-Durán MD, Gallego-Pinazo R, García-Medina JJ, Zanón-Moreno V, Nucci C, Dolz-Marco R, et al. Oxidative stress and its downstream signaling in aging eyes. Clin Interv Aging. 2014;9:637–52. https://doi.org/10.2147/cia.S52662.
Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc). 2005;70:200–14. https://doi.org/10.1007/s10541-005-0102-7.
Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. 2016;2016:1245049. https://doi.org/10.1155/2016/1245049.
Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715S–24S. https://doi.org/10.1093/ajcn/57.5.715S.
Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Asp Med. 2009;30:1–12. https://doi.org/10.1016/j.mam.2008.08.006.
Gianazza E, Brioschi M, Fernandez AM, Banfi C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019;23:101119. https://doi.org/10.1016/j.redox.2019.101119.
Pamplona R. Advanced lipoxidation end-products. Chem-Biol Interact. 2011;192:14–20. https://doi.org/10.1016/j.cbi.2011.01.007.
Gianazza E, Brioschi M, Martinez Fernandez A, Casalnuovo F, Altomare A, Aldini G, et al. Lipid peroxidation in atherosclerotic cardiovascular diseases. Antioxid Redox Signal. 2021;34:49–98. https://doi.org/10.1089/ars.2019.7955.
Aldini G, Vistoli G, Stefek M, Chondrogianni N, Grune T, Sereikaite J, et al. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic Res. 2013;47:93–137. https://doi.org/10.3109/10715762.2013.792926.
Mol M, Regazzoni L, Altomare A, Degani G, Carini M, Vistoli G, et al. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: methodological aspects and biological consequences. Free Radic Biol Med. 2017;111:328–44. https://doi.org/10.1016/j.freeradbiomed.2017.01.036.
Jové M, Mota-Martorell N, Pradas I, Martín-Gari M, Ayala V, Pamplona R. The advanced lipoxidation end-product malondialdehyde-lysine in aging and longevity. Antioxidants (Basel). 2020;9:1132. https://doi.org/10.3390/antiox9111132.
Martín-Sierra C, Laranjeira P, Domingues MR, Paiva A. Lipoxidation and cancer immunity. Redox Biol. 2019;23:101103. https://doi.org/10.1016/j.redox.2019.101103.
Peña-Bautista C, Vento M, Baquero M, Cháfer-Pericás C. Lipid peroxidation in neurodegeneration. Clin Chim Acta. 2019;497:178–88. https://doi.org/10.1016/j.cca.2019.07.037.
Castro JP, Jung T, Grune T, Siems W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic Biol Med. 2017;111:309–15. https://doi.org/10.1016/j.freeradbiomed.2016.10.497.
Mas-Bargues C, Escrivá C, Dromant M, Borrás C, Viña J. Lipid peroxidation as measured by chromatographic determination of malondialdehyde. Human plasma reference values in health and disease. Arch Biochem Biophys. 2021;709:108941. https://doi.org/10.1016/j.abb.2021.108941.
Niki E. Lipid peroxidation products as oxidative stress biomarkers. Biofactors. 2008;34:171–80. https://doi.org/10.1002/biof.5520340208.
Grigolo B, Roseti L, Fiorini M, Facchini A. Enhanced lipid peroxidation in synoviocytes from patients with osteoarthritis. J Rheumatol. 2003;30:345–7.
Shah R, Raska K Jr, Tiku ML. The presence of molecular markers of in vivo lipid peroxidation in osteoarthritic cartilage: a pathogenic role in osteoarthritis. Arthritis Rheum. 2005;52:2799–807. https://doi.org/10.1002/art.21239.
Cracowski JL, Durand T, Bessard G. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol Sci. 2002;23:360–6. https://doi.org/10.1016/s0165-6147(02)02053-9.
Basu S, Whiteman M, Mattey DL, Halliwell B. Raised levels of F(2)-isoprostanes and prostaglandin F(2alpha) in different rheumatic diseases. Ann Rheum Dis. 2001;60:627–31. https://doi.org/10.1136/ard.60.6.627.
Franz A, Joseph L, Mayer C, Harmsen JF, Schrumpf H, Fröbel J, et al. The role of oxidative and nitrosative stress in the pathology of osteoarthritis: novel candidate biomarkers for quantification of degenerative changes in the knee joint. Orthop Rev (Pavia). 2018;10:7460. https://doi.org/10.4081/or.2018.7460.
Tiku ML, Shah R, Allison GT. Evidence linking chondrocyte lipid peroxidation to cartilage matrix protein degradation. Possible role in cartilage aging and the pathogenesis of osteoarthritis. J Biol Chem. 2000;275:20069–76. https://doi.org/10.1074/jbc.M907604199.
Hines M, Gomez-Contreras PC, Rodman S, Liman S, Coleman M. Lipid peroxidation regulates chondrocyte mitochondrial dynamics and cartilage injury response. Free Radic Biol Med. 2022;180:s22. https://doi.org/10.1016/j.freeradbiomed.2021.12.041.
Zubavlenko R, Belova SV, Gladkova ЕV, Matveeva OV, Ulyanov VY. Morphological changes in articular cartilage and free-radical lipid peroxidation in rats with posttraumatic osteoarthrosis. Bull Exp Biol Med. 2021;172:214–7. https://doi.org/10.1007/s10517-021-05365-3.
Gladkova ЕV. Role of imbalance of lipid peroxidation and articular cartilage remodeling in the pathogenesis of early primary and post-traumatic gonarthrosis in rats. Bull Exp Biol Med. 2022;172:415–8. https://doi.org/10.1007/s10517-022-05405-6.
Ozgocmen S, Kaya H, Fadillioglu E, Aydogan R, Yilmaz Z. Role of antioxidant systems, lipid peroxidation, and nitric oxide in postmenopausal osteoporosis. Mol Cell Biochem. 2007;295:45–52. https://doi.org/10.1007/s11010-006-9270-z.
Yin G, Wang Y, Cen XM, Yang M, Liang Y, Xie QB. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NF-κB pathway in rheumatoid arthritis synovial cells. Mediators Inflamm. 2015;2015:460310. https://doi.org/10.1155/2015/460310.
Santin Y, Fazal L, Sainte-Marie Y, Sicard P, Maggiorani D, Tortosa F, et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020;27:1907–23. https://doi.org/10.1038/s41418-019-0470-y.
Cindrić M, Čipak Gašparović A, Milković L, Bujak IT, Mihaljević B, Žarković N, et al. 4-Hydroxynonenal modulates blood-brain barrier permeability in vitro through changes in lipid composition and oxidative status of endothelial cells and astrocytes. Int J Mol Sci. 2022;23:14373. https://doi.org/10.3390/ijms232214373.
Wang Y, Wang W, Yang H, Shao D, Zhao X, Zhang G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic Biol Med. 2019;131:237–42. https://doi.org/10.1016/j.freeradbiomed.2018.11.037.
Lou B, Boger M, Bennewitz K, Sticht C, Kopf S, Morgenstern J, et al. Elevated 4-hydroxynonenal induces hyperglycaemia via Aldh3a1 loss in zebrafish and associates with diabetes progression in humans. Redox Biol. 2020;37:101723. https://doi.org/10.1016/j.redox.2020.101723.
Khan F, Moinuddin, Mir AR, Islam S, Abidi M, Husain MA, et al. Unsaturated aldehyde, 4-hydroxynonenal (HNE) alters the structural integrity of HSA with consequences in the immuno-pathology of rheumatoid arthritis. Int J Biol Macromol. 2018;112:306–14. https://doi.org/10.1016/j.ijbiomac.2018.01.188.
Cucci MA, Compagnone A, Daga M, Grattarola M, Ullio C, Roetto A, et al. Post-translational inhibition of YAP oncogene expression by 4-hydroxynonenal in bladder cancer cells. Free Radic Biol Med. 2019;141:205–19. https://doi.org/10.1016/j.freeradbiomed.2019.06.009.
Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193–9. https://doi.org/10.1016/j.redox.2014.12.011.
Guéraud F. 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free Radic Biol Med. 2017;111:196–208. https://doi.org/10.1016/j.freeradbiomed.2016.12.025.
Sonowal H, Ramana KV. 4-Hydroxy-trans-2-nonenal in the regulation of anti-oxidative and pro-inflammatory signaling pathways. Oxid Med Cell Longev. 2019;2019:5937326. https://doi.org/10.1155/2019/5937326.
Chacko BK, Wall SB, Kramer PA, Ravi S, Mitchell T, Johnson MS, et al. Pleiotropic effects of 4-hydroxynonenal on oxidative burst and phagocytosis in neutrophils. Redox Biol. 2016;9:57–66. https://doi.org/10.1016/j.redox.2016.06.003.
Uchida K. HNE as an inducer of COX-2. Free Radic Biol Med. 2017;111:169–72. https://doi.org/10.1016/j.freeradbiomed.2017.02.004.
Milkovic L, Cipak Gasparovic A, Zarkovic N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic Res. 2015;49:850–60. https://doi.org/10.3109/10715762.2014.999056.
Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20:1615–30. https://doi.org/10.1038/cdd.2013.138.
Jana AK, Agarwal S, Chatterjee SN. The induction of lipid peroxidation in liposomal membrane by ultrasound and the role of hydroxyl radicals. Radiat Res. 1990;124:7–14.
Bhatia R, Thompson CM, Clement EJ, Ganguly K, Cox JL, Rauth S, et al. Malondialdehyde-acetaldehyde extracellular matrix protein adducts attenuate unfolded protein response during alcohol and smoking-induced pancreatitis. Gastroenterology. 2022;163:1064–78.e1010. https://doi.org/10.1053/j.gastro.2022.06.071.
Duryee MJ, Clemens DL, Opperman PJ, Thiele GM, Duryee LM, Garvin RP, et al. Malondialdehyde-acetaldehyde modified (MAA) proteins differentially effect the inflammatory response in macrophage, endothelial cells and animal models of cardiovascular disease. Int J Mol Sci. 2021;22:12948. https://doi.org/10.3390/ijms222312948.
Thiele GM, Duryee MJ, Anderson DR, Klassen LW, Mohring SM, Young KA, et al. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2015;67:645–55. https://doi.org/10.1002/art.38969.
Duryee MJ, Ahmad R, Eichele DD, Hunter CD, Mitra A, Talmon GA, et al. Identification of immunoglobulin G autoantibody against malondialdehyde-acetaldehyde adducts as a novel serological biomarker for ulcerative colitis. Clin Transl Gastroenterol. 2022;13:e00469. https://doi.org/10.14309/ctg.0000000000000469.
Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011;111:5997–6021. https://doi.org/10.1021/cr200131e.
Benedetti A, Comporti M, Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta. 1980;620:281–96. https://doi.org/10.1016/0005-2760(80)90209-x.
Parvez S, Long MJC, Poganik JR, Aye Y. Redox signaling by reactive electrophiles and oxidants. Chem Rev. 2018;118:8798–888. https://doi.org/10.1021/acs.chemrev.7b00698.
Morquette B, Shi Q, Lavigne P, Ranger P, Fernandes JC, Benderdour M. Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006;54:271–81. https://doi.org/10.1002/art.21559.
El-Bikai R, Welman M, Margaron Y, Cote JF, Macqueen L, Buschmann MD, et al. Perturbation of adhesion molecule-mediated chondrocyte-matrix interactions by 4-hydroxynonenal binding: implication in osteoarthritis pathogenesis. Arthritis Res Ther. 2010;12:R201. https://doi.org/10.1186/ar3173.
Vaillancourt F, Morquette B, Shi Q, Fahmi H, Lavigne P, Di Battista JA, et al. Differential regulation of cyclooxygenase-2 and inducible nitric oxide synthase by 4-hydroxynonenal in human osteoarthritic chondrocytes through ATF-2/CREB-1 transactivation and concomitant inhibition of NF-kappaB signaling cascade. J Cell Biochem. 2007;100:1217–31. https://doi.org/10.1002/jcb.21110.
Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. 2021;9:789948. https://doi.org/10.3389/fcell.2021.789948.
Campo GM, Avenoso A, Campo S, D’Ascola A, Traina P, Calatroni A. Chondroitin-4-sulphate inhibits NF-kB translocation and caspase activation in collagen-induced arthritis in mice. Osteoarthr Cartil. 2008;16:1474–83. https://doi.org/10.1016/j.joca.2008.04.002.
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. https://doi.org/10.1155/2019/5080843.
Vaillancourt F, Fahmi H, Shi Q, Lavigne P, Ranger P, Fernandes JC, et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 2008;10:R107. https://doi.org/10.1186/ar2503.
Shi Q, Abusarah J, Zaouter C, Moldovan F, Fernandes JC, Fahmi H, et al. New evidence implicating 4-hydroxynonenal in the pathogenesis of osteoarthritis in vivo. Arthritis Rheumatol. 2014;66:2461–71. https://doi.org/10.1002/art.38704.
Zhao Y, Wang B, Zhang J, He D, Zhang Q, Pan C, et al. ALDH2 (aldehyde dehydrogenase 2) protects against hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2019;39:2303–19. https://doi.org/10.1161/atvbaha.119.312946.
Unguryte A, Bernotiene E, Bagdonas E, Garberyte S, Porvaneckas N, Jorgensen C. Human articular chondrocytes with higher aldehyde dehydrogenase activity have stronger expression of COL2A1 and SOX9. Osteoarthr Cartil. 2016;24:873–82. https://doi.org/10.1016/j.joca.2015.11.019.
Pan L, Ding W, Li J, Gan K, Shen Y, Xu J, et al. Aldehyde dehydrogenase 2 alleviates monosodium iodoacetate-induced oxidative stress, inflammation and apoptosis in chondrocytes via inhibiting aquaporin 4 expression. Biomed Eng Online. 2021;20:80. https://doi.org/10.1186/s12938-021-00917-0.
Geib T, Iacob C, Jribi R, Fernandes J, Benderdour M, Sleno L. Identification of 4-hydroxynonenal-modified proteins in human osteoarthritic chondrocytes. J Proteom. 2021;232:104024. https://doi.org/10.1016/j.jprot.2020.104024.
Niu J, Wan X, Yu GY, Jiang S, Yi RN, Wu YP, et al. Phospholipid peroxidation-driven modification of chondrogenic transcription factor mediates alkoxyl radicals-induced impairment of embryonic bone development. Redox Biol. 2022;56:102437. https://doi.org/10.1016/j.redox.2022.102437.
Pan J, Zhou X, Li W, Novotny JE, Doty SB, Wang L. In situ measurement of transport between subchondral bone and articular cartilage. J Orthop Res. 2009;27:1347–52. https://doi.org/10.1002/jor.20883.
Yajun W, Jin C, Zhengrong G, Chao F, Yan H, Weizong W, et al. Betaine attenuates osteoarthritis by inhibiting osteoclastogenesis and angiogenesis in subchondral bone. Front Pharm. 2021;12:723988. https://doi.org/10.3389/fphar.2021.723988.
Shi Q, Vaillancourt F, Cote V, Fahmi H, Lavigne P, Afif H, et al. Alterations of metabolic activity in human osteoarthritic osteoblasts by lipid peroxidation end product 4-hydroxynonenal. Arthritis Res Ther. 2006;8:R159. https://doi.org/10.1186/ar2066.
Cighetti G, Debiasi S, Paroni R, Allevi P. Free and total malondialdehyde assessment in biological matrices by gas chromatography-mass spectrometry: what is needed for an accurate detection. Anal Biochem. 1999;266:222–9. https://doi.org/10.1006/abio.1998.2952.
Tiku ML, Allison GT, Naik K, Karry SK. Malondialdehyde oxidation of cartilage collagen by chondrocytes. Osteoarthr Cartil. 2003;11:159–66. https://doi.org/10.1016/s1063-4584(02)00348-5.
Goranov NV. Serum markers of lipid peroxidation, antioxidant enzymatic defense, and collagen degradation in an experimental (Pond-Nuki) canine model of osteoarthritis. Vet Clin Pathol. 2007;36:192–5. https://doi.org/10.1111/j.1939-165x.2007.tb00208.x.
Watari T, Naito K, Sakamoto K, Kurosawa H, Nagaoka I, Kaneko K. Evaluation of the effect of oxidative stress on articular cartilage in spontaneously osteoarthritic STR/OrtCrlj mice by measuring the biomarkers for oxidative stress and type II collagen degradation/synthesis. Exp Ther Med. 2011;2:245–50. https://doi.org/10.3892/etm.2011.196.
Suantawee T, Tantavisut S, Adisakwattana S, Tanavalee A, Yuktanandana P, Anomasiri W, et al. Oxidative stress, vitamin E, and antioxidant capacity in knee osteoarthritis. J Clin Diagn Res. 2013;7:1855–9. https://doi.org/10.7860/JCDR/2013/5802.3333.
Surapaneni KM, Venkataramana G. Status of lipid peroxidation, glutathione, ascorbic acid, vitamin E and antioxidant enzymes in patients with osteoarthritis. Indian J Med Sci. 2007;61:9–14.
Valenzuela A. The biological significance of malondialdehyde determination in the assessment of tissue oxidative stress. Life Sci. 1991;48:301–9. https://doi.org/10.1016/0024-3205(91)90550-u.
Olszewska-Slonina DM, Jung S, Olszewski KJ, Cwynar A, Drewa G. Evaluation of selected parameters of lipid peroxidation and paraoxonase activity in blood of patients with joint osteoarthritis. Protein Pept Lett. 2018;25:853–61. https://doi.org/10.2174/0929866525666180821120050.
Mikuls TR, Duryee MJ, England BR, Anderson DR, Hearth-Holmes M, Su K, et al. Malondialdehyde-acetaldehyde antibody concentrations in rheumatoid arthritis and other rheumatic conditions. Int Immunopharmacol. 2018;56:113–8. https://doi.org/10.1016/j.intimp.2018.01.022.
Freeman TL, Haver A, Duryee MJ, Tuma DJ, Klassen LW, Hamel FG, et al. Aldehydes in cigarette smoke react with the lipid peroxidation product malonaldehyde to form fluorescent protein adducts on lysines. Chem Res Toxicol. 2005;18:817–24. https://doi.org/10.1021/tx0500676.
Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic Biol Med. 2002;32:303–8. https://doi.org/10.1016/s0891-5849(01)00742-0.
Duryee MJ, Klassen LW, Schaffert CS, Tuma DJ, Hunter CD, Garvin RP, et al. Malondialdehyde-acetaldehyde adduct is the dominant epitope after MDA modification of proteins in atherosclerosis. Free Radic Biol Med. 2010;49:1480–6. https://doi.org/10.1016/j.freeradbiomed.2010.08.001.
Grönwall C, Amara K, Hardt U, Krishnamurthy A, Steen J, Engström M, et al. Autoreactivity to malondialdehyde-modifications in rheumatoid arthritis is linked to disease activity and synovial pathogenesis. J Autoimmun. 2017;84:29–45. https://doi.org/10.1016/j.jaut.2017.06.004.
Sakuraba K, Krishnamurthy A, Sun J, Zheng X, Xu C, Peng B, et al. Autoantibodies targeting malondialdehyde-modifications in rheumatoid arthritis regulate osteoclasts via inducing glycolysis and lipid biosynthesis. J Autoimmun. 2022;133:102903. https://doi.org/10.1016/j.jaut.2022.102903.
Zimmerman MC, Clemens DL, Duryee MJ, Sarmiento C, Chiou A, Hunter CD, et al. Direct antioxidant properties of methotrexate: Inhibition of malondialdehyde-acetaldehyde-protein adduct formation and superoxide scavenging. Redox Biol. 2017;13:588–93. https://doi.org/10.1016/j.redox.2017.07.018.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81. https://doi.org/10.1080/15548627.2020.1810918.
Sun K, Guo Z, Hou L, Xu J, Du T, Xu T, et al. Iron homeostasis in arthropathies: from pathogenesis to therapeutic potential. Ageing Res Rev. 2021;72:101481. https://doi.org/10.1016/j.arr.2021.101481.
Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75. https://doi.org/10.1016/j.jare.2022.01.004.
Lv M, Cai Y, Hou W, Peng K, Xu K, Lu C, et al. The RNA-binding protein SND1 promotes the degradation of GPX4 by destabilizing the HSPA5 mRNA and suppressing HSPA5 expression, promoting ferroptosis in osteoarthritis chondrocytes. Inflamm Res. 2022;71:461–72. https://doi.org/10.1007/s00011-022-01547-5.
Lv Z, Han J, Li J, Guo H, Fei Y, Sun Z, et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. EBioMedicine. 2022;84:104258. https://doi.org/10.1016/j.ebiom.2022.104258.
Chang SH, Mori D, Kobayashi H, Mori Y, Nakamoto H, Okada K, et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat Commun. 2019;10:1442. https://doi.org/10.1038/s41467-019-09491-5.
Xu X, Liu S, Liu H, Ru K, Jia Y, Wu Z, et al. Piezo channels: awesome mechanosensitive structures in cellular mechanotransduction and their role in bone. Int J Mol Sci. 2021;22:6429. https://doi.org/10.3390/ijms22126429.
Karim A, Bajbouj K, Shafarin J, Qaisar R, Hall AC, Hamad M. Iron overload induces oxidative stress, cell cycle arrest and apoptosis in chondrocytes. Front Cell Dev Biol. 2022;10:821014. https://doi.org/10.3389/fcell.2022.821014.
Jing X, Du T, Li T, Yang X, Wang G, Liu X, et al. The detrimental effect of iron on OA chondrocytes: Importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J Cell Mol Med. 2021;25:5671–80. https://doi.org/10.1111/jcmm.16581.
Jing X, Wang Q, Du T, Zhang W, Liu X, Liu Q, et al. Calcium chelator BAPTA-AM protects against iron overload-induced chondrocyte mitochondrial dysfunction and cartilage degeneration. Int J Mol Med. 2021;48:196. https://doi.org/10.3892/ijmm.2021.5029.
Pan Z, He Q, Zeng J, Li S, Li M, Chen B, et al. Naringenin protects against iron overload-induced osteoarthritis by suppressing oxidative stress. Phytomedicine. 2022;105:154330. https://doi.org/10.1016/j.phymed.2022.154330.
Jenei-Lanzl Z, Meurer A, Zaucke F. Interleukin-1β signaling in osteoarthritis – chondrocytes in focus. Cell Signal. 2019;53:212–23. https://doi.org/10.1016/j.cellsig.2018.10.005.
Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. https://doi.org/10.1038/nrrheum.2010.196.
Wang X, Liu Z, Peng P, Gong Z, Huang J, Peng H. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chem-Biol Interact. 2022;366:110148. https://doi.org/10.1016/j.cbi.2022.110148.
Sun K, Hou L, Guo Z, Wang G, Guo J, Xu J, et al. JNK-JUN-NCOA4 axis contributes to chondrocyte ferroptosis and aggravates osteoarthritis via ferritinophagy. Free Radic Biol Med. 2023;200:87–101. https://doi.org/10.1016/j.freeradbiomed.2023.03.008.
Kawai Y, Kubota E, Okabe E. Reactive oxygen species participation in experimentally induced arthritis of the temporomandibular joint in rats. J Dent Res. 2000;79:1489–95. https://doi.org/10.1177/00220345000790071001.
Tchetina EV, Markova GA, Poole AR, Zukor DJ, Antoniou J, Makarov SA, et al. Deferoxamine suppresses collagen cleavage and protease, cytokine, and COL10A1 expression and upregulates AMPK and krebs cycle genes in human osteoarthritic cartilage. Int J Rheumatol. 2016;2016:6432867. https://doi.org/10.1155/2016/6432867.
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. 2019;14:532–41. https://doi.org/10.4103/1673-5374.245480.
Lin J, Guo Z, Zheng Z, Hou L, Xu J, Liu Q, et al. Desferoxamine protects against hemophilic arthropathy through the upregulation of HIF-1α-BNIP3 mediated mitophagy. Life Sci. 2023;312:121172. https://doi.org/10.1016/j.lfs.2022.121172.
Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G, et al. Deferoxamine alleviates osteoarthritis by inhibiting chondrocyte ferroptosis and activating the Nrf2 pathway. Front Pharmacol. 2022;13:791376.
Hu W, Chen Y, Dou C, Dong S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann Rheum Dis. 2021;80:413–22. https://doi.org/10.1136/annrheumdis-2020-218089.
He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2022;157:113915. https://doi.org/10.1016/j.biopha.2022.113915.
Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15:259–66. https://doi.org/10.1038/nm.1910.
Zhang H, Wang A, Li G, Zhai Q, Huang Z, Wang X, et al. Osteoporotic bone loss from excess iron accumulation is driven by NOX4-triggered ferroptosis in osteoblasts. Free Radic Biol Med. 2023;198:123–36. https://doi.org/10.1016/j.freeradbiomed.2023.01.026.
Zhang J, Hu W, Ding C, Yao G, Zhao H, Wu S. Deferoxamine inhibits iron-uptake stimulated osteoclast differentiation by suppressing electron transport chain and MAPKs signaling. Toxicol Lett. 2019;313:50–9. https://doi.org/10.1016/j.toxlet.2019.06.007.
Cheng Q, Chen M, Liu M, Chen X, Zhu L, Xu J, et al. Semaphorin 5A suppresses ferroptosis through activation of PI3K-AKT-mTOR signaling in rheumatoid arthritis. Cell Death Dis. 2022;13:608. https://doi.org/10.1038/s41419-022-05065-4.
Xia L, Gong N. Identification and verification of ferroptosis-related genes in the synovial tissue of osteoarthritis using bioinformatics analysis. Front Mol Biosci. 2022;9:992044. https://doi.org/10.3389/fmolb.2022.992044.
Xu W, Wang X, Liu D, Lin X, Wang B, Xi C, et al. Identification and validation of hub genes and potential drugs involved in osteoarthritis through bioinformatics analysis. Front Genet. 2023;14:1117713. https://doi.org/10.3389/fgene.2023.1117713.
Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthr Cartil. 2020;28:555–61. https://doi.org/10.1016/j.joca.2020.01.007.
Neha K, Haider MR, Pathak A, Yar MS. Medicinal prospects of antioxidants: a review. Eur J Med Chem. 2019;178:687–704. https://doi.org/10.1016/j.ejmech.2019.06.010.
Ighodaro OM, Akinloye OA. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alex J Med. 2019;54:287–93. https://doi.org/10.1016/j.ajme.2017.09.001.
Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69:1502–10. https://doi.org/10.1136/ard.2009.119966.
Koike M, Nojiri H, Kanazawa H, Yamaguchi H, Miyagawa K, Nagura N, et al. Superoxide dismutase activity is significantly lower in end-stage osteoarthritic cartilage than non-osteoarthritic cartilage. PLoS One. 2018;13:e0203944. https://doi.org/10.1371/journal.pone.0203944.
Alfonso-Prieto M, Biarnes X, Vidossich P, Rovira C. The molecular mechanism of the catalase reaction. J Am Chem Soc. 2009;131:11751–61. https://doi.org/10.1021/ja9018572.
Collins JA, Wood ST, Nelson KJ, Rowe MA, Carlson CS, Chubinskaya S, et al. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signaling in aging chondrocytes. J Biol Chem. 2016;291:6641–54. https://doi.org/10.1074/jbc.M115.693523.
Olszewska-Słonina DM, Matewski D, Drewa G, Woźniak A, Czajkowski R, Rajewski P, et al. Oxidative equilibrium in the prophylaxis of degenerative joint changes: an analysis of pre- and postoperative activity of antioxidant enzymes in patients with hip and knee osteoarthritis. Med Sci Monit. 2010;16:Cr238–245.
Li S, Yang X, Feng Z, Wang P, Zhu W, Cui S. Catalase enhances viability of human chondrocytes in culture by reducing reactive oxygen species and counteracting tumor necrosis factor-α-induced apoptosis. Cell Physiol Biochem. 2018;49:2427–42. https://doi.org/10.1159/000493841.
Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M. Glutathione peroxidase family – an evolutionary overview. FEBS J. 2008;275:3959–70. https://doi.org/10.1111/j.1742-4658.2008.06542.x.
Brigelius-Flohe R, Flohe L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid Redox Signal. 2020;33:498–516. https://doi.org/10.1089/ars.2019.7905.
Ostalowska A, Birkner E, Wiecha M, Kasperczyk S, Kasperczyk A, Kapolka D, et al. Lipid peroxidation and antioxidant enzymes in synovial fluid of patients with primary and secondary osteoarthritis of the knee joint. Osteoarthr Cartil. 2006;14:139–45. https://doi.org/10.1016/j.joca.2005.08.009.
Sun J, Wei X, Lu Y, Cui M, Li F, Lu J, et al. Glutaredoxin 1 (GRX1) inhibits oxidative stress and apoptosis of chondrocytes by regulating CREB/HO-1 in osteoarthritis. Mol Immunol. 2017;90:211–8. https://doi.org/10.1016/j.molimm.2017.08.006.
Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, et al. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol. 2022;18:91–100. https://doi.org/10.1038/s41589-021-00915-2.
Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z, Wei X, et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med. 2020;160:552–65. https://doi.org/10.1016/j.freeradbiomed.2020.08.029.
Xiong Y, Xiao C, Li Z, Yang X. Engineering nanomedicine for glutathione depletion-augmented cancer therapy. Chem Soc Rev. 2021;50:6013–41. https://doi.org/10.1039/d0cs00718h.
Desideri E, Ciccarone F, Ciriolo MR. Targeting glutathione metabolism: partner in crime in anticancer therapy. Nutrients. 2019;11:1926. https://doi.org/10.3390/nu11081926.
Lu SC. Regulation of glutathione synthesis. Mol Asp Med. 2009;30:42–59. https://doi.org/10.1016/j.mam.2008.05.005.
Someya S, Kim MJ. Cochlear detoxification: role of alpha class glutathione transferases in protection against oxidative lipid damage, ototoxicity, and cochlear aging. Hear Res. 2021;402:108002. https://doi.org/10.1016/j.heares.2020.108002.
Yang KC, Wu CC, Chen WY, Sumi S, Huang TL. l-Glutathione enhances antioxidant capacity of hyaluronic acid and modulates expression of pro-inflammatory cytokines in human fibroblast-like synoviocytes. J Biomed Mater Res A. 2016;104:2071–9. https://doi.org/10.1002/jbm.a.35729.
Zhu S, Makosa D, Miller B, Griffin TM. Glutathione as a mediator of cartilage oxidative stress resistance and resilience during aging and osteoarthritis. Connect Tissue Res. 2020;61:34–47. https://doi.org/10.1080/03008207.2019.1665035.
Setti T, Arab MGL, Santos GS, Alkass N, Andrade MAP, Lana J. The protective role of glutathione in osteoarthritis. J Clin Orthop Trauma. 2021;15:145–51. https://doi.org/10.1016/j.jcot.2020.09.006.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85. https://doi.org/10.1016/j.freeradbiomed.2020.02.027.
Chiu PR, Hu YC, Huang TC, Hsieh BS, Yeh JP, Cheng HL, et al. Vitamin C protects chondrocytes against monosodium iodoacetate-induced osteoarthritis by multiple pathways. Int J Mol Sci. 2016;18:38. https://doi.org/10.3390/ijms18010038.
Burger MG, Steinitz A, Geurts J, Pippenger BE, Schaefer DJ, Martin I, et al. Ascorbic acid attenuates senescence of human osteoarthritic osteoblasts. Int J Mol Sci. 2017;18:2517. https://doi.org/10.3390/ijms18122517.
Sutipornpalangkul W, Morales NP, Charoencholvanich K, Harnroongroj T. Lipid peroxidation, glutathione, vitamin E, and antioxidant enzymes in synovial fluid from patients with osteoarthritis. Int J Rheum Dis. 2009;12:324–8. https://doi.org/10.1111/j.1756-185X.2009.01430.x.
Bhatti FU, Mehmood A, Latief N, Zahra S, Cho H, Khan SN, et al. Vitamin E protects rat mesenchymal stem cells against hydrogen peroxide-induced oxidative stress in vitro and improves their therapeutic potential in surgically-induced rat model of osteoarthritis. Osteoarthr Cartil. 2017;25:321–31. https://doi.org/10.1016/j.joca.2016.09.014.
Hassan WN, Bin-Jaliah I, Haidara MA, Eid RA, Heidar EHA, Dallak M, et al. Vitamin E ameliorates alterations to the articular cartilage of knee joints induced by monoiodoacetate and diabetes mellitus in rats. Ultrastruct Pathol. 2019;43:126–34. https://doi.org/10.1080/01913123.2019.1627446.
Tantavisut S, Tanavalee A, Honsawek S, Suantawee T, Ngarmukos S, Adisakwatana S, et al. Effect of vitamin E on oxidative stress level in blood, synovial fluid, and synovial tissue in severe knee osteoarthritis: a randomized controlled study. BMC Musculoskelet Disord. 2017;18:281. https://doi.org/10.1186/s12891-017-1637-7.
Saito Y. Lipid peroxidation products as a mediator of toxicity and adaptive response – the regulatory role of selenoprotein and vitamin E. Arch Biochem Biophys. 2021;703:108840. https://doi.org/10.1016/j.abb.2021.108840.
Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12:706. https://doi.org/10.1038/s41419-021-04008-9.
Veen L, Hantikainen E, Bellocco R, Ye W, Serafini M, Ponzano M, et al. Dietary antioxidants, non-enzymatic antioxidant capacity and the risk of osteoarthritis in the Swedish National March Cohort. Eur J Nutr. 2021;60:169–78. https://doi.org/10.1007/s00394-020-02239-8.
Xu C, Wang S, Ti W, Yang J, Yasen Y, Memetsidiq M, et al. Role of dietary patterns and factors in determining the risk of knee osteoarthritis: a meta-analysis. Mod Rheumatol. 2022;32:815–21. https://doi.org/10.1093/mr/roab059.
Crane FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr. 2001;20:591–8. https://doi.org/10.1080/07315724.2001.10719063.
Li X, Zhan J, Hou Y, Hou Y, Chen S, Luo D, et al. Coenzyme Q10 regulation of apoptosis and oxidative stress in H(2)O(2) induced BMSC death by modulating the Nrf-2/NQO-1 signaling pathway and its application in a model of spinal cord injury. Oxid Med Cell Longev. 2019;2019:6493081. https://doi.org/10.1155/2019/6493081.
Beyer RE. Inhibition by coenzyme Q of ethanol- and carbon tetrachloride-stimulated lipid peroxidation in vivo and catalyzed by microsomal and mitochondrial systems. Free Radic Biol Med. 1988;5:297–303. https://doi.org/10.1016/0891-5849(88)90100-1.
Abd El-Gawad HM, Khalifa AE. Quercetin, coenzyme Q10, and L-canavanine as protective agents against lipid peroxidation and nitric oxide generation in endotoxin-induced shock in rat brain. Pharm Res. 2001;43:257–63. https://doi.org/10.1006/phrs.2000.0781.
Schmelzer C, Lindner I, Rimbach G, Niklowitz P, Menke T, Doring F. Functions of coenzyme Q10 in inflammation and gene expression. Biofactors. 2008;32:179–83. https://doi.org/10.1002/biof.5520320121.
Bauerova K, Paulovicova E, Mihalova D, Drafi F, Strosova M, Mascia C, et al. Combined methotrexate and coenzyme Q10 therapy in adjuvant-induced arthritis evaluated using parameters of inflammation and oxidative stress. Acta Biochim Pol. 2010;57:347–54.
Lee J, Hong YS, Jeong JH, Yang EJ, Jhun JY, Park MK, et al. Coenzyme Q10 ameliorates pain and cartilage degradation in a rat model of osteoarthritis by regulating nitric oxide and inflammatory cytokines. PLoS One. 2013;8:e69362. https://doi.org/10.1371/journal.pone.0069362.
Na HS, Woo JS, Kim JH, Lee JS, Um IG, Cho KH, et al. Coenzyme Q10 encapsulated in micelles ameliorates osteoarthritis by inhibiting inflammatory cell death. PLoS One. 2022;17:e0270351. https://doi.org/10.1371/journal.pone.0270351.
Li X, Guo Y, Huang S, He M, Liu Q, Chen W, et al. Coenzyme Q10 prevents the interleukin-1 beta induced inflammatory response via inhibition of MAPK signaling pathways in rat articular chondrocytes. Drug Dev Res. 2017;78:403–10. https://doi.org/10.1002/ddr.21412.
Quinzii CM, Emmanuele V, Hirano M. Clinical presentations of coenzyme q10 deficiency syndrome. Mol Syndromol. 2014;5:141–6. https://doi.org/10.1159/000360490.
Chang PS, Yen CH, Huang YY, Chiu CJ, Lin PT. Associations between coenzyme Q10 status, oxidative stress, and muscle strength and endurance in patients with osteoarthritis. Antioxidants (Basel). 2020;9:1275. https://doi.org/10.3390/antiox9121275.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8. https://doi.org/10.1038/s41586-019-1707-0.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92. https://doi.org/10.1038/s41586-019-1705-2.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82172498).
Author information
Authors and Affiliations
Contributions
XZ: writing original draft and literature search; LH, ZG, GW, JX, and ZZ: edit and revisions; KS: conceptualization and revisions of the manuscript; FG: conceptualization. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All the authors agree for the publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Hou, L., Guo, Z. et al. Lipid peroxidation in osteoarthritis: focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 9, 320 (2023). https://doi.org/10.1038/s41420-023-01613-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01613-9
- Springer Nature Limited
This article is cited by
-
Research progress on nanotechnology of traditional Chinese medicine to enhance the therapeutic effect of osteoarthritis
Drug Delivery and Translational Research (2024)
-
The Role of Ferroptosis in Amyotrophic Lateral Sclerosis Treatment
Neurochemical Research (2024)
-
Artemisia herba-alba: antioxidant capacity and efficacy in preventing chronic arthritis in vivo
Inflammopharmacology (2024)