
Abstract
DDRGK domain-containing protein 1 (DDRGK1) is an important component of the newly discovered ufmylation system and its absence has been reported to induce extensive endoplasmic reticulum (ER) stress. Recently, emerging evidence indicates that the ufmylation system is correlated with autophagy, although the exact mechanism remains largely unknown. To explore the regulation mechanism of DDRGK1 on autophagy, in this study, we established an immortalized mouse embryonic fibroblast (MEF) cell lines harvested from the DDRGK1F/F:ROSA26-CreERT2 mice, in which DDRGK1 depletion can be induced by 4-hydroxytamoxifen (4-OHT) treatment. Here, we show that DDRGK1 deficiency in MEFs has a dual effect on autophagy, which leads to a significant accumulation of autophagosomes. On one hand, it promotes autophagy induction by impairing mTOR signaling; on the other hand, it blocks autophagy degradation by inhibiting autophagosome–lysosome fusion. This dual effect of DDRGK1 depletion on autophagy ultimately aggravates apoptosis in MEFs. Further studies reveal that DDRGK1 loss is correlated with suppressed lysosomal function, including impaired Cathepsin D (CTSD) expression, aberrant lysosomal pH, and v-ATPase accumulation, which might be a potential trigger for impairment in autophagy process. Hence, this study confirms a crucial role of DDRGK1 as an autophagy regulator by controlling lysosomal function. It may provide a theoretical basis for the treatment strategies of various physiological diseases caused by DDRGK1 deficiency.
Similar content being viewed by others


Introduction
The ubiquitin-fold modifier 1 (Ufm1) conjugation system is a novel ubiquitin-like modification system that consists of a set of ubiquitin-like proteins (UBLs), including Ufm1, Ufm1-activating E1 enzyme (Uba5), Ufm1-conjugating E2 enzyme (Ufc1), and Ufm1-specific E3 ligase (Ufl1, also known as RACD, NLBP and Maxer)1,2,3. DDRGK domain-containing protein 1 (DDRGK1, also known as C20orf116, and Dashurin), another critical component of the Ufm1 conjugation system, was found as a Ufm1 target whose ufmylation was conducted by Ufl1 (ref. 2). It contains a proteasome component (PCI) domain that is generally associated with protein interactions and a putative signal peptide at its N-terminus that contributes to its endoplasmic reticulum (ER) anchoring4. Studies show that DDRGK1, together with several other Ufm1 system components Ufl1, C53/LZAP (Cdk5 activator-binding protein Cdk5rap3)4,5 and Ufm1, forms a large complex that is anchored at the ER and each of these proteins is indispensable for maintaining ER homeostasis2,6,7. Recent studies have shown that DDRGK1 deficiency can cause a variety of physiological and pathological diseases, including hematopoietic dysfunction8, impaired intestinal homeostasis9, and compromised plasma cell development10. This evidence indicates a critical role of DDRGK1 in maintaining normal physiological processes, though the underlying mechanism is poorly understood.
Macrophagy (hereafter referred to as autophagy), an evolutionary conserved biological process in eukaryotes, is considered to maintain cell homeostasis by engulfing the cytoplasmic proteins or damaged organelles in double-membrane-bound structures, known as autophagosomes, and then deliver them to the lysosome for degradation11,12. Autophagy is rapidly induced by nutrient deprivation13, oxidative stress14, hypoxia15,16, ER stress17,18, and other cellular stresses and diseases19,20. The stimulation of autophagy is a complex dynamic process, which requires plenty of autophagy-related genes (ATGs), including two vital ubiquitin-like conjugation systems21, Atg5–Atg12–Atg16L1 complex and microtubule-associated protein light chain 3 (LC3, a homolog of yeast Atg8)22. Atg7 is considered to be involved in both of these two conjugation systems, and its deficiency may impair the autophagy induction. Autophagosome–lysosome fusion is another essential step in autophagy process. It drives autophagic degradation, which is generally achieved by soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins (SNAREs). Especially the STX17 (syntaxin 17)–SNAP29 (synaptosome associated protein 29)–VAMP8 (vesicle-associated membrane protein 8) complex is widely considered to mediate autophagosome–lysosome fusion23,24.
As emerging evidence indicates that ER stress-induced unfolded protein reaction promotes autophagy stimulation25,26,27,28, it seems that the DDRGK1 is implicated in autophagy regulation. In fact, a previous study had shown that Ufl1 loss leads to impaired autophagic degradation29, providing evidence for the involvement of ufmylation system in autophagy regulation. However, it remains elusive whether DDRGK1 is required for this process.
Therefore, in this study, immortalized mouse embryonic fibroblast (MEF) cell lines harvested from the DDRGK1F/F:ROSA26-CreERT2 mice (with thorough DDRGK1 knockout efficiency) were used as a research model to investigate the role of DDRGK1 in autophagy regulation. Here, for the first time, we demonstrated that DDRGK1 deletion suppressed the autophagosome–lysosome fusion, thus leading to an impaired autophagic degradation and increased apoptosis in MEFs. Collectively, our findings reveal a DDRGK1-dependent mechanism in the regulation of autophagic flux.
Results
Result 1: 4-OHT-induced DDRGK1 deficiency promotes apoptosis in MEFs
To investigate the function of DDRGK1, immortalized MEFs derived from the DDRGK1F/F:ROSA26-CreERT2 mice8 were treated with 4-hydroxytamoxifen (4-OHT) to induce the deficiency of DDRGK1. As is shown in Fig. 1a, DDRGK1 mRNA was significantly decreased in a time-dependent manner and was almost undetectable on the third and fourth days. Consistently, >80% of DDRGK1 protein was depleted after treating with 4-OHT for 4 days (Fig. 1b, c). Our previous study suggests that DDRGK1 deletion leads to cell death of hematopoietic stem cells (HSCs)/progenitor cells8. Likewise, we found that DDRGK1 deficiency led to cell apoptosis, for a subset of proapoptotic genes, including Noxa, Bim, and Bax were significantly upregulated and Bcl-2, an antiapoptotic gene were remarkably downregulated in MEFs (Fig. 1d). Furthermore, flow cytometry analysis showed a significant increased apoptosis in 4-OHT group compared with the control group (Fig. 1e, f). We also measured the mRNA level of Ufl1 and C53/LZAP, as they are other two novel components of Ufm1 system, which have a crosstalk with DDRGK1 (ref. 4). Interestingly, the mRNA levels of Ufl1 and C53/LZAP were not affected by DDRGK1 deletion (Fig. 1g), suggesting the aforementioned results were indeed caused by DDRGK1 deletion. Taken together, these data suggest that 4-OHT mediates a sufficient deletion of DDRGK1, and DDRGK1 is required for maintaining the normal physiological balance in MEFs.

a MEFs were treated with 4-OHT (2 μM) for 0, 1, 2, 3, and 4 days. The indicated samples were then collected for qRT-PCR analysis to determine the transcriptional levels of DDRGK1. Data were shown as mean ± SEM, n = 3. **P < 0.01; ***P < 0.001. b MEFs were treated with EtOH or 4-OHT (2 μM) for 4 days. Cell lysates were collected to determine the DDRGK1 levels using western blotting. Asterisk indicates nonspecific bands. c Quantification of DDRGK1 levels (normalized to β-actin) in EtOH- and 4-OHT-treated cells. Data were shown as mean ± SEM, n = 3. **P < 0.01. d MEFs were cultured in EtOH or 4-OHT (2 μM) for 4 days. qRT-PCR assay was performed to detect the mRNA levels of apoptosis-related genes, including Bcl-2, Noxa, Bim, and Bak. Data were shown as mean±SEM, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001. e Flow cytometry analysis of apoptosis in MEFs incubated with EtOH or 4-OHT for 4 days. f Quantification of apoptosis rate in control and DDRGK1-deficient MEFs. Data were shown as mean ± SEM, n = 3. ***P < 0.001. g MEFs treated with EtOH or 2 μM 4-OHT for 4 days were then analyzed by qRT-PCR to determine the mRNA levels of DDRGK1, Ufl1, and C53/LZAP. Data were presented as mean ± SEM, n = 3. ***P < 0.001; ns not significant, P > 0.05.
Result 2: DDRGK1 deficiency is involve in autophagy–lysosome-related regulation
Although DDRGK1 has been reported to regulate numerous physiological processes, ranging from HSC development8, plasma cell development10 to intestinal inflammation9, the underlying mechanism remains poorly understood. Thus, we performed iTRAQ-based liquid chromatography–mass spectrometry (LC–MS) to identify differentially expressed proteins between the control and DDRGK1-deleted MEFs. The hierarchical clustering analysis showed a high biological reproducibility both in control and 4-OHT groups (Fig. S1a and Table S3).
A total of 5092 proteins were detected, of which 167 were differentially expressed (Fig. 2a and Table S3). Consistent with what we expected, a large number of differential expressed proteins were enriched in KEGG pathways related to ER homeostasis and protein processing, such as “protein processing in ER” and “protein digestion and absorption” terms (Fig. 2b and Table S4; Fig. S1b and Table S6). Importantly, a considerable differential expressed proteins were enriched in lysosome-related pathways, including “lysosome”, “regulation of autophagy”, and “collecting duct acid secretion” terms (Fig. 2b and Table S4; Fig. S1b and Table S6), suggesting that DDRGK1 might participate in autophagy–lysosome-related degradation process. Therefore, we further analyzed the 15 differentially expressed proteins in these three terms and found that a majority of proteins were upregulated with only CTSD and ASAH1 downregulated (Fig. 2c and Table S5). Changes were validated by western blot analysis, which showed a significant increase in Atp6v0d1 and Atp6v1a, and a decrease in CTSD (Fig. 2d, e). These data suggest that DDRGK1 have a potential regulation on autophagy–lysosome-related pathway.

a Distribution of differential expressed proteins between DDRGK1-deleted and the control groups detected by LC–MS. The number of changed proteins in each category of log(FC) is indicated above each bar. b Specific significantly enriched KEGG pathways in 4-OHT- or EtOH-treated groups. Number of upregulated and downregulated proteins is shown on the bar plot. The P values is shown for each pathway based on hypergeometric distribution. c Expression patterns of proteins enriched in lysosome- and autophagy-related pathways. d Western blot analysis for indicated proteins. MEFs treated with EtOH or 4-OHT for 4 days were then subjected to western blotting to validate the expression changes of selected representatives—Atp6v0d1, Atp6v1a, and CTSD. e Quantificational expressions of Atp6v0d1, Atp6v1a, and CTSD protein standardized by β-actin expression. Data represent mean ± SEM, n = 3. *P < 0.05.
Result 3: DDRGK1 deletion causes a significant accumulation of autophagosomes in vitro and in vivo
To validate the effects of DDRGK1 on MEF autophagy, we first examined the expression of LC3 protein, a typical biomarker of autophagy, and SQSTM1, a substrate of autophagic degradation, which would be degraded by autolysosome during autophagic degradation30,31. As is shown in Fig. 3a–d, both conversion of LC3-I to LC3-II and SQSTM1 expression was significantly promoted, while SQSTM1 transcript level was not affected in MEFs after treating with 4-OHT. Accordingly, immunofluorescence assay with LC3 staining showed an elevated number of LC3-positive puncta in DDRGK1-deleted cells (Fig. 3e, f), suggesting an accumulation of autophagosomes. Using transmission electron microscopy (TEM), we detected a significant accumulation of autophagic vesicles (both autophagosomes and autolysosomes). These data further confirm that DDRGK1 deletion facilitates accumulation of autophagosomes in MEFs (Fig. 3g–i).

a Western blot analysis of LC3 and SQSTM1 protein in cell lysates from control and DDRGK1-deficient MEFs. b, c Quantification of conversion of LC3-I to LC3-II and SQSTM1 expression standardized by β-actin level. Data represent mean ± SEM; n = 3. *P < 0.05; ***P < 0.001. d The mRNA level of SQSTM1 determined by qRT-PCR analysis. Data represent mean ± SEM; n = 3. ns not significant, P > 0.05. e Immunofluorescence staining of endogenous LC3 protein in EtOH- and 4-OHT-treated MEFs. Each red puncta represents a autophagosome accumulated in the cytoplasm of MEFs. Laser confocal-scanning microscopy was employed to observe the fluorescent puncta in MEFs. Bar, 10 μm. f Quantification of the punctate LC3 per cell. Data represent mean ± SEM; n = 3. **P < 0.01. g TEM images of autophagic vesicle in control and DDRGK1-deleted MEFs. Red arrowheads represent autophagosomes and yellow ones represent autolysosomes. Bar, 0.5 μm. h, i Quantification of autophagic vesicle numbers per imaging area. Data represent mean ± SEM; n = 3. **P < 0.01; ***P < 0.001. j Western blot analysis of LC3, SQSTM1, and DDRGK1 proteins in liver tissues of wild-type and DDRGK1-deficient mice. k–m Quantification of conversion of LC3-I to LC3-II (k), SQSTM1 (l), and DDRGK1 (m) expression standardized by β-actin expression. Data represent mean ± SEM; n = 3. **P < 0.01; ***P < 0.001.
To determine if the regulation of DDRGK1 on autophagy was conserved in vivo, we detected the LC3 and SQSTM1 level in liver tissues of wild-type and DDRGK1F/F: ALB-cre+ mice. Likewise, DDRGK1 loss led to a dramatic accumulation of SQSTM1 and LC3-II conversion in DDRGK1-deleted liver tissues (Fig. 3j–m), indicating the function of DDRGK1 in autophagy regulation is conserved in vivo. Taken together, these data suggest that DDRGK1 deletion lead to a significant accumulation of autophagosomes both in vitro and in vivo.
To further assess whether the accumulation of autophagosomes is caused by enhanced autophagosome formation or attenuated autolysosome degradation, we next detected LC3 expression in the presence of bafilomycin A1 (Baf-A1), an autophagy inhibitor that blocks fusion of autophagosomes with lysosomes. Compared with the control group, the LC3-II level was significantly increased after Baf-A1 addition. However, the addition of Baf-A1 brought no further change in LC3-II level in 4-OHT-treated groups, indicating a potential suppression of autophagic degradation during DDRGK1 deficiency. Notably, on the basis of Baf-A1 supplement (serves as an inhibition of autophagic degradation), the expression of LC3-II was further dramatically elevated in DDRGK1-deleted group when compared with the control group (Fig. 4a, b), suggesting that DDRGK1 deficiency might concomitantly promote autophagosome formation in addition to suppressing autophagic degradation.

a Western blot analysis of LC3 protein in control and DDRGK1-deficient MEFs treated with or without 10 nM Baf-A1. b Quantification of LC3-II level that normalized to β-actin content. Data represent mean ± SEM; n = 3. **P < 0.01; ***P < 0.001; ns not significant, P > 0.05. c Western blot for phosphorylation of AMPK, ULK1, and 4E-BP1 in cell lysates from EtOH- and 4-OHT-treated MEFs. d–f Quantification of p-AMPK/AMPK (d), p-ULK1/β-Actin (e), and p-4E-BP1/4E-BP1 (f) ratios, respectively. Data were presented as mean ± SEM; n = 3. *P < 0.05; **P < 0.01.
Result 4: autophagy induction is associated with an impaired mTOR signaling in DDRGK1-deficient cells
mTORC1 is an important regulator of autophagy, whose dephosphorylation promotes autophagy induction32,33. To verify if DDRGK1 deletion promotes autophagy initiation, we tested the activity of mTORC1 by detecting the phosphorylation status of mTORC1 targets, ULK1 (refs. 34,35) and 4E-BP1. As is shown in Fig. 4c, e, f, DDRGK1 deletion suppressed the phosphorylation of ULK1 (Ser757) and 4E-BP1, indicating a decreased activity of mTORC1. Moreover, the increased phosphorylation of AMPK (another upstream regulator of autophagy)36,37 in DDRGK1-deficient group further confirmed the enhanced autophagy induction (Fig. 4c, d). These data suggest that DDRGK1 deficiency promotes autophagy induction by controlling the activity of autophagy upstream kinases.
Result 5: DDRGK1 is indispensable in maintaining autophagic flux
To further determine whether DDRGK1 loss-induced inhibition of autophagic degradation was correlated with impaired autophagic flux, we transfected MEFs with a tandem-tagged mCherry-EGFP-LC3 plasmid, which exhibits yellow fluorescence in autophagosomes due to the combination of mCherry and EGFP signals. However, when autophagosomes are fused with lysosomes, the EGFP signal is quenched due to the acidic environment of lysosomes and only shows the mCherry signal38,39. Compared with the control group, DDRGK1 loss led to a significant increase of yellow puncta, while the only mCherry puncta that represent autolysosomes were severely decreased (Fig. 5a, b), suggesting that DDRGK1 deficiency causes an impaired autophagic flux in MEFs.

a Representative images of control and DDRGK1-deleted MEFs transfected with mCherry-EGFP-LC3 plasmid for 36 h. Enlarged images of the yellow box were indicated on the right side. Laser confocal-scanning microscopy was employed to observe the fluorescent puncta. Bar, 10 μm. b Quantification of only mCherry-labeled and EGFP + mCherry-labeled puncta. Data represent mean ± SEM; n = 3. *P < 0.05. c Colocalization of endogenous LC3 and LAMP2 proteins in control and DDRGK1-deficient MEFs treated with or without rapamycin. Enlarged images of the yellow box were indicated on the right side. Images were detected by laser confocal-scanning microscopy. Bar, 10 μm. d Quantification of the number of LC3 and LAMP2 colocalized puncta per cell. Data represent mean ± SEM; n = 3. **P < 0.01; ***P < 0.001.
To more directly visualize the fusion of autophagosomes and lysosomes, we assessed the immunofluorescence colocalization of LC3 and LAMP2 proteins. Rapamycin was added to induce autophagy level, as well as autophagy flux. Compared with the EtOH group, both rapamycin and 4-OHT treatment induced a dramatic accumulation of autophagosomes. However, the colocalization of LC3 and LAMP2 in the rapamycin-treated group was robustly enhanced, while that in the 4-OHT group was dramatically decreased (Fig. 5c, d). These data indicate that DDRGK1 loss blocked the fusion of autophagosomes and lysosomes, thus leading to an impaired autophagic degradation.
Result 6: DDRGK1 deficiency-induced apoptosis is associated with autophagy defect in MEFs
To determine whether DDRGK1 deficiency-induced apoptosis was related to autophagy defect, we tested the apoptosis level in MEFs after siAtg7 transfection. Firstly, we designed three small interfering RNAs (siRNAs) targeting Atg7 and selected the one exhibiting the highest knockdown efficiency, siAtg7-3, for subsequent experiments (Fig. 6a). As is shown in Fig. 6b, c, Atg7 protein expression was significant decreased both in control and DDRGK1-deleted MEFs when transfected with Atg7 siRNA. Moreover, Atg7 knockdown also significantly suppressed the accumulation of autophagosomes, as indicated with a remarkable decrease of LC3 protein in both control and DDRGK1-deleted groups, and a remarkable decrease of SQSTM1 protein in DDRGK1-deleted groups (Fig. 6d–f).

a MEFs were transfected with siNC oligos, siAtg7-1, siAtg7-2, and siAtg7-3 (selected for the following experiments and hereafter referred to as siAtg7) siRNAs for 48 h, respectively. Cell lysates were collected to determine the protein levels of Atg7 by western blot. b Western blot for Atg7 protein from EtOH- or 4-OHT-treated MEFs transfected with or without siAtg7 for 48 h. c Quantification of Atg7 levels normalized to β-actin levels using densitometric analysis. Data represent mean ± SEM; n = 3. **P < 0.01; ***P < 0.001. d Western blot analysis of LC3 and SQSTM1 proteins in EtOH- and 4-OHT-treated MEFs transfected with or without siAtg7 for 48 h. e, f Quantification of LC3 and SQSTM1 levels normalized to β-actin level. Data represent mean ± SEM; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ns not significant, P > 0.05. g Flow cytometry analysis for apoptosis in EtOH- and 4-OHT-treated MEFs transfected with or without siAtg7 for 48 h. h Quantification of apoptosis rate in EtOH- and 4-OHT-treated MEFs. Data represent mean ± SEM; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001.
Flow cytometry analysis showed that loss of DDRGK1 significantly increased the apoptosis rate in MEFs (Fig. 6g, h). Notably, the enhanced apoptosis rate in DDRGK1-deleted MEFs was remarkably alleviated by siAtg7 transfection, though apoptosis level was also decreased in EtOH-treated MEFs, probably because there was a certain degree of basal autophagy activity (Fig. 6g, h). These data suggest that DDRGK1 deficiency-induced apoptosis in MEFs is associated with aberrant accumulation of autophagosomes in some way.
Result 7: DDRGK1 deficiency leads to dysfunction in lysosomes
The fusion of autophagosomes and lysosomes is a complex dynamic process conducted by various factors, including proper acidic environment, normal function of lysosomal enzyme, and SNAREs. To investigate the specific mechanism of how autophagosome–lysosome fusion is suppressed, we detected the above factors separately. The results displayed that DDRGK1 loss increased the VAMP8 protein level without altering the level of Stx17 and SNAP29 proteins (Fig. 7a, b), which was consistent with the transcriptional level (Fig. 7c). Moreover, lysosomal acidification was detected by using the Lyso-Tracker Red probe, a lysosomal-specific fluorescent probe that contains a weakly basic group so that it can selectively stay in acidic lysosomes and exhibit red fluorescence. To our surprise, DDRGK1 loss significantly increased the fluorescence signals, suggesting the lysosomal pH was reduced rather than alkalinized (Fig. 7d, e). However, the deletion of DDRGK1 led to a compromised expression of CTSD (Fig. 7f, g), which was consistent with our proteomics results. Taken together, these data suggest that DDRGK1 loss leads to an impairment in CTSD level and lysosomal function.

a Western blot analysis for SNAP29, Stx17, and VAMP8 proteins in EtOH- and 4-OHT-treated MEFs. b Quantitative expressions of SNAP29, Stx17, and VAMP8 proteins normalized to β-actin. Data represent mean ± SEM; n = 3. ns not significant, P > 0.05. c The transcriptional levels of Snap29, Stx17, and Vamp8 in EtOH- and 4-OHT-induced MEFs. Data represent mean ± SEM; n = 3. *P < 0.05; ns not significant, P > 0.05. d Lyso-Tracker Red signals in control and DDRGK1-deleted MEFs. MEFs cultured in EtOH or 4-OHT for 4 days were then incubated with diluted Lyso-Tracker Red dye (diluted in cell culture medium in a ratio of 1:20,000) for another 20 min. Images were detected by laser confocal-scanning microscopy. Bar, 20 μm. e Quantification of normalized number of Lyso-Tracker Red puncta. Data were shown as mean ± SEM; n = 3. **P < 0.01. f Western blot analysis for CTSD expression in EtOH- and 4-OHT-induced MEFs. g Quantification of CTSD level normalized to β-actin expression. Data represent mean ± SEM; n = 3. *P < 0.05.
Result 8: DDRGK1 is involved in proteasome-mediated degradation of v-ATPase subunits
To investigate the mechanism by which DDRGK1 affects lysosomal function, we then assessed the relationship between DDRGK1 and lysosome. We thus examined the subcellular localization of DDRGK1, and found a remarkable colocalization of DDRGK1 and LAMP2 (Fig. 8a), indicating DDRGK1 might be localized on the lysosomal surface. A previous study of Mümine Şentürk et al.40 has reported that ubiquilin, a ubiquitin-binding protein, mediates lysosome acidification by interacting with subunits of lysosomal proton pump, v-ATPase, thus facilitating autophagic flux. Considering the presence of v-ATPase subunits on our LC–MS data, we wonder if they have an interaction with DDRGK1. The co-immunoprecipitation assay showed that DDRGK1 indeed interact with Atp6v0d1 (Fig. 8b). To determine whether the enhancement of v-ATPase subunits during DDRGK1 deficiency was correlated with proteasome-dependent degradation, we performed MG132 incubation for different period of time in cells that pretreated with cycloheximide (CHX, an inhibitor of protein synthesis). Interestingly, MG132 treatment induced an accumulation of Atp6v0d1 and Atp6v1a in control MEFs, whereas such accumulation was not observed in DDRGK1-deficient cells (Fig. 8c–e). Especially after 9 h treatment with MG132, both Atp6v0d1 and Atp6v1a increased to a level similar to that of the DDRGK1-deleted groups (Fig. 8c–e), indicating that DDRGK1 accelerates proteasome-mediated degradation of v-ATPase subunits.

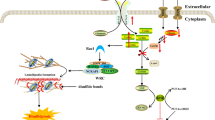
a Colocalization of endogenous DDRGK1 and LAMP2 proteins in MEFs. Magnification is shown on the left side. Bar (upper), 40 μm; bar (lower), 10 μm. b Co-immunoprecipitation of DDRGK1 and Atp6v0d1 in MEFs without any treatment. Total cell lysates (input) were collected for immunoprecipitation with anti-Atp6v0d1, followed by immunoblotting with anti-DDRGK1. IP indicates the immunoprecipitation antibody; IB indicates the immunoblotting antibody. c Western blot for Atp6v0d1 and Atp6v1a proteins. MEFs were treated with EtOH or 4-OHT for 4 days. Before harvested, MEFs were pretreated with 20 μg/mL cycloheximide (CHX) for 12 h accompanied by 2 μM MG132 treatment for the indicated times. Cell lysates were then collected for western blotting. d, e Quantification of Atp6v0d1 (d) and Atp6v1a (e) levels normalized to β-actin levels. Data represent mean ± SEM; n = 3. *P < 0.05; **P < 0.01. f Schematic model for regulation of DDRGK1 on autophagy. Under normal conditions, DDRGK1 regulates ubiquitin–proteasome-mediated degradation of v-ATPase subunits (including Atp6v0d1 and Atp6v1a) and maintains the stable expression of them, which leads to a normal function of lysosomes, promotes the fusion of autophagosomes and lysosomes, and the subsequent autophagic degradation. However, DDRGK1 deficiency induces an aberrant accumulation of Atp6vd1 and Atp6v1a due to the impaired ubiquitin–proteasome degradation pathway, thus resulting in dysregulation of lysosome, blocked fusion of autophagosomes and lysosomes, and suppressed autophagic degradation.
Discussion
In the present study, we show that DDRGK1 deficiency in MEFs leads to an aberrant accumulation of autophagosomes due to the enhanced autophagy induction (as indicated with activation of mTORC1 signaling) and impaired autophagic degradation (as indicated with impaired autophagic flux), which ultimately aggravates apoptosis in MEFs. Furthermore, DDRGK1 is required for maintaining a proper expression of v-ATPase subunits by modulating ubiquitin–proteasome degradation pathway, as its deficiency induces aberrant accumulation of Atp6vod1 and Atp6v1a, which might be a trigger for lysosomal dysfunction and impaired autophagic flux. Taken together, our findings demonstrate an important role of DDRGK1 as an autophagy regulator by controlling lysosomal function, providing a novel insight into the molecular and cellular mechanism of DDRGK1 function.
The MEFs in our study were harvested from DDRGK1F/F:ROSA26-CreERT2 mice, so its DDRGK1 depletion can be efficiently induced by 4-OHT treatment. This makes it a perfect model to investigate the mechanism of DDRGK1. First, DDRGK1 seems to participate in autophagy process as we identified an abundant number of differentially expressed proteins enriched in lysosome- and autophagy-related pathways, during DDRGK1 deletion. This hypothesis was then confirmed by the enhanced accumulation of autophagosomes in DDRGK1-deleted MEFs. N’Diaye et al. argue that loss of UBQLN1 and 2 (two ubiquitin-binding proteins) causes a dual effect on autophagy, with increased autophagosome formation on the one hand, but decreased delivery of autophagosomes to lysosomes on the other hand41. Similarly, our data also validate a dual effect of DDRGK1 deficiency on autophagy, showing enhanced autophagy initiation by inactivating mTOR and activating AMPK kinase, but suppressed autophagic degradation with impaired autophagy flux. Based on these data, we hypothesize that, the impaired autophagic turnover induced by DDRGK1 loss may causes physiological imbalance in MEFs.
It is reported that excessive accumulation of autophagosomes induces apoptosis and cell death42,43. Indeed, in our study, the dual effect of DDRGK1 loss on autophagy aggravates autophagosomes accumulation and apoptosis, which could be attenuated by siAtg7 transfection (serves as a way to block autophagy induction). Interestingly, Atg7 knockdown also alleviated the apoptosis in EtOH-treated groups, although the decline was lower than that of the DDRGK1-deleted groups, probably because of a certain degree of basal autophagy level in MEFs.
As autophagic degradation is closely related to autophagosome–lysosome fusion and lysosomal function44,45, we analyzed the function of lysosome during DDRGK1 loss. Though there was no significant change on STX17, SNAP29, and VAMP8 protein levels in our data, the interactions of them still remains to be verified in the future researches. Instead, the significantly decreased CTSD protein during DDRGK1 loss indicates a decreased lysosomal degradation capacity, which may in turn block the fusion of autophagosomes and lysosomes. In fact, reduced CTSD level has been demonstrated to impair the fusion of autophagosomes and lysosomes in the previous study46. Moreover, the aberrant accumulation of v-ATPase subunits also indicates a dysfunction in lysosomes. Given that v-ATPase is necessary for translocation of mTORC1 to lysosome surface, where mTORC1 is activated47, it is likely that the impaired mTORC1 activity is somewhat caused by v-ATPase dysfunction, which would explain why DDRGK1 deficiency has a dual effect of on autophagy regulation.
As DDRGK1 loss impairs lysosomal function, there might be a potential interaction between DDRGK1 and lysosome. To our surprise, we detected a colocalization of DDRGK1 with LAMP2, indicating a localization of DDRGK1 on lysosomal membrane or an interaction of DDRGK1 with lysosomal proteins. In fact, previous findings of Mümine Şentürk et al. have demonstrated an interaction between ubiquilin and v-ATPase, while ubiquilin loss impairs v-ATPase units levels and lysosomal acidification40. Consistently, DDRGK1 interacts with Atp6v0d1, one of the v-ATPase subunits presented in our proteomic data. Nevertheless, the acidification of lysosome was enhanced in our DDRGK1-deleted MEFs as indicated with an increased signal of Lyso-Tracker Red. Based on the contrary results, we speculate that lysosomes need to maintain an appropriate range of pH for normal function, either too high or too low may affect its degradation efficiency.
Recent studies show that DDRGK1 inhibits proteasomal degradation and is indispensable for maintaining the stability of related proteins, such as SOX9 (ref. 48) and IRE1α (ref. 49). Interestingly, in the present study, we found DDRGK1 is required for ubiquitin–proteasome-mediated v-ATPase subunits degradation, while DDRGK1 deletion blocked the proteasomal degradation, leading to elevated expression of Atp6v0d1 and Atp6v1a. This difference may be due to the different binding forms between DDRGK1 and target proteins. Therefore, we propose that DDRGK1 serves as a chaperone for v-ATPase units, and promotes normal lysosomal function by maintaining the stable expression of v-ATPase units.
In conclusion, our findings put forward a novel role of DDRGK1 in autophagy regulation via modulating lysosomal function, which provide a critical insight into DDRGK1 function. Thus, the study may provide a theoretical basis for the treatment strategies of various physiological diseases caused by DDRGK1 deficiency.
Materials and methods
Antibodies
Primary antibodies used in this study included DDRGK1 rabbit polyclonal antibody (Proteintech, 21445–1-AP, Wuhan, China); β-actin rabbit monoclonal antibody (Cell Signaling Technology, #4970, MA, USA); LC3B rabbit polyclonal antibody (Sigma, L7543, MO, USA); SQSTM1 mouse monoclonal antibody (Abcam, ab56416, Cambridge, UK); LAMP2 mouse monoclonal antibody (Santa Cruz, sc-18822, CA, USA); SNAP29 rabbit polyclonal antibody (Proteintech, 12704-1-AP); Stx17 rabbit polyclonal antibody (Proteintech, 17815-1-AP); VAMP8 rabbit polyclonal antibody (Proteintech, 15546-1-AP); Atg7 mouse monoclonal antibody (Proteintech, 67341-1-lg); Atp6v0d1 rabbit polyclonal antibody (Proteintech, 18274-1-AP); Atp6v1a rabbit polyclonal antibody (Proteintech, 17115-1-AP); CTSD rabbit polyclonal antibody (Proteintech, 21327-1-AP); 4E-BP1 rabbit monoclonal antibody (Cell Signaling Technology, #9644); phospho-4E-BP1 (Thr37/46) rabbit monoclonal antibody (Cell Signaling Technology, #2855); AMPK rabbit monoclonal antibody (Cell Signaling Technology, #5832); phospho-AMPKα (Thr172) rabbit monoclonal antibody (Cell Signaling Technology, #2535); and phospho-ULK1 (Ser757) rabbit monoclonal antibody (Cell Signaling Technology, #4202).
HRP-conjugated secondary antibodies, goat anti-mouse IgG (Abcam, ab6789) and goat anti-rabbit IgG (Abcam, ab6721), were all purchased from Abcam. Fluorophore-conjugated secondary antibodies, TRITC-conjugated goat anti-rabbit IgG (H + L; ZF-0316) and Alexa Fluor 488-conjugated goat anti-mouse IgG (H + L; ZF-0512) were purchased from Zhongshan Golden Bridge Biotechnology (Beijing, China).
Animals, tissues, cell culture, and treatment
All procedures with mouse were conducted in accordance with the guidelines of the Animal Research Institute Committee at Nanjing Agricultural University (SYXK-2017-0027). Immortalized MEFs were derived from the DDRGK1F/F:ROSA26-CreERT2 mice. Briefly, harvest mouse embryos (genotyping, DDRGK1F/F:ROSA26-Cre/ERT2+/+ or DDRGK1F/F:ROSA26-Cre/ERT2+/−) at E13 or E14. Dissect head and red organs of embryos, wash in PBS and place all embryos in a clean Petri dish. Finely mince the tissue using a sterile razor blade until it becomes possible to pipette. Add 1 mL of 0.05% trypsin/EDTA (Gibco, NY, USA). After each 5 min of incubation, dissociate cells by pipetting up and down thoroughly. The fibroblasts are the only cells that have the ability to attach to the flasks with the DMEM medium (HyClone, Utah, USA). Ideally, cells are 80–90% confluent after 24 h and at this stage, then transfect with large T lentivirus, select the positive immortalized cells with puromycin after 2 days, expand the positive cells. We also generated DDRGK1 hepatocyte-specific knockout mice (DDRGK1F/F:Alb-Cre+) by crossing DDRGK1 floxed mice with Alb-cre+ mice (C57BL/6 background), and harvested liver tissues from mice at age of 8–12 weeks to investigate the relationship between DDRGK1 and autophagy.
The MEFs were cultured in DMEM medium supplemented with 10% fetal bovine serum (Sigma) and 50 μg/mL penicillin/streptomycin at 37 °C with 5% CO2. MEFs were treated with 4-OHT (2 μM, Sigma) or ethanol (EtOH) for 4 days to induce DDRGK1 deletion or as a control. Baf-A1 (10 nM, Selleck, S1413, TX, USA) was added to the indicated medium for 2 h before the MEFs were harvested. For autophagy induction, rapamycin (200 nM, Selleck, S1039) was supplemented to control and DDRGK1-deleted MEFs for 24 h. To test the stability of proteins, MEFs were pretreated with 20 μg/mL CHX (Sigma, A6185) for 12 h to inhibit protein synthesis, then incubated with 2 μM MG132 (Sigma, M7449) for the indicated times.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from cultured MEFs using Trizol (Invitrogen, CA, USA). cDNA was synthesized using the PrimeScriptTM RT Master Mix (TAKARA, Shiga, Japan). qRT-PCR was performed using SYBR Green Master Mix (Vazyme, Nanjing, China) on a Step-One PCR system (Applied Biosystems, CA, USA). Primers used for the qRT-PCR were listed in the Supplementary Table S1 and all the expression data were normalized against β-actin as a control.
Plasmid transfection and RNA interference
For transfection of DNA plasmid, recombinant mCherry-EGFP-LC3 plasmid was transfected into cells using Lipofectamine 3000 reagent (Invitrogen), according to the manufacturer’s instruction. Approximately 1 μg of plasmid with 2 μL P3000 reagent and 2 μL of Lipofectamine 3000 reagent (according to 12-well plate) were separately added into 50 μL of Opti-MEM (Gibco) medium, then mixed together for 10 min at room temperature (RT) and added to cells in culture medium. For RNA interference, MEFs were transfected with Atg7 siRNA or negative control siRNA (GenePharma, Shanghai, China; see Supplementary Table S2 for sequences) via Lipofectamine RNAiMAX reagent (Invitrogen), according to the manufacturer’s instructions. Briefly, ~50 pmol of siRNA and 3 μL of Lipofectamine RNAiMAX reagent (according to 12-well plate) were separately added into 50 μL of Opti-MEM medium, then mixed together for 5 min and transfected into MEFs.
Immunoprecipitation and western blotting
The cells or tissues were lysed in IP lysis buffer or RIPA lysis buffer (Beyotime, Beijing, China) containing a complete protease inhibitor cocktail (Roche, Basel, Switzerland). Whole-cell lysates were centrifuged with 12,000 × g for 10 min at 4 °C and the supernatant were used for immunoprecipitation via incubation with Atp6v0d1 antibody at 4 °C overnight, followed by incubation with Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific, 88802, MA, USA) for 1 h to affiliate the antigen sample/antibody mixture. The magnetic beads were then washed three times with IP buffer and one time with purified water. After being eluted, immunoprecipitates were boiled in SDS loading buffer for 5 min at 95 °C and separated by SDS–PAGE Gel (Genscript, Nanjing, China), and transferred onto polyvinylidene fluoride membrane (Millipore, MA, USA). Western blot experiments were performed with the indicated antibodies and visualized by WesternBrightTM ECL kit (Advansta, CA, USA).
Flow cytometry with Annexin V-FITC/PI staining
Flow cytometry analysis using the Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme) was performed, according to the manufacturer’s instruction. In brief, harvested MEFs were incubated with FITC and PI for 10 min at RT and sorted by FACScan Flow Cytometer (Becton Dickinson, NJ, USA) afterward. Apoptotic events were recorded as a combination of Annexin V+/PI− (early apoptotic) and Annexin V+/PI+ (late apoptotic) events, and results were displayed as percentage of annexin V-positive cells analyzed using FlowJo software. All experiments were repeated in triplicate independently.
Immunofluorescent staining
MEFs seeded on coverslips in 12-well plates were fixed in 4% paraformaldehyde for 20 min at RT, then permeabilized by PBS with 0.5% Triton X-100 for 20 min at RT. MEFs were washed three times with PBS, then blocked with 5% BSA in PBS for 1 h at RT. Then, cells were incubated with anti-LAMP2 mouse antibody mixed with anti-LC3 or DDRGK1 rabbit antibodies at 4 °C overnight. After washing three times with PBS, cells were then incubated with TRITC-conjugated anti-rabbit IgG and Alexa Fluor 488-conjugated anti-mouse IgG antibody for 1 h, and counterstained with DAPI for 10 min. Finally, the coverslips were mounted on glass slides and observed under a laser-scanning confocal microscope (Carl Zeiss, Zeiss LSM 900, Jena, Germany) and images were photographed at random positions for each group.
Detection of autophagic flux
MEFs were seeded on coverslips in 12-well plates and transfected with the mCherry-EGFP-LC3 plasmid for ~32 h. Then, the living cells were quickly imaged with Zeiss LSM 900.
Lyso-Tracker Red staining
After 4-day treatment with EtOH or 4-OHT, MEFs seeded on coverslips in 12-well plates were then incubated with Lyso-Tracker Red (Beyotime) at a dilution of 1:20,000 for 15 min, and then counterstained with DAPI for 10 min. Images were photographed by Zeiss LSM 900.
Transmission electron microscope
MEFs treated with EtOH or 4-OHT for 4 days were harvested by trypsin digestion and fixed with 2.5% glutaraldehyde for 24 h at 4 °C. Samples were then postfixed with 1% osmium tetroxide for 2 h at 4 °C, washed, and stained with 2% aqueous uranyl acetate for 2 h. Then, samples were dehydrated with sequential gradient ethanol, embedded in Epon812, sectioned to ~50 nm, and mounted on carbon-coated copper grids. Ultrathin sections were counterstained with uranyl acetate and lead citrate and observed, using a transmission electron microscope (JEOL, JEM-1010, Tokyo, Japan).
Statistical analysis
Statistical analysis and graphs were performed and drawn with GraphPad Prism v.7 Software. Statistical significance was assessed by unpaired and two-tailed Student’s t test, and data were presented as means ± SEM of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
References
Komatsu, M. et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. Embo J. 23, 1977–1986 (2004).
Tatsumi, K. et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 285, 5417–5427 (2010).
Daniel, J. & Liebau, E. The ufm1 cascade. Cells 3, 627–638 (2014).
Wu, J., Lei, G., Mei, M., Tang, Y. & Li, H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J. Biol. Chem. 285, 15126–15136 (2010).
Quintero, M. et al. Cdk5rap3 is essential for intestinal Paneth cell development and maintenance. Cell Death Dis 12, 131 (2021).
Lemaire, K. et al. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE 6, e18517 (2011).
Kwon, J. et al. A novel LZAP-binding protein, NLBP, inhibits cell invasion. J. Biol. Chem. 285, 12232–12240 (2010).
Cai, Y. F. et al. UFBP1, a key component of the Ufm1 conjugation system, is essential for ufmylation-mediated regulation of erythroid development. PLoS Genet. 11, e1005643 (2015).
Cai, Y. et al. Indispensable role of the ubiquitin-fold modifier 1-specific E3 ligase in maintaining intestinal homeostasis and controlling gut inflammation. Cell Discov. 5, 7 (2019).
Zhu, H. et al. Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat. Commun. 10, 1084 (2019).
Levine, B. & Klionsky, D. J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 (2004).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Mortimore, G. E. & Poso, A. R. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu. Rev. Nutr. 7, 539–564 (1987).
Filomeni, G., De Zio, D. & Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388 (2015).
DePavia, A., Jonasch, E. & Liu, X. D. Autophagy degrades hypoxia inducible factors. Mol. Cell Oncol. 3, e1104428 (2016).
Mazure, N. M. & Pouyssegur, J. Hypoxia-induced autophagy: cell death or cell survival? Curr. Opin. Cell Biol. 22, 177–180 (2010).
Deegan, S., Saveljeva, S., Gorman, A. M. & Samali, A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol. Life Sci. 70, 2425–2441 (2013).
B’chir, W. et al. The eIF2 alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 (2013).
Bravo-San Pedro, J. M., Kroemer, G. & Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120, 1812–1824 (2017).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Mizushima, N., Yoshimori, T. & Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132 (2011).
Kliosnky, D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy 12, 443–443 (2016).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Li, Y. et al. Decoding three distinct states of the Syntaxin17 SNARE motif in mediating autophagosome-lysosome fusion. Proc. Natl Acad. Sci. USA 117, 21391–21402 (2020).
Ogata, M. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 26, 9220–9231 (2006).
Rashid, H. O., Yadav, R. K., Kim, H. R. & Chae, H. J. ER stress: autophagy induction, inhibition and selection. Autophagy 11, 1956–1977 (2015).
Hoyer-Hansen, M. & Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14, 1576–1582 (2007).
Senft, D. & Ronai, Z. A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148 (2015).
Zhang, M. et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 22, 1922–1934 (2015).
Bjorkoy, G. et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 (2005).
Komatsu, M. et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 (2007).
Kamada, Y. et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150, 1507–1513 (2000).
Matsuura, A., Tsukada, M., Wada, Y. & Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192, 245–250 (1997).
Ganley, I. G. et al. ULK1 center dot ATG13 center dot FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 (2009).
Jung, C. H. et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 (2009).
Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–U171 (2011).
Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007).
Leeman, D. S. et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283 (2018).
Senturk, M. et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 21, 384 (2019). -+.
N’Diaye, E. N. et al. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. Embo Rep. 10, 173–179 (2009).
Pan, H. et al. Autophagic flux disruption contributes to Ganoderma lucidum polysaccharide-induced apoptosis in human colorectal cancer cells via MAPK/ERK activation. Cell Death Dis. 10, 456 (2019).
Chen, S. et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy 11, 2346–2357 (2015).
Lieberman, A. P. et al. Autophagy in lysosomal storage disorders. Autophagy 8, 719–730 (2012).
Wang, C. et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 9, 3492 (2018).
Zheng, W. et al. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 59, 651–660 (2020).
Zoncu, R. et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683 (2011).
Egunsola, A. T. et al. Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J. Clin. Investig. 127, 1475–1484 (2017).
Liu, J. et al. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nat. Commun. 8, 14186 (2017).
Acknowledgements
We thank Prof. Honglin Li, Dr. Chunwan Lu, and Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta University for technical support.
Funding
This study was supported by the National Natural Science Foundation of China (grant Nos. 31970413, 81802858, and 31630072); NIH/NIDDK (grant No. 1R01DK113409); National Key R&D Program of China (grant No. 2018YFC1200201); and Start-up grant from Nanjing Agricultural University (grant No. 804090).
Author information
Authors and Affiliations
Contributions
Yan Cao designed the study, performed the experiments, analyzed the data, and wrote the manuscript. R.L. contributed a lot to research design and experimental operation. M.S. revised the manuscript. Chengyu Li, Y.Z., Q.J., S.L., and Chunwan Lu contributed to some technical support. Honglin Li provided part of materials. Honglin Liu and Yafei Cai conceived the project, discussed the results, and supervised the study. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics statement
All procedures with mice were conducted in accordance with the guidelines of the Animal Research Institute Committee at Nanjing Agricultural University (SYXK-2017-0027).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by X. Wang
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, Y., Li, R., Shen, M. et al. DDRGK1, a crucial player of ufmylation system, is indispensable for autophagic degradation by regulating lysosomal function. Cell Death Dis 12, 416 (2021). https://doi.org/10.1038/s41419-021-03694-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03694-9
- Springer Nature Limited
This article is cited by
-
Decreased eggshell strength caused by impairment of uterine calcium transport coincide with higher bone minerals and quality in aged laying hens
Journal of Animal Science and Biotechnology (2024)
-
A neuroprotective role of Ufmylation through Atg9 in the aging brain of Drosophila
Cellular and Molecular Life Sciences (2023)
-
Ubiquitin-like modifier 1 ligating enzyme 1 relieves cisplatin-induced premature ovarian failure by reducing endoplasmic reticulum stress in granulosa cells
Reproductive Biology and Endocrinology (2022)