
Abstract
Including apolipoprotein E-ε4 (APOE-ε4) status and older age into consideration may increase the accuracy of plasma Aβ42/Aβ40 detecting Aβ+ individuals, but the rationale behind this remains to be fully understood. Besides, both Aβ pathology and vascular diseases are related to neurodegeneration and cognitive decline, but it is still not fully understood how APOE-ε4 modulates these relationships. In this study, we examined 241 non-demented Alzheimer’s Disease Neuroimaging Initiative participants to investigate the associations among age, white matter hyperintensities (WMH), hypertension, hyperlipidemia, body mass index (BMI), plasma Aβ42/Aβ40 measured by liquid chromatography tandem mass spectrometry, and 18F-florbetapir Aβ PET as well as their prediction of longitudinal adjusted hippocampal volume (aHCV) and cognition in APOE-ε4 carriers and non-carriers. We found older age predicted faster WMH increase (p = 0.024) and cortical Aβ accumulation (p = 0.043) in APOE-ε4 non-carriers only, whereas lower plasma Aβ42/Aβ40 predicted faster cortical Aβ accumulation (p < 0.018) regardless of APOE-ε4 status. While larger WMH and underweight predicted (p < 0.05) faster decreases in aHCV and cognition in APOE-ε4 non-carriers, lower plasma Aβ42/Aβ40 predicted (p < 0.031) faster decreases in aHCV and cognition in APOE-ε4 carriers. Higher Aβ PET also predicted faster rates of aHCV (p = 0.010) in APOE-ε4 carriers only, but was related to faster rates of cognitive decline (p < 0.022) regardless of APOE-ε4 status. These findings may provide novel insights into understanding different mechanisms underlie neurodegeneration and cognitive decline in non-demented elderly adults with and without APOE-ε4 allele, which may help the design of anti-Alzheimer’s clinical trials.
Similar content being viewed by others


Introduction
β-amyloid(Aβ) pathology of Alzheimer’s disease (AD) [1, 2] can be evaluated by either PET imaging [3,4,5,6] or cerebrospinal fluid (CSF) [7]. However, the highly-cost and limited-availability of PET imaging, and the side effect of invasive lumbar puncture limit their use in screening Aβ positive (Aβ+) individuals. Recent studies suggested that plasma Aβ42/Aβ40 measured by liquid chromatography tandem mass spectrometry (LC-MS/MS) [8,9,10] or enzyme-linked immunosorbent assay (ELISA) [11, 12] or ultrasensitive single molecule array (SIMOA) [13,14,15] techniques may be of advantage for screening individuals with high risk of AD [16]. Apolipoprotein E-ε4 (APOE-ε4) is the most important genetic risk factor of sporadic AD [17]. Combining older age, APOE-ε4 allele and plasma Aβ42/Aβ40 may increase the accuracy of detecting Aβ PET or CSF Aβ42/Aβ40 positive individuals or predicting future diagnosis [9,10,11,12,13,14], but the rationale behind this remains to be fully understood.
The Dominantly Inherited Alzheimer Network group [18] have suggested that white matter hyperintensities (WMH) may be closely linked to AD progression. A few studies [19,20,21,22,23,24,25,26,27] reported significant relationship between WMH and Aβ pathology, whereas other groups [28,29,30,31,32,33] observed opposite results. Besides, two studies [34, 35] demonstrated that APOE-ε4 carriers have higher WMH than APOE-ε4 non-carriers, whereas other studies [36,37,38] found that APOE-ε4 allele might be independent of cerebrovascular disease. Furthermore, both WMH [26, 39,40,41,42,43] and plasma Aβ42/Aβ40 [44,45,46,47] may be associated with neurodegeneration or cognitive decline. However, it remains unclear how APOE-ε4 modulates the relationships between Aβ and WMH as well as their association with neurodegeneration and cognitive decline. Rather than focus on the differences between APOE-ε4 carriers and non-carriers, we aim to determine the association among age, vascular diseases, plasma Aβ and cortical Aβ plaques in addition to how Aβ pathologies and vascular diseases predict longitudinal neurodegeneration and cognitive decline in APOE-ε4 carriers and non-carriers separately. We assume that Aβ pathologies and vascular diseases may play different roles in neurodegeneration and cognitive decline in APOE-ε4 carriers and non-carriers.
In this study, we examined non-demented Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants to investigate the associations among age, vascular risk factors, WMH and plasma Aβ42/Aβ40, Aβ PET and their prediction of longitudinal WMH, cortical Aβ deposition, hippocampal atrophy, and cognitive decline in APOE-ε4 carriers and non-carriers. Our goal is to determine whether APOE-ε4 modulates the association between Aβ pathology and WMH, and their prediction of neurodegeneration and cognitive decline in non-demented elderly adults.
Participants and methods
Participants
Data used in this study were obtained from the ADNI database (ida.loni.usc.edu). The ADNI study was approved by institutional review boards of all participating centers, and written informed consent was obtained from all participants or their authorized representatives. In this study,126 cognitively unimpaired (CU) participants, and 115 participants with mild cognitive impairment (MCI) who had concurrent (acquisition intervals within 1 year) LC-MS/MS plasma Aβ42 and Aβ40, 18F-florbetapir (FBP) Aβ PET, structural MRI, WMH measurements, vascular risk factors, APOE-ε4 genotyping, and the cognitive test battery were included. Among 241 participants, 188 and 166 participants had at least 2-year’s longitudinal measurements of WMH and Aβ PET respectively, and 165 participants with at least two-year’s longitudinal MRI and cognitive tests measurements.
Vascular risk factors
Body mass index (BMI) was calculated according to the formula: BMI = (body weight in kg)/(body height in meters [2]). Hyperlipidemia (HLD) (key words “hyperlipidemia” or “‘cholesterol”) and hypertension (HTN) (key words “hypertension” or “HTN” or “high blood pressure”) histories were defined as present or absent by searching text fields within the participants’ self-reported medical history (RECMHIST.csv and INITHEALTH.csv files downloaded from ADNI website at March 23, 2021).
Plasma Aβ42 and Aβ40
LC-MS/MS plasma Aβ40 and Aβ42 were analyzed by the Washington University School of Medicine, St. Louis group. Briefly, targeted Aβ isoforms were immunoprecipitated with an anti-Aβ middomain antibody (HJ5.1) using a KingFisher (Thermo) automated immunoprecipitation platform. Immuno-enriched fractions were subsequently digested with Lys-N protease and subjected to LC-MS/MS as previously described [48] and also on the ADNI website (ida.loni.usc.edu). Absolute Aβ isoform concentrations were determined with a 15N-labeled internal standard for each isoform. The plasma Aβ42/Aβ40 ratio was calculated by dividing each plasma Aβ42 by plasma Aβ40.
PET imaging and analysis
Details on FBP PET image acquisition and analysis are given elsewhere (http://adni-info.org). Briefly, PET data were acquired in five-min frames from 50–70 min post-injection (http://adni-info.org). Pre-processed FBP PET and structural MRI scans were downloaded from the LONI website (ida.loni.usc.edu). Cross-sectional (at the baseline timepoint) FBP standardized uptake value ratios (SUVRs) were calculated by dividing uptake across frontal, cingulate, parietal and temporal regions by that in the whole cerebellum to generate cortical summary COMPOSITE SUVRs [49]. Individuals with COMPOSITE FBP SUVR ≥1.11 were defined as Aβ+ as we described previously [7]. Considering that a composite reference region (made up of brainstem, whole cerebellum, and eroded white matter) [49] has shown superior stability in longitudinal analyses of Aβ PET, SUVRs that referred to the composite reference were used to investigate longitudinal changes of FBP SUVR.
Hippocampal volume and white matter hyperintensities
Hippocampal volume (HCV) (cm3) was calculated across hemispheres from the structural MRI scans using Freesurfer, and adjusted by estimated total intracranial volume (TIV) using the approach employed by Jack et al. [50]. The adjusted hippocampal volume (aHCV) was calculated as the difference between the raw HCV and the expected HCV as described previously [51]. WMH was calculated at the University of California, Davis based on a Bayesian approach to segmentation of high resolution T1-weighted and FLAIR images as described previously [39] and also on the ADNI website. In order to compensate for individual variance in brain size and non-normal distribution, WMH was normalized to TIV and log10 transformed prior to analysis (Log10(WMH/TIV)).
Preclinical Alzheimer cognitive composite scores
Preclinical Alzheimer’s Cognitive Composite (PACC) scores [52] were calculated by combing the standard z scores (using the mean values of all the ADNI CU participants) of the Delayed Recall portion of the Alzheimer’s Disease Assessment Scale, the delayed recall score on the logical memory IIa subtest from the Wechsler Memory Scale, the digit symbol substitution test score from the Wechsler Adult Intelligence Scale–Revised and the MMSE total score as we previously described [51].
Statistical analysis
Normality of distributions was tested using the Shapiro-Wilk test and visual inspection of data. Data are presented as median (interquartile range (IQR)) or number (%) unless otherwise noted. Baseline characteristics were compared between APOE-ε4 carriers and non-carriers by using a two-tailed Mann-Whitney test or Fisher’s exact test.
In order to investigate how APOE-ε4 status affects the associations among age, vascular disease risk factors, WMH, and plasma Aβ42/Aβ40, we used generalized linear model (GLM) to examine the relationships of WMH and plasma Aβ42/Aβ40 with age, sex, HTN, HLD and BMI in APOE-ε4 non-carriers and carriers, adjusting for the diagnosis status. Afterwards, we first studied the associations of Aβ PET with age, plasma Aβ42/Aβ40 and WMH using Pearson’s correlation test, and further used GLM models to determine the cross-sectional relation of Aβ PET with plasma Aβ42/Aβ40 and WMH in APOE-ε4 non-carriers and carriers, adjusting for age, sex, and diagnosis status.
Subsequently, we used linear mixed-effect (LME) models to investigate the prediction of longitudinal WMH changes over time by baseline plasma Aβ42/Aβ40 and Aβ PET, and the prediction of longitudinal Aβ PET changes over time by baseline plasma Aβ42/Aβ40 and WMH in APOE-ε4 non-carriers and carriers, adjusting for age, sex, HTN, HLD, and BMI as well as their interaction with time, diagnosis status, and including a random slope and intercept for each participant.
In order to investigate how APOE-ε4 status affects the predictive effects of baseline plasma Aβ42/Aβ40, Aβ PET and WMH on perspective neurodegeneration and cognitive decline, we used LME models to study how baseline plasma Aβ42/Aβ40, Aβ PET and WMH predict longitudinal changes of aHCV and PACC over time in APOE-ε4 non-carriers and carriers, including the interaction of HTN and time, HLD and time, BMI and time, and adjusting for age, sex, education and diagnosis status.
Finally, we used LME models (including a random slope and intercept for each participant) to estimate: (1) annual rate of aHCV (ΔaHCV), adjusting for sex and diagnosis; (2) annual rate of PACC change (ΔPACC), adjusting for sex, education and diagnosis. Considering that WMH and plasma Aβ42/Aβ40 were related to ΔaHCV and ΔPACC in APOE-ε4 non-carriers and APOE-ε4 carriers respectively (See Figs. 3, 4 in Results), we then conducted the mediation analyses among age, WMH and ΔaHCV, and among WMH, ΔaHCV and ΔPACC in APOE-ε4 non-carriers, and the mediation analyses among plasma Aβ42/Aβ40, Aβ PET and ΔaHCV, and among plasma Aβ42/Aβ40, ΔaHCV and ΔPACC in APOE-ε4 carriers using latent variable modeling [53] (R; Lavaan package).
We selected two-sided p < 0.05 as the significance level unless otherwise noted. In the mediation analyses, all the variables were converted to standard z scores. Total, direct, and indirect associations were calculated via a 5000-iteration bootstrapping procedure. Longitudinal data of biomarkers were defined as the data that was closest in time to, and after, the baseline plasma Aβ42/Aβ40. Statistical analyses were performed in the statistical program R (v4.0.2, The R Foundation for Statistical Computing) unless otherwise noted.
Results
Demographics
Data in this study were acquired in ADNI between July 2010 and March 2021. The characteristics of 241 participants analyzed in this study can be found in Table 1. In total, 92 (38.2%) individuals were APOE-ε4 carriers. At baseline, APOE-ε4 carriers had slightly younger age, lower plasma Aβ42/Aβ40, higher FBP SUVR, and higher percentages of Aβ PET positivity than APOE-ε4 non-carriers, while no other difference was found. Notably, the percentage of MCI individuals between APOE-ε4 carriers and non-carriers was not significantly different from each other. Longitudinal data of different biomarkers were shown in Table 1 as well.
The cross-sectional association among age, vascular risk disease, plasma Aβ42/Aβ40, and Aβ PET
Greater WMH was related to older age in both APOE-ε4 non-carriers (standardized β value (βstd) = 0.46 [95% ci, 0.28, 0.64], p < 0.001) and APOE-ε4 carriers (βstd = 0.42 [95% ci, 0.18, 0.66], p = 0.001), and was related to HTN (βstd = 0.53 [95% ci, 0.13, 0.92], p = 0.010) in APOE-ε4 non-carriers only (Supplemental Fig. 1A, B). No significant association was found among age, vascular risk factors and plasma Aβ42/Aβ40 (Supplemental Fig. 1C, D). Older age, lower plasma Aβ42/Aβ40 and greater WMH were significantly associated with higher FBP SUVR regardless of APOE-ε4 status (Supplemental Fig. 2), whereas lower plasma Aβ42/Aβ40 but not greater WMH was significantly related to higher FBP SUVR after adjusting for age and sex (Supplemental Fig. 3).
Prediction of longitudinal WMH and Aβ PET
At follow-up, older age was associated with faster WMH increase over time in APOE-ε4 non-carriers (βstd = 0.0625 [95% CI, 0.0083, 0.1167], p = 0.024) but not in APOE-ε4 carriers (Fig. 1A, B, E, H). No other significant predictor of longitudinal WMH changes was found regardless of APOE-ε4 status (Fig. 1). Lower plasma Aβ42/Aβ40 but not greater WMH at baseline was related to faster rates of FBP SUVR increase in both APOE-ε4 non-carriers (βstd = −0.0887 [95% CI, −0.1265, −0.0509], p < 0.001) and APOE-ε4 carriers (βstd = −0.0631 [95% CI, −0.1152, −0.0111], p = 0.017) (Fig. 2A–C, F). Older age also predicted faster increases in FBP SUVR (βstd = 0.0430 [95% CI, 0.0013, 0.0847], p = 0.043) in APOE-ε4 non-carriers but not in APOE-ε4 carriers (Fig. 2E, H).

The estimated β values of each predictor for longitudinal WMH changes in linear mixed effect models in (A) APOE-ε4 non-carriers and (B) APOE-ε4 carriers. Prediction of longitudinal WMH changes over time by baseline plasma Aβ42/Aβ40, Aβ PET and age in APOE-ε4 non-carriers (C–E) and carriers (F–H). Note: lower, median and upper represent the value that cuts off the first 25%, 50% and 75% of the data when it is sorted in ascending order.

The estimated β values of each predictor for longitudinal Aβ PET changes in linear mixed effect models in (A) APOE-ε4 non-carriers and (B) APOE-ε4 carriers. Prediction of longitudinal Aβ PET changes over time by baseline plasma Aβ42/Aβ40, WMH and age in APOE-ε4 non-carriers (C–E) and APOE-ε4 carriers (F–H). Note: lower, median and upper represent the value that cuts off the first 25%, 50% and 75% of the data when it is sorted in ascending order.
Prediction of longitudinal hippocampal atrophy and cognitive decline
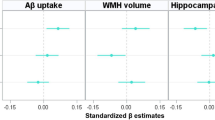
The predictors of longitudinal aHCV changes over time in APOE-ε4 non-carriers and carriers were summarized in Fig. 3A, B. In APOE-ε4 non-carriers, greater WMH (Fig. 3E, βstd = −0.051 [95% CI, −0.080, −0.023], p < 0.001) but not lower plasma Aβ42/Aβ40 (Fig. 3C) and higher FBP SUVR (Fig. 3D) at baseline predicted faster decreases in aHCV. Lower BMI (Fig. 3G, βstd = 0.031 [95% CI, 0.002, 0.060], p = 0.035) was also associated with faster rates of aHCV decreases. In contrast, lower plasma Aβ42/Aβ40 (Fig. 3H, βstd = 0.063 [95% CI, 0.012, 0.113], p = 0.016) and higher FBP SUVR (Fig. 3I, βstd = −0.067 [95% CI, −0.118, −0.016], p = 0.010) but not greater WMH (Fig. 3J) and lower BMI (Fig. 3L) at baseline predicted faster rates of aHCV decreases in APOE-ε4 carriers. No other significant predictor was found.

Associations of longitudinal adjusted hippocampal volume (aHCV) with baseline plasma Aβ42/Aβ40, Aβ PET, WMH, hypertension (HTN), hyperlipidemia (HLD) and body mass index (BMI) in (A) APOE-ε4 non-carriers and (B) APOE-ε4 non-carriers. Prediction of longitudinal aHCV changes over time by baseline plasma Aβ42/Aβ40, Aβ PET, WMH, HTN and BMI in APOE-ε4 non-carriers (C–G) and APOE-ε4 carriers (H–L). Notes: APOE-ε4 non-carriers (HTN−: n = 60, HTN+: n = 43), APOE-ε4 carriers (HTN−: n = 44, HTN+: n = 18); lower, median and upper represent the value that cuts off the first 25%, 50% and 75% of the data when it is sorted in ascending order.
The predictors of longitudinal PACC changes over time in APOE-ε4 non-carriers and carriers were summarized in Fig. 4A, B. In APOE-ε4 non-carriers, higher FBP SUVR (Fig. 4D, βstd = −0.106 [95% CI, −0.192, −0.019], p = 0.017), greater WMH (Fig. 4E, βstd = −0.084 [95% CI, −0.167, −0.002], p = 0.046) and lower BMI (Fig. 4G, βstd = 0.116 [95% CI, 0.032, 0.201], p = 0.007) but not lower plasma Aβ42/Aβ40 (Fig. 4C) at baseline predicted faster PACC decline. In contrast, lower plasma Aβ42/Aβ40 (Fig. 4H, βstd = 0.097[95% CI, 0.009, 0.184], p = 0.030), higher FBP SUVR (Fig. 4I, βstd = −0.104 [95% CI, −0.193, −0.016], p = 0.021) and HTN (Fig. 4K, βstd = −0.304 [95% CI, −0.477, −0.132], p < 0.001) but not greater WMH (Fig. 4J) and lower BMI (Fig. 4L) at baseline predicted faster PACC decline (Fig.4F, G, I) in APOE-ε4 carriers.

Associations of longitudinal PACC changes over time by baseline plasma Aβ42/Aβ40, Aβ PET, WMH, hypertension (HTN), Hyperlipidemia (HLD) and body mass index (BMI) in (A) APOE-ε4 non-carriers and (B) APOE-ε4 non-carriers. Prediction of longitudinal PACC changes over time by baseline plasma Aβ42/Aβ40, Aβ PET (FBP SUVR), WMH, HTN and BMI in APOE-ε4 non-carriers (C–G) and carriers (H–L). Notes: APOE-ε4 non-carriers (HTN−: n = 60, HTN+: n = 43), APOE-ε4 carriers (HTN−: n = 44, HTN+: n = 18); lower, median and upper represent the value that cuts off the first 25%, 50% and 75% of the data when it is sorted in ascending order.
The mediation analyses among age, WMH, Aβ pathology, neurodegeneration and cognitive decline
In the mediation analyses among age, WMH, ΔaHCV, and ΔPACC in APOE-ε4 non-carriers, WMH explained 24.4% (−0.11/(−0.45) = 0.244) of the association between age and ΔaHCV (Supplemental Fig. 4A), which explained 46.9% (−0.15/(−0.32) = 0.469) of the association between WMH and ΔPACC in APOE-ε4 non-carriers (Supplemental Fig. 4B).
In the mediation analyses among plasma Aβ42/Aβ40, Aβ PET, ΔaHCV, and ΔPACC in APOE-ε4 carriers, Aβ PET explained 31.5% (0.17/0.54 = 0.315) of the association between plasma Aβ42/Aβ40 and ΔaHCV (Supplemental Fig. 4C), which explained 74.4% (0.32/0.43 = 0.744) of the association between plasma Aβ42/Aβ40 and ΔPACC in APOE-ε4 carriers (Supplemental Fig. 4D).
Discussion
In this study, we investigated the relationships among age, vascular risk diseases, plasma Aβ42/Aβ40, Aβ PET, neurodegeneration and cognitive decline in non-demented elderly adults with and without APOE-ε4 allele respectively. Older age predicted faster rates of WMH increase and Aβ accumulation in APOE-ε4 non-carriers only, whereas lower plasma Aβ42/Aβ40 but not greater WMH predicted faster rates of Aβ accumulation regardless of APOE-ε4 status. Importantly, we found lower plasma Aβ42/Aβ40 predicted faster rates of hippocampal atrophy and cognitive decline in APOE-ε4 carriers only independent of cortical Aβ burden. Higher Aβ PET also predicted faster hippocampal atrophy over time in APOE-ε4 carriers only, but was related to faster cognitive decline regardless of APOE-ε4 status. In contrast, greater WMH and lower BMI predicted faster hippocampal atrophy and cognitive decline in APOE-ε4 non-carriers, implying vascular risk factors play an important role in neurodegeneration and cognitive decline in non-demented elderly adults without APOE-ε4 allele. These findings support our hypothesis that Aβ pathologies and vascular diseases play distinct roles in hippocampal atrophy and cognitive decline in non-demented elderly adults with and without APOE-ε4 allele.
Consistent with a few recent literatures [9,10,11,12,13,14], we found APOE-ε4 carriers showed lower plasma Aβ42/Aβ40, higher cortical Aβ deposition and larger probability of Aβ PET positivity compared to APOE-ε4 non-carriers. One genome-wide association study [54] found that APOE-ε4 allele had the strongest association with Aβ42 levels but not with Aβ40 levels in plasma measured by enzyme-linked immunosorbent assay (ELISA), and was significantly related to lower plasma Aβ42/Aβ40. However, we did not find significant relation among age, vascular risk factors and plasma Aβ42/Aβ40 regardless of APOE-ε4 status, which was in agreement with one previous ADNI study [28] in which plasma Aβ was measured by ELISA approach without considering APOE-ε4 status. Longitudinally, we found lower plasma Aβ42/Aβ40 predicted faster rates of Aβ accumulation regardless of APOE-ε4 status, which was consistent with the recent findings reported by the BIOFINER group [47]. In addition, we also found older age was related to faster Aβ accumulation rates in the absence of APOE-ε4 allele, providing further evidence for explaining why combing lower plasma Aβ42/Aβ40 and older age can improve the accuracy of detecting amyloid positivity defined by CSF [11, 14] or PET [9].
Previous studies [44,45,46,47] have reported significant association between plasma Aβ42/Aβ40 and neurodegeneration or cognitive decline, although none of them investigated how APOE-ε4 affects these relationships. Importantly, we further found that lower plasma Aβ42/Aβ40 predicted longitudinal neurodegeneration and cognitive decline in APOE-ε4 carriers only, but did not show significant predictive effect in APOE-ε4 non-carriers over around 5–6 years of median follow-up. These findings indicate that plasma Aβ42/Aβ40 may be useful for screening APOE-ε4 carriers (such as Alzheimer’s Prevention Initiative (API) Generation Study) as the potential participants for anti-AD clinical trials with neurodegeneration or cognitive decline as the ending points, whereas its application may be limited in non-demented elderly adults without APOE-ε4 allele.
Furthermore, we noticed that plasma Aβ42/Aβ40 and cortical Aβ burden independently predicted longitudinal hippocampal atrophy in APOE-ε4 carriers but not in APOE-ε4 non-carriers, suggesting that APOE-ε4 allele may probably modulate the association between Aβ pathology and hippocampal atrophy. The mediation analyses provided further evidence that cortical Aβ burden only partially explained the association between plasma Aβ42/Aβ40 and longitudinal hippocampal atrophy, which fully mediated the association between plasma Aβ42/Aβ40 and cognitive decline in APOE-ε4 carriers. In contrast, we found elevated cortical Aβ deposition significantly predicted longitudinal cognitive decline regardless of APOE-ε4 status, which may be explained by that increased cortical Aβ burden may be related to other aspect of neurodegeneration [55] that resulting in cognitive decline in addition to hippocampal atrophy in APOE-ε4 non-carriers. Together, these findings suggest that plasma Aβ42/Aβ40 may only detect one aspect of Aβ pathology even in the presence of APOE-ε4 allele, but lower plasma Aβ42/Aβ40 may be related to hippocampal atrophy independent of cortical Aβ burden in APOE-ε4 carriers.
In line with one recent study [33], we found greater WMH was related to higher cortical Aβ burden in univariate regression analyses, but this association disappeared after adjusting for age. Unlike a few reports [21, 23, 26], we found greater baseline WMH did not predict longitudinal cortical Aβ accumulation, neither lower baseline plasma Aβ42/Aβ40 nor higher cortical Aβ deposition predicted longitudinal WMH increase regardless of APOE-ε4 status. Consistent with our findings, several cross-sectional studies [28,29,30,31,32,33] did not find relation between WMH and Aβ pathology measured by CSF, PET imaging or immunohistochemistry. The distinct clinical status of participants may be related with the discrepancy, because previous studies reported no significant association between Aβ pathology and WMH in non-demented elderly adults [29, 32] but did find they related to each other in cohorts with demented elderly adults [26, 32]. Besides, the discordance may be also due to the fact that we did the analyses in APOE-ε4 non-carriers and carriers separately.
Older age showed strong correlation with greater WMH at baseline and predicted longitudinal WMH increases in APOE-ε4 non-carriers, suggesting older age may be tightly linked to white matter lesions in the absence of APOE-ε4 allele. Another key finding of this study was that age-related WMH increase predicted longitudinal hippocampal atrophy and cognitive decline in APOE-ε4 non-carriers. Consistent with our findings, the Mayo clinic group [43] and one previous ADNI study [39] also found older age was one significant predictor of vascular health, which showed direct correlation with AD pattern neurodegeneration or cognitive decline. One recent study [26] found APOE-ε4 only affected the associations of Aβ pathology with neurodegeneration and cognition, whereas did not modulate the associations of WMH with longitudinal neurodegeneration and cognitive decline. This suggests that greater WMH may affect brain atrophy and cognitive decline independent of APOE-ε4 allele. The mediation analyses between WMH-related neurodegeneration and cognitive decline in APOE-ε4 non-carriers showed that greater WMH partially explained the age-related hippocampal atrophy, which mediated the association between WMH and cognitive decline in APOE-ε4 non-carriers. In accordance with the findings of one previous study [56] that being underweight could increase the risk of dementia, we further demonstrated lower BMI predicted faster rates of hippocampal atrophy and cognitive decline in APOE-ε4 non-carriers, implying underweight may be also a risk factor of AD in non-demented elder adults without APOE-ε4 allele. Altogether, it is likely that older age-associated vascular diseases and underweight may contribute to faster neurodegeneration and cognitive decline in elderly adults without APOE-ε4 allele.
The strength of this study is that we analyzed how APOE-ε4 allele modulates the cross-sectional and longitudinal associations between plasma Aβ42/Aβ40, Aβ PET and white matter lesions as well as their prediction of longitudinal hippocampal atrophy and cognitive decline simultaneously up to 9 years’ follow-up. However, this study also has limitations. First, the inclusion criteria of ADNI excluded subjects with important vascular pathology, thus our analyses on vascular risk factors should be taken cautiously. Second, ADNI did not have longitudinal LC-MS/MS plasma Aβ42/Aβ40 data at this moment, so further longitudinal data and other techniques (ELISA [11, 12] or ultrasensitive single molecule array [13,14,15]) would be useful to validate our findings. Third, we noticed that APOE-ε4 carriers with hypertension showed faster rates of cognitive decline than those without hypertension, but the interpretation of this findings may be limited due to the relatively small sample size (n = 18) of APOE-ε4 carriers with hypertension. Considering that the academic community becomes increasingly concerned about the overuse and misinterpretation of significance testing and p values [57], thus the 95% ci values may be more informative than their corresponding p values in our statistical analysis.
In conclusions, this study suggests that lower plasma Aβ42/Aβ40 may predict hippocampal atrophy and cognitive decline in APOE-ε4 carriers, whereas the white matter lesion and underweight are more involved in hippocampal atrophy and cognitive decline of APOE-ε4 non-carriers. These findings are important for understanding different mechanisms related to neurodegeneration and cognitive decline in APOE-ε4 carriers and non-carriers, providing significant reference for anti-AD clinical trials.
References
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7:263–9.
Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. 2020;15:40.
Guo T, Brendel M, Grimmer T, Rominger A, Yakushev I. Predicting regional pattern of longitudinal β-amyloid accumulation by baseline PET. J Nucl Med. 2017;58:639–45.
Guo T, Dukart J, Brendel M, Rominger A, Grimmer T, Yakushev I. Rate of β-amyloid accumulation varies with baseline amyloid burden: Implications for anti-amyloid drug trials. Alzheimer’s Dement. 2018;14:1387–96.
Márquez F, Yassa MA. Neuroimaging biomarkers for Alzheimer’s disease. Mol Neurodegener. 2019;14:21.
Guo T, Landau SM, Jagust WJ. Detecting earlier stages of amyloid deposition using PET in cognitively normal elderly adults. Neurology. 2020;94:e1512–e1524.
Guo T, Shaw LM, Trojanowski JQ, Jagust WJ, Landau SM. Association of CSF Aβ, amyloid PET, and cognition in cognitively unimpaired elderly adults. Neurology. 2020;95:e2075–e2085.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54.
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93: https://doi.org/10.1212/WNL.0000000000008081.
West T, Kirmess KM, Meyer MR, Holubasch MS, Knapik SS, Hu Y, et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener. 2021;16:30.
Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related β-amyloid status. JAMA Neurol. 2019;76:1060.
Stocker H, Nabers A, Perna L, Möllers T, Rujescu D, Hartmann A, et al. Prediction of Alzheimer’s disease diagnosis within 14 years through Aβ misfolding in blood plasma compared to APOE4 status, and other risk factors. Alzheimer’s Dement. 2020;16:283–91.
De Meyer S, Schaeverbeke JM, Verberk IMW, Gille B, De Schaepdryver M, Luckett ES, et al. Comparison of ELISA- and SIMOA-based quantification of plasma Aβ ratios for early detection of cerebral amyloidosis. Alzheimers Res Ther. 2020;12:162.
Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, van Berckel BNM, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol. 2018;84:648–58.
Vergallo A, Mégret L, Lista S, Cavedo E, Zetterberg H, Blennow K, et al. Plasma amyloid β 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer’s disease. Alzheimer’s Dement. 2019;15:764–75.
Zetterberg H, Blennow K. Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol Neurodegener. 2021;16:10.
Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20:68–80.
Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TLS, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79:929–39.
Osborn KE, Liu D, Samuels LR, Moore EE, Cambronero FE, Acosta LMY, et al. Cerebrospinal fluid β-amyloid42 and neurofilament light relate to white matter hyperintensities. Neurobiol Aging. 2018;68:18–25.
Weaver NA, Doeven T, Barkhof F, Biesbroek JM, Groeneveld ON, Kuijf HJ, et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol Aging. 2019;84:225–34.
Moscoso A, Rey-Bretal D, Silva-Rodríguez J, Aldrey JM, Cortés J, Pías-Peleteiro J, et al. White matter hyperintensities are associated with subthreshold amyloid accumulation. Neuroimage. 2020;218:116944.
Hilal S, Akoudad S, Van Duijn CM, Niessen WJ, Verbeek MM, Vanderstichele H, et al. Plasma amyloid-β levels, cerebral small vessel disease, and cognition: the rotterdam study. J Alzheimer’s Dis. 2017;60:977–87.
Scott JA, Braskie MN, Tosun D, Maillard P, Thompson PM, Weiner M, et al. Cerebral amyloid is associated with greater white-matter hyperintensity accrual in cognitively normal older adults. Neurobiol Aging. 2016;48:48–52.
Marnane M, Al-Jawadi OO, Mortazavi S, Pogorzelec KJ, Wang BW, Feldman HH, et al. Periventricular hyperintensities are associated with elevated cerebral amyloid. Neurology. 2016;86:535–43.
Caballero MÁA, Song Z, Rubinski A, Duering M, Dichgans M, Park DC, et al. Age‐dependent amyloid deposition is associated with white matter alterations in cognitively normal adults during the adult life span. Alzheimer’s Dement. 2020;16:651–61.
Dadar M, Camicioli R, Duchesne S, Collins DL. The temporal relationships between white matter hyperintensities, neurodegeneration, amyloid beta, and cognition. Alzheimer’s Dement Diagnosis, Assess Dis Monit. 2020;12:7250–7.
Grimmer T, Faust M, Auer F, Alexopoulos P, Förstl H, Henriksen G, et al. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging. 2012;33:2766–73.
Toledo JB, Vanderstichele H, Figurski M, Aisen PS, Petersen RC, Weiner MW, et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011;122:401–13.
Rutten-Jacobs LCA, de Leeuw F-E, Geurts-van Bon L, Gordinou de Gouberville MC, Schepens-Franke AN, Dederen PJ, et al. White Matter Lesions Are Not Related to β-Amyloid Deposition in an Autopsy-Based Study. Curr Gerontol Geriatr Res. 2011;2011:1–5.
Marchant NL, Reed BR, DeCarli CS, Madison CM, Weiner MW, Chui HC, et al. Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol Aging. 2012;33:1006.e25–1006.e36.
Lo RY, Jagust WJ. Alzheimer’s Disease Neuroimaging Initiative. Vascular burden and Alzheimer disease pathologic progression. Neurology. 2012;79:1349–55.
Arfanakis K, Evia AM, Leurgans SE, Cardoso LFC, Kulkarni A, Alqam N, et al. Neuropathologic correlates of white matter hyperintensities in a community-based cohort of older adults. J Alzheimer’s Dis. 2020;73:333–45.
Garnier-Crussard A, Bougacha S, Wirth M, André C, Delarue M, Landeau B, et al. White matter hyperintensities across the adult lifespan: relation to age, Aβ load, and cognition. Alzheimers Res Ther. 2020;12:127.
Brickman AM, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Provenzano FA, et al. APOE ε4 and risk for Alzheimer’s disease: do regionally distributed white matter hyperintensities play a role? Alzheimer’s Dement. 2014;10:619–29.
Sudre CH, Cardoso MJ, Frost C, Barnes J, Barkhof F, Fox N, et al. APOE ε4 status is associated with white matter hyperintensities volume accumulation rate independent of AD diagnosis. Neurobiol Aging. 2017;53:67–75.
Kim HJ, Ye BS, Yoon CW, Cho H, Noh Y, Kim GH, et al. Effects of APOE ε4 on brain amyloid, lacunar infarcts, and white matter lesions: Astudy among patients with subcortical vascular cognitive impairment. Neurobiol Aging. 2013;34:2482–7.
Hong YJ, Yoon B, Shim YS, Cho AH, Shin HE, Kim YI, et al. APOE ε4 allele status in Korean dementia patients with severe white matter hyperintensities. J Alzheimer’s Dis. 2011;24:519–24.
Kandel BM, Avants BB, Gee JC, McMillan CT, Erus G, Doshi J, et al. White matter hyperintensities are more highly associated with preclinical Alzheimer’s disease than imaging and cognitive markers of neurodegeneration. Alzheimer’s Dement Diagnosis. Assess Dis Monit. 2016;4:18–27.
Carmichael O, Schwarz C, Drucker D, Fletcher E, Harvey D, Beckett L, et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol. 2010;67:1370–8.
Lindemer ER, Greve DN, Fischl B, Salat DH, Gomez-Isla T. White matter abnormalities and cognition in patients with conflicting diagnoses and CSF profiles. Neurology. 2018;90:e1461–e1469.
Wang YL, Chen W, Cai WJ, Hu H, Xu W, Wang ZT, et al. Associations of white matter hyperintensities with cognitive decline: a longitudinal study. J Alzheimer’s Dis. 2020;73:759–68.
Fiford CM, Manning EN, Bartlett JW, Cash DM, Malone IB, Ridgway GR, et al. White matter hyperintensities are associated with disproportionate progressive hippocampal atrophy. Hippocampus. 2017;27:249–62.
Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Lowe VJ, Graff-Radford J, et al. Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann Neurol. 2017;82:706–18.
Okereke OI, Xia W, Selkoe DJ, Grodstein F. Ten-year change in plasma amyloid beta levels and late-life cognitive decline. Arch Neurol. 2009;66:1247–53.
Verberk IMW, Hendriksen HMA, van Harten AC, Wesselman LMP, Verfaillie SCJ, van den Bosch KA, et al. Plasma amyloid is associated with the rate of cognitive decline in cognitively normal elderly: the SCIENCe project. Neurobiol Aging. 2020;89:99–107.
Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, et al. Plasma amyloid‐β and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimer’s Dement. 2015;11:249–57.
Pereira JB, Janelidze S, Stomrud E, Palmqvist S, van Westen D, Dage JL et al. Plasma markers predict changes in amyloid, tau, atrophy and cognition in non-demented subjects. Brain. 2021;144:2826–2836.
Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017;13:841–9.
Landau SM, Fero A, Baker SL, Koeppe R, Mintun M, Chen K, et al. Measurement of longitudinal β-amyloid change with 18 f-florbetapir pet and standardized uptake value ratios. J Nucl Med. 2015;56:567–74.
Jack CR, Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, et al. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol. 2012;71:765–75.
Guo T, Korman D, Baker SL, Landau SM, Jagust WJ. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in alzheimer’s disease pathophysiology. Biol Psychiatry. 2021;89:786–94.
Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71:961–70.
Rosseel Y. lavaan: an R package for structural equation modeling. J Stat Softw. 2012;48:1–93.
Damotte V, Lee SJ, Chouraki V, Grenier‐Boley B, Simino J, Adams H et al. Plasma amyloid β levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: A genome‐wide association study in over 12,000 non‐demented participants. Alzheimer’s Dement. 2021;17:1663–1674.
Bozoki AC, Zdanukiewicz M, Zhu DC. The effect of β-amyloid positivity on cerebral metabolism in cognitively normal seniors. Alzheimer’s Dement. 2016;12:1250–8.
Qizilbash N, Gregson J, Johnson ME, Pearce N, Douglas I, Wing K, et al. BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. 2015;3:431–6.
Harrington D, D’Agostino RB, Gatsonis C, Hogan JW, Hunter DJ, Normand SLT, et al. New guidelines for statistical reporting in the journal. N. Engl J Med. 2019;381:285–6.
Acknowledgements
The authors would like to thank William Jagust and Susan Landau for their comments on data interpretation, and all the ADNI participants and staff for their contributions to data acquisition. The Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding
This work was supported by the National Natural Science Foundation of China (82171197) and Shenzhen Bay Laboratory Open Project (SZBL2020090501014).
Author information
Authors and Affiliations
Consortia
Contributions
DS: study concept and design, data processing, statistical analysis, interpretation of the results, and writing and revising the manuscript. SX and AL: data processing, interpretation of the results and critical revision of the manuscript. QW, HG, YH, HX, WG: interpretation of the results and critical revision of the manuscript. LZ: interpretation of the results, critical revision of the manuscript and providing supervision. T.G.: study concept and design, interpretation of the results, writing and revising the manuscript, obtaining funding and providing supervision. ADNI provided all data used for this study.
Corresponding author
Ethics declarations
Competing interests
Dr. Guo services in Alzheimer’s Association as the ISTAART “PIA to Elevate Early Career Researchers (PEERs)” Asia lead. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, D., Xie, S., Li, A. et al. APOE-ε4 modulates the association among plasma Aβ42/Aβ40, vascular diseases, neurodegeneration and cognitive decline in non-demented elderly adults. Transl Psychiatry 12, 128 (2022). https://doi.org/10.1038/s41398-022-01899-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01899-w
- Springer Nature Limited