
Abstract
Currently, over five million Americans suffer with Alzheimer’s disease (AD). In the absence of a cure, this number could increase to 13.8 million by 2050. A critical goal of biomedical research is to establish indicators of AD during the preclinical stage (i.e. biomarkers) allowing for early diagnosis and intervention. Numerous advances have been made in developing biomarkers for AD using neuroimaging approaches. These approaches offer tremendous versatility in terms of targeting distinct age-related and pathophysiological mechanisms such as structural decline (e.g. volumetry, cortical thinning), functional decline (e.g. fMRI activity, network correlations), connectivity decline (e.g. diffusion anisotropy), and pathological aggregates (e.g. amyloid and tau PET). In this review, we survey the state of the literature on neuroimaging approaches to developing novel biomarkers for the amnestic form of AD, with an emphasis on combining approaches into multimodal biomarkers. We also discuss emerging methods including imaging epigenetics, neuroinflammation, and synaptic integrity using PET tracers. Finally, we review the complementary information that neuroimaging biomarkers provide, which highlights the potential utility of composite biomarkers as suitable outcome measures for proof-of-concept clinical trials with experimental therapeutics.
Similar content being viewed by others

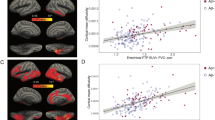
Alzheimer’s disease and the need for biomarkers
Alzheimer’s disease (AD) is the most common cause for dementia [1]. Although there are various subtypes, the most common form is amnestic and severely impacts episodic memory [2]. With the exception of AD cases caused by genetic mutations (i.e. familial AD), age is the greatest risk factor. Currently, one in ten people 65 years of age or older have AD. In less than 60 years, life expectancy in the United States has increased by 9 years and the population of people 65 years of age and above has increased by 34 million people (16 million to 50 million). An estimated 5.5 million Americans currently suffer with AD and in the absence of effective treatment or a cure, this number could increase to 13.8 million by 2050 [1].
A critical goal of biomedical research is to establish indicators of AD during the preclinical stage (i.e. biomarkers) allowing for early diagnosis and intervention. These biomarkers are quantifiable characteristics of biological processes related to Alzheimer’s disease that are linked to clinical endpoints and thus can be used as surrogates for the disease process. Over the last decade, numerous advances have been made in developing biomarkers for AD using neuroimaging approaches. These approaches offer tremendous versatility in terms of understanding and targeting pathophysiological mechanisms such as structural decline (e.g. loss in volume, cortical thinning), functional decline (e.g. fMRI hyperactivity, altered network connectivity), white matter decline (e.g. diffusion anisotropy reduction, white matter pathology), and pathology aggregation (e.g. amyloid and tau PET).
In this review, we survey the state of the literature on neuroimaging approaches to developing novel biomarkers for AD, focusing on amnestic, late-onset (LOAD). We discuss advantages and limitations of each method and suggest that combining imaging modalities to create “composite biomarkers” may be a productive approach. These biomarkers may provide utility as potential outcomes for proof-of-concept clinical trials with experimental therapeutics.
Pathology and spatiotemporal spread
Neuropathological staging criteria of AD-related changes originally indicated that although the distribution of beta-amyloid (Aβ) neuritic plaques varies widely, neurofibrillary tangles and neuropil threads show a distribution pattern that allow for the differentiation of six stages [3]. Stages I-II show alterations that are confined to the transentorhinal region, which spread to limbic (Stage III-IV), and finally to isocortical regions (Stage V-VI).
More recently, pathology studies have indicated that intraneuronal aggregations of the protein tau seem to precede the extracellular deposition of Aβ by approximately a decade [4, 5]. Notably, non-argyrophillic tau lesions are thought to first appear in the locus coeruleus prior to the appearance of argyrophillic tau lesions caused by neurofibrillary tangles (NFTs) within the transentorhinal region of the cerebral cortex [6]. Intraneuronal inclusions consisting of aggregated protein tau appear in selectively vulnerable cell types that appear to spread in a regionally and temporally specific manner that is independent of proximity to affected area [7].
A key advantage of using brain imaging techniques is that they operate at a higher level of spatiotemporal sensitivity than fluid biomarkers, thereby offering an opportunity to stage progression of the disease. Thus far, imaging using combinations of in vivo PET and MRI techniques have shown progression patterns that largely recapitulate staging based on post-mortem histology [8].
Biomarker-based staging of preclinical Alzheimer’s disease
Identifying early biomarkers prior to the onset of disease symptoms is of critical importance to the field. It is thought that early intervention (i.e. during the pre-symptomatic stage) will be far more effective than later intervention, once the neurodegenerative cascade has set in. Historically, AD has been viewed as a disease of clinical symptoms in the clinical setting. By classifying AD in this manner, its diagnosis would likely include a considerable amount of non-AD cases as defined by its pathological characteristics. In 2011, the National Institute on Aging and the Alzheimer’s Association (NIA-AA) Working Group put forth staging criteria that incorporate neuroimaging biomarkers [9]. The authors presented a conceptual framework and operational research criteria for preclinical AD where Stage 1 is characterized by the presence of asymptomatic β-amyloidosis, or increased amyloid burden. Stage 2 includes neuronal injury and evidence of neurodegenerative change. Lastly, stage 3 additionally includes evidence of subtle cognitive decline, which is not yet sufficient for clinical diagnosis. The new research framework proposed by the NIA-AA defines AD pathologically with the use of biomarkers, which could potentially differentiate cases that clinically resemble AD such as hippocampal sclerosis. This framework additionally allows for staging using either fluid or neuroimaging biomarkers. However, certain features, which may be critical for the pathophysiology of the disease, could only be detected using imaging techniques. Hippocampal hyperactivity on task-activated functional MRI is one such example. Ewers et al. [10] and Leal and Yassa [11] include this feature in staging the disease and highlight that it seems to appear within a temporally constrained window.
Jack and Holtzman [12] proposed several time-dependent models of AD that take into consideration varying age of onset as well as co-morbid pathologies. Out of the five biomarkers proposed, three were imaging biomarkers (amyloid PET, structural MRI, and FDG PET). Importantly, anatomical information from imaging biomarkers provides crucial disease-staging information. This implies an advantage for imaging biomarkers over fluid biomarkers, because imaging can distinguish the different phases of the disease both temporally and anatomically.
The NIA-AA research framework has since been updated [13, 14] to focus on A/T/N criteria, first proposed by Jack and colleagues [15] and pave the path to more personalized diagnosis and treatment. The new framework highlights the value of positive amyloid biomarkers (A) to specifically indicate AD-related processes. Pathological tau (T) is only taken to indicate an AD-related process in the presence of amyloid positivity. Finally, (N) biomarkers are thought to provide nonspecific information about neuronal injury and neurodegenerative change.
The combination of amyloid with other biomarkers can then be used to stage AD progression. Additionally, according to this new framework, the presence of tau and neurodegeneration in the absence of amyloidosis is considered evidence for non-AD pathological processes. An important aspect of the 2018 NIA-AA working group framework is the flexibility to include additional biomarkers in future iterations. In our survey of neuroimaging methods, we will make the case that there are several methods for measuring A, T, and N pathologies, but also discuss new approaches to imaging additional biomarkers which may be integrated in biomarker models in the future (e.g. neuroinflammation).
Imaging Amyloid Burden
Given the critical importance of identifying amyloid pathology in the brain as an early stage of AD progression, positron emission tomography (PET) scans with radiolabeled tracers specific to Aβ have become fairly commonplace in the research setting. The pathological Aβ peptide is generated by abnormal proteolytic processing of a physiological constituent of the nerve cell membrane, the amyloid precursor protein (APP). PET scans operate on the principle that positron-emitting radioligands accumulate in a region of interest. The positively-charged positrons encounter negatively-charged electrons, which results in annihilation releasing gamma photons that are detected by scintillation detectors [16]. This method can be used to image Aβ in vivo via radiolabeled tracers, which are injected via a bolus injection, followed by a waiting period to allow for uptake by brain tissue.
Amyloid tracers were developed via the modification of the histological dye, thioflavin-T, which has a high affinity to fibrillar and cerebrovascular amyloid, is cleared rapidly from normal brain tissue, and crosses the blood-brain barrier in sufficient amounts to be imaged in vivo [17]. Amyloid burden imaging was first explored with carbon-based tracers [11C] such as Pittsburgh Compound B (PiB), but the development of fluorine-based tracers [18F] has allowed for a wider availability of these longer lasting tracers facilitating widespread use. These tracers include florbetapir, florbetaben, and flutmetamol, which have an extended half-life (~110 minutes) as compared to [11C] tracers (20 minutes).
Most amyloid imaging studies point to the parietal cortices as the earliest sites of amyloid deposition [18]. Notably, these regions (posterior cingulate, restrosplenial cortex, precuneus) are heavily interconnected with the medial temporal lobes (MTL) [19], which are sites for early aggregation of tau pathology. Thus, the progression of the disease may be influenced by the anatomical and functional connectivity between the posterior cortices and the MTL. Amyloid tracers additionally bind to cerebrovascular amyloid. Cerebral Amyloid Angiopathy (CAA) is a feature of AD, is characterized by cortical vascular amyloid deposits, and is associated with cortical tissue loss, vascular dysfunction and cognitive decline [20, 21]. CAA severity is also associated with allocortical microinfarcts located in the hippocampal CA1 subfield [21]. Therefore, combining amyloid-PET with other imaging modalities may provide clues into the pathological sequence of events.
While amyloid tracers have produced similar qualitative findings across studies, institutions, and tracers, they vary in quantitative outcome measures of tracer retention. In an effort to standardize quantitative amyloid imaging measures, the Centiloid Project Working Group was formed in 2012 during the Alzheimer’s Imaging Consortium pre-meeting to the Alzheimer’s Association International Conference. The group works to harmonize across [11C] PiB and [18F] tracers using a percentile-based normalized system to address the variability of the method and tracer of each analysis (scales the outcome to a 0 to 100 scale). Despite these efforts, there still remain differences among tracers and in their sensitivity. Other factors that differ across studies include acquisition duration, target and reference region choice, partial volume correction, scanner differences, as well as differences in reconstruction algorithms and methods of attenuation correction.
A major limitation to amyloid imaging and studies of amyloid burden in general is a poorly understood relationship with cognition. It has been argued that since changes in amyloid may occur earlier than cognitive symptoms, such a relationship may not be expected. However, in the absence of a strong relationship, it remains unclear whether amyloid burden, in and of itself, is pathological or whether it is a sentinel for another pathology that may have more severe consequence on neural integrity. Understanding the latter is critical, especially as numerous clinical trials have targeted amyloid pathology as an attempt to modify the disease process.
Several studies have recently attempted to shed light on the relationship between amyloid and cognitive impairment [22,23,24]. These studies have found evidence for a link between Aβ accumulation and cognitive outcomes that appears to be mediated by neurodegenerative changes (e.g. cortical thinning and hippocampal volume loss). However, Aβ accumulation does not appear to be a precondition for neurodegenerative decline. For example, Wirth et al. [25] demonstrated that neurodegenerative changes could significantly predict cognitive performance in the absence of Aβ pathology. Most recently, it has been shown that subthreshold amyloid deposition predicts tau deposition in aging [26] suggesting that amyloid binding varies on a continuum. Despite its limitations, amyloid PET has been a tremendously informative tool in AD biomarker research, not only for staging disease progression, but additionally to select individuals for participation in biomarker-based clinical trials during the asymptomatic phase (secondary prevention trials), such as the A4 trial [27].
Imaging Tau Burden
Tau is a neuronal protein that is produced throughout the nervous system and promotes self-assembly of axonal microtubules and stabilizes them [28]. Homeostatic shifts between a less highly phosphorylated state, where tau is bound to axonal microtubules, and a more highly phosphorylated state, where tau is soluble in the axoplasm, are enabled by axonal kinases and phosphates [29]. Changes in the equilibrium can give rise to conformational changes that lead to aggregation and changes in solubility that alters the functional role of tau and allowing for it to become resistant to autophagy and other mechanisms that regulate the removal of tau [30, 31].
Soluble hyperphosphorylated tau aggregates into spherical units of nucleation that then assembles linearly and forms ribbons of protofibrils with a β-sheet core. The absence or recurrence of twists enables classification of hyperphosphorylated tau into straight filaments, paired helical filaments (PHF) with regular twists, or irregularly twisted filaments. Recent evidence also suggests that tau pathology may spread trans-synaptically, in a prion-like fashion [32, 33] and that a critical component of the pathological cascade may be the conversion of tau monomers from an inert to a seed-competent form [34].
Development of selective tau PET tracers started as early as 2002 with quinolone and benzimidazole derivatives for their affinity to bind to PHFs. The cooccurrence of PHFs with Aβ provided an additional challenge as Aβ has the potential to also bind to the ligand, but a 25 fold selectivity for PHF over Aβ has been achieved [35]. Notably, the presence of other tauopathies have been described [36]. Although the 3R/4R isoform of tau that these tracers binds to overlaps with other tauopathies, the spatial distribution of tracer binding may help discriminate between pathologies [35].
Unlike Aβ plaque deposition, human post-mortem studies indicate that NFT density correlates with neurodegeneration and cognitive impairment [37, 38]. Several tau PET studies have shown a close relationship between patterns of tau deposition and atrophy measures [39,40,41] and recent work has shown memory scores to be strongly correlated with medial temporal tau tracer uptake, whereas whole-brain measures showed weak associations with memory and MTL atrophy, supporting the notion that regional tau measures have greater sensitivity to early neurodegeneration and memory decline compared to global measures of tau [39, 42]. Older age is also associated with binding in the medial temporal lobe (MTL), the extent of which is associated with memory deficits [43]. Consistent with prior histopathological reports, PET detection of tau outside the MTL is associated with the presence of cortical Aβ binding even at a subthreshold level [26, 44]. Overall, tau imaging appears capable of detecting regionally specific patterns of tau deposition that follow Braak and Braak staging of NFT pathology [8, 39, 42]. However, disentangling primary age-related tauopathy (PART) and AD will be challenging since there is considerable overlap in the medial temporal lobe.
Of the available tracers, [18F]-1451 (or T-807) has been characterized the most extensively and has demonstrated increased uptake and signal detection in patients with prodromal AD [35].
While tau PET’s reliability is still under investigation, test-retest reliability of the tracer was recently examined in a sample of 21 subjects (including MCI and AD patients) and showed low variability within subject. Intra-class correlation of SUVR’s was above 0.92 across all regions tested, which indicates high test-retest reliability and suggests that this method can be used to detect changes in tau burden over time [45]. Despite its many advantages, [18F]-1451 and similar tracers appear to bind to some dense core plaques [46], melanin-containing structures [47, 48], and minimal binding to TDP-43 [47]. This has called into question the utility of these first-generation tracers to specifically bind to tau pathology. Second generation tracers, such as [18F] MK-6240 fare better in terms of off-target binding, but large-scale studies with these tracers are still lacking [49].
A major strength of tau PET imaging is the ability to recapitulate histology-based Braak and Braak staging of tau pathology. While longitudinal studies remain necessary to validate the approach, initial cross-sectional data suggest a medial temporal to isocortical progression [50]. Limitations of tau PET imaging are largely similar to amyloid PET imaging and include issues with harmonization across studies and tracers and choice of reference region.
Imaging Neural Injury and Neurodegeneration
Synaptic Integrity and Circuit Connectivity – Resting state fMRI
Functional MRI techniques are based on blood-oxygenation-level-dependent (BOLD) contrast which is associated with neural activity at the population level. Resting-state functional magnetic resonance imaging (rs-fMRI) studies examine the temporal correlation of the BOLD signal between the regions of interest (or functional connectivity) by analyzing task-independent spontaneous fluctuations in brain networks [51, 52]. An emerging systems-based model of AD considers the large-scale disruptions across the course of AD. In preclinical AD, studies have generally noted that resting state fMRI (rsfMRI) is linked to metabolic changes (indexed by PET imaging) and precedes neurodegeneration (review by [53]). Most analyses have focused on the default mode network (DMN) [54, 55] - a network that involves the medial prefrontal cortex, posterior cingulate cortex, precuneus, anterior cingulate cortex, parietal cortex, and the medial temporal lobe, including the hippocampus [56, 57]. As regions within the DMN are highly overlapping with the spatial distribution of both amyloid and tau pathology [57], resting state fMRI can offer important information on the integrity of these circuits and the degree to which their synaptic connectivity may be affected by the disease process. While some studies have found that alterations to DMN connectivity become more dramatic with disease progression, others have found dynamic changes that relate to Aß and tau-specific profiles [40, 58,59,60,61,62,63].
In addition to changes in the DMN, some studies have suggested that connectivity within the MTL is also disrupted with aging and AD. For example, Yassa et al. [64] showed an age-related decrease in connectivity between the entorhinal cortex and the dentate and CA3 regions of the hippocampus, the extent of which was correlated with memory deficits. Connectivity changes in other networks have also been reported [65]. For example, the interaction between the DMN and the salience network, which consists of anterior insula, dorsal anterior cingulate cortex, is associated with increased connectivity in amyloid-positive individuals with low neocortical tau, and decreased connectivity as a function of elevated Tau-PET signal [62]. Functional connectivity is thought to be an early marker of synaptic pathology that may be associated with isolation of the hippocampus from its cortical input.
Reduced Inhibition and Hippocampal Hyperactivity – Task activated fMRI
Numerous studies have used task-activated fMRI to examine functional changes in MCI and early AD. Dickerson and colleagues [66] found increased hippocampal activity during learning in individuals with MCI compared to normal controls and individuals with AD. Another study [67] using an independent component analysis found that less impaired MCI patients showed this increase, while more impaired MCI patients showed a decrease in activity similar to mild AD cases [66, 68]. These results suggested that hippocampal hyperactivation was temporally constrained. Additional data from [69] showed that the extent of hippocampal hyperactivation at baseline predicted cognitive decline as measured by the CDR-SB scores over four years after scanning. High-resolution fMRI studies have shown that this hippocampal hyperactivity is specific to the dentate and CA3 subregions of the hippocampus [70]. Recent work has also shown that this effect is noted in cognitively intact APOE ε4 carriers [71]. Studies in aged rodents with memory deficits suggest that CA3 hyperactivity may be due, at least in part, to the loss of GABAergic drive in hilar inhibitory interneurons, particularly somatostatin-positive (SOM+) interneurons [72].
Using domain-selective tasks that differentially engage the anterolateral (aLEC) and posteromedial entorhinal cortex (pMEC), Reagh et al. [73] found an age-related imbalance in the aLEC-DG/CA3 circuit characterized by reduced signaling in the aLEC that is coupled with increased signaling in DG/CA3 in the absence of structural thinning of the regions. These findings suggest that hyperactivity in the DG/CA3 region may, in part, be due to disruptions in the aLEC-DG/CA3 circuit via degeneration of the perforant path. Recent evidence also suggests that hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline [74].
This elevation in hippocampal activity can be targeted with pharmacological manipulations such as low-dose levetiracetam (LEV; an antiepileptic), which has shown positive results in a proof-of-concept trial. The drug successfully reduced hyperactivity in the hippocampus and reduced memory deficits in patients with amnestic MCI [75]. Later work showed that this effect was limited to the lower dosage of the drug and disappeared when higher doses were used [76] suggesting an alternative mechanism at higher doses. Interestingly, LEV targets synaptic vesicle protein SV2A which can now be imaged using a novel PET tracer (see last section on new approaches). Additionally, low dose LEV fully restores hilar SOM expression in aged, memory-impaired rats [72], suggesting that restoring inhibition may be a critical therapeutic path and that high-resolution functional MRI may be a suitable method to assess target engagement and therapeutic efficacy in clinical trials.
Reduced white matter integrity – Diffusion MRI
Diffusion tensor imaging (DTI) has been used to investigate the microstructural features of white matter [77]. The majority of DTI studies assess white matter integrity using voxel-wise values such as fractional anisotropy (FA), which is a scalar quantity that measures the anisotropy (i.e. directionality) of the diffusion signal in any given voxel. There are many factors that affect FA including axonal degeneration, demyelination, disorganization, packing density, and other microstructural features, but it is often measured as an indirect proxy to white matter integrity. Although the neural basis of anisotropy is still not completely understood, it has been used as an index of white matter integrity in thousands of studies across humans and animals. Typically the higher the FA value, the more intact a fiber pathway is thought to be.
A number of DTI studies have shown white matter loss with aging (see review by Chua et al. [78]), most likely due to thin myelinated fiber degeneration [79,80,81,82]. DTI studies of MCI and AD show widespread declines in white matter integrity throughout the brain with the most reliable changes reported in the temporal lobes [78, 83,84,85,86].
Investigations of white matter connectivity changes in aging and AD have focused on the fornix and the cingulum, as they are the major links between the limbic system and the rest of the brain. The fornix is the largest input/output fiber bundle of the hippocampus and connects it to the hypothalamus, while the cingulum connects the cingulate and the parahippocampal gyri to the septal cortex. Damage to the fornix has been found to reproduce learning and memory deficits resulting from hippocampal lesions in rats [87, 88] and in monkeys [89,90,91]. DTI fiber tracking studies show reduced fractional anisotropy in the fornix in AD [92, 93]. Several studies have found white matter changes in the cingulum in MCI and mild AD cases [94,95,96].
The perforant path connects EC layer II neurons to the hippocampal DG and CA3 [97] and is critical for normal hippocampal function [98]. This pathway’s integrity is reduced in aged rats with memory loss [99, 100]. Perforant path lesions also result in EC layer II neuronal loss [101], one of the earliest hallmark features of AD. Thus, attempts to evaluate perforant path alterations are critical to understanding early AD pathophysiology.
Numerous studies have shown changes in parahippocampal white matter in aging and MCI using structural MRI and diffusion tensor imaging (DTI) [102,103,104,105]. However, since there are many crossing fibers in the region and the perforant path is only ~ 2-3 mm thick fiber sheet, it was not possible to uniquely ascribe these changes to the perforant path itself. More recent work used an ultrahigh resolution (submillimeter) DTI technique to assess the perforant path [106, 107], which was validated against post-mortem data [108]. This method more specifically allowed for imaging the perforant path and documented loss of integrity with aging in a manner that was related to the extent of memory deficits.
Traditional DTI approaches are limited by the inability to resolve intra-voxel complexities such as fiber bending, crossing, and twisting [109]. High angular resolution diffusion imaging (HARDI) addresses this limitation by sampling the diffusion signal along many more gradient directions and providing adequate information to model diffusion with an orientation distribution function (ODF), a more versatile diffusion representation that captures multiple orientations in a voxel [110]. Given the complexity of white matter and the specific patterns of atrophy related to AD, HARDI may offer an improved approach to biomarker discovery.
Cortical thinning and volume loss – Structural MRI
Compared with images from other modalities, MR images provide excellent anatomical detail and additionally provide a strong grey/white matter contrast. Processes believed to be pathological in nature are often described in terms of anatomical location, cortical thickness, volumetry, and morphological characteristics.
Coronal T1-weighted, three dimensional, high resolution images are often used in cross-sectional and longitudinal studies to measure the hippocampal volume and to assess changes in hippocampal volume over time in AD [111, 112]. They have also been used to reveal many age-related changes in the brain. There is a decrease in total brain volume resultant from cortical thinning and gyral atrophy [113]. Specifically, the prefrontal cortex and the hippocampal formation display volume loss in advanced aging that significantly accelerates from normal aging to MCI to AD [114, 115].
Volume and shape changes in the hippocampus have been shown with healthy aging and preclinical AD [115,116,117,118]. Some MRI studies have also shown that the extent of hippocampal and entorhinal volume decline with increasing age predicted performance on memory tasks [119, 120]. Despite these studies, it is not clear whether any of these changes are actually the result of frank cell loss with age, or perhaps are secondary to synaptic and dendritic loss. Studies in aged rodents and non-human primates have reliably demonstrated the absence of frank cell loss in the hippocampus with age [121,122,123], but regions in the prefrontal cortex are found to undergo cell loss [124,125,126].
Although dramatic neuronal loss is not observed in preclinical AD or MCI, several studies have shown mild hippocampal atrophy during these stages. Hippocampal atrophy has been linked to cognitive impairment suggestive of AD [127,128,129]. Several human structural MRI studies have used very-high-dimension transformation techniques to observe changes in the shape of the hippocampus associated with AD. Consistent with the histological data, changes in the area of the CA1 fields in the hippocampus have been reported [130, 131]. Notably, in one of these studies, the same region of CA1 identified as differing in shape between non-demented and mildly demented patients also varied in the non-demented patients as a function of whether or not they later converted to a CDR (Clinical Dementia Rating) of 0.5 [130]. More recent work by the same group suggests that surface deflections across all hippocampal subfields (CA1 lateral zone, dentate gyrus/CA2-4 superior zone, and subiculum inferior medial zone) differentiate non-demented controls from early AD patients [132].
Recent high-resolution structural imaging studies in MCI patients where subfields of the hippocampus were manually segmented have suggested that specific subfields are more vulnerable than others. Yassa et al. [70] found that the CA1 and CA3/dentate gyrus regions both show volumetric loss, with left-lateralized changes in both subregions. The subiculum and other medial temporal regions were no different in MCI patients and controls. Similar techniques showed that the subiculum, CA1, and entorhinal cortex are further affected in AD [133, 134]. Mueller and Weiner [133] also found that APOE ε4 status was associated with volumetric decline in the CA3/dentate subregions, suggesting that early risk for AD may selectively affect this region, and is consistent with the loss of synaptic input reported in animal studies.
Subfield-specific patterns of atrophy are complex and require improved segmentation of hippocampal subfields that are both reliable and histologically validated. Current efforts by the Hippocampal Subfield Group (HSG: http://hippocampalsubfields.com) is making advances in this direction [135, 136]. Higher resolution scans at increased MRI strength (7T) have also shown promise in examining changes in particular layers of the hippocampal region that may be vulnerable at early stages of the disease. The apical dendrites of hippocampal CA1 pyramidal neurons, in the stratum radiatum/stratum lacunosum-moleculare (SRLM), are targeted by tau pathology early in the course of disease. Several studies have shown that using high-resolution (~200 micron) T2-weighted scans at 7T allows for identification and assessment of SRLM and demonstrate AD-related atrophy [137]. Similar changes have also been noted in nondemented older adults [138, 139] and in APOE ε4 carriers [140].
In recent years, cortical thinning in the entorhinal cortex (EC) has been identified as a highly sensitive measure of structural change both in MCI and AD [141]. EC thickness diminishes prior to, and predicts, hippocampal atrophy [142,143,144,145]. Several recent studies using the Alzheimer’s Disease Neuroimaging Initiative (ADNI) data have shown evidence of EC thinning in older adults with CSF pathological markers of AD (Aβ and p-Tau) [144, 146]. Another recent study by Ewers et al. [147] suggested that EC loss was one of the best predictors of MCI conversion to AD, even surpassing multimarker models.
Thus, results from structural MRI studies have generally shown that both the entorhinal cortex and the hippocampus show robust volumetric declines in MCI and AD (with the entorhinal change occurring earlier) and may be used as an early diagnostic feature. Limitations of the methods include differences in spatial resolution across scans, susceptibility to movement, and difficulties in determining the neural source of volume or thickness loss (cell loss vs. dendritic and synaptic loss) without exceptionally high-resolution scanning that is not feasible for most institutions.
Cerebral glucose hypometabolism – FDG-PET
PET methods have been used for over three decades to examine alterations in brain glucose metabolism in aging, MCI and AD [148]. Regional cerebral metabolism can be assessed with 18F-2fluoro-2-deoxy-D-glucose (FDG) as a metabolic marker. In particular, findings of reduced hippocampal metabolism in MCI and AD have been reported [149]. Cerebral glucose hypometabolism on FDG-PET appears to be a downstream marker of neuronal injury and neurodegeneration. In particular, it appears reliably in temporal, parietal (and possibly frontal) lobes but spares sensorimotor cortices, visual cortices, basal ganglia, thalamic nuclei and the cerebellum [150].
Importantly, age-related patterns of cerebral glucose metabolism differ substantially from patterns observed in AD, which has led to the utility of this technique in aiding clinical diagnosis. While classic studies (e.g. [151]) have shown that average cerebral glucose metabolism decreases with age, the regions showing the least age-related change include the medial temporal lobes, the posterior cingulate cortex and the precuneus. Those are the same regions expressing significant hypometabolism in AD. Thus, FDG-PET can be used to determine if the pattern of cerebral hypometabolism is normal or abnormal. Mosconi et al. [152] showed that it can be used to differentiate AD patients from healthy subjects with 99% sensitivity and 98% specificity.
Studies have also suggested that FDG-PET can be quite accurate at differentially diagnosing AD from other dementias and has a high concordance rate with clinical diagnosis [153]. That said, recent results have also suggested that hypometabolism in one of the key regions implicated in AD, the posterior cingulate cortex, cannot be used in isolation for differential diagnosis, as a subset of patients with the behavioral variant of frontotemporal dementia also show this pattern of hypometabolism [154].
While it has been suggested that structural MRI and FDG-PET can be used interchangeably to index neurodegenerative processes, more recent data suggest that they offer complementary and non-overlapping information. For example, Benvenutto et al. [155] showed that the extent of glucose hypometabolism can be used to track clinical severity, whereas structural MRI markers had higher associations with higher educational attainment (higher cognitive reserve). Other work has also shown that FDG-PET can be used to predict conversion from MCI to AD (odds ratio of 84.9%) [156].
Recent work by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) 2 PET Core have examined the combined utility of FDG-PET and amyloid PET at tracking progression of the disease. For example, they demonstrate that amyloid PET (using florbetapir uptake) is negatively associated with temporoparietal metabolism [157]. In healthy controls, florbetapir was associated with cognitive change, whereas in MCI patients FDG-PET metabolism was associated with cognitive change [158]. This is consistent with the biomarker model in which amyloid aggregation precedes neurodegeneration.
Limitations of FDG-PET include all of the limitations previously discussed for other PET-based approaches including harmonization of procedures and analyses. However, given the long history of FDG-PET scanning, these methods are far more standardized than amyloid or tau imaging.
Emerging Methods
The final section discusses some of the most exciting emerging methods that may potentially allow us to add new and informative biomarkers to the AT(N) criteria. In addition to protein aggregation and cellular injury/neurodegeneration, AD is characterized by increased inflammation, epigenetic dysfunction, and synaptic loss. The three emerging methods we discuss below attempt to break new ground in imaging and tracking these pathologies in vivo.
Imaging Neuroinflammation (TSPO-PET)
Translocator protein (TSPO) is an outer mitochondrial membrane protein that is expressed in many tissues throughout the body [159]. In the healthy brain, TSPO is only expressed at low levels and its expression is upregulated in activated and proliferating microglia and astrocytes following brain injury and neuroinflammation [160,161,162]. The differential expression of TSPO in activated glia enables for it to be exploited with PET to observe and quantify neuroinflammatory changes. Thus, PET tracers for TSPO were developed over the past two decades as markers for glial activation and neuroinflammation in AD. The attempts have had mixed results.
The first PET study with a TSPO tracer in AD patients was published by Cagnin et al. [163] and showed an increased uptake of the [11C]-based tracer PK11195. Later reports provided mixed results with some studies showing weak links between microglial activation and AD progression [164] and a poorly understood relationship with amyloid beta deposition [165, 166]. It became clear that TSPO tracers had limitations including a modest binding affinity, high non-specific binding, and low signal-to-noise ratio [167].
Second generation tracers were subsequently developed to improve these limitations. However, they were affected by genetic variability of the TSPO binding site due to the rs6971 single-nucleotide polymorphism, which resulted in high-affinity, mixed-affinity, and low-affinity binders [168]. This effectively limited the use of the tracer to studies only in high and mixed-affinity binders, and required a genetic test prior to the scan. One recent study with the [11C]-PBR28 PET tracer has also found significant widespread clusters positively correlated between levels of microglial activation and tau aggregation via [18F]-AV1451 PET imaging in MCI and AD subjects [169]. The correlations were stronger in AD than MCI. However, levels of microglial activation and amyloid deposition were also correlated, and the correlations were stronger in MCI than AD. This would suggest that microglial activation can correlate with both tau aggregation and amyloid deposition.
Third generation tracers, such as GE-180 were produced with the intent of TSPO quantification regardless of genotype, [170, 171]. Early data suggests that increased TSPO binding is associated with various dementias, but more studies are needed in AD patients. Although, TSPO imaging may potentially serve as a biomarker for neuroinflammation, future development of these tracers and enhancing their specificity and sensitivity will be needed [172].
Imaging Epigenetics (11C-Martinostat PET)
Epigenetics refers to a set of molecular mechanisms that are involved in regulating gene expression, but which do not involve alterations to the genetic code itself. They include modifications to the structure of the DNA (methylation) or modifications of the chromatin (acetylation). Chromatin includes DNA and the histone proteins that help package genomic DNA into the nucleus of a cell. Epigenetic modifications are thought to be involved in the dynamic process of learning and memory and are altered by aging and AD pathology.
Whether epigenetic alterations contribute causally to AD or are a consequence of upstream events still remains subject to debate [173]. Certain epigenetic changes may arise before AD pathology presents [174] and some may be more downstream [175, 176]. In both cases, understanding the changes to the epigenetic landscape that occur prior to, and during, the progression of AD can significantly enrich our understanding of disease pathophysiology.
Histone acetylation is a particular type of epigenetic modification controlled by histone acetyltransferases (HATs), which add acetyl groups to histone proteins, and histone deacetylase (HDACs), which remove acetyl groups from histone proteins. Imaging this process in vivo in humans would allow for a means to assess the epigenetic landscape. The novel radiotracer [11C] Martinostat allows for imaging HDAC density with high specific binding of a subset of class I HDAC enzymes (isoforms 1, 2, and 3), favorable kinetics, and high affinity [177]. In human studies, HDAC expression was higher in cortical gray matter than white matter and was generally lowest in the amygdala and hippocampus [178]. Follow-up work by the same group developed a fluorinated variant of the tracer [18F] MGS3 [179], which exhibits specific binding, comparable brain uptake and regional distribution to [11C] Martinostat, however the radiosynthesis process remains highly inefficient precluding complete validation using blocking experiments in nonhuman primates and subsequent use in humans. Epigenetic imaging may soon offer a unique look into gene regulatory processes that are implicated in AD, however, it is still too early at this time to determine its utility as a biomarker for AD.
Imaging Synapses (11C-UCB-J PET)
Synapse loss is an important feature of neurodegeneration, and it precedes cellular degeneration in most cases. Observing synaptic loss in humans has not been possible until recently with the advent of novel PET tracers for synaptic vesicle proteins. The synaptic vesicle protein 2A (SV2A) found in neurons as well as endocrine cells is essential for synaptic neurotransmitter release and is targeted by anti-epileptics such as levetiracetam. Thus, it can potentially serve as a biomarker for synaptic density. The recent development of the SV2A PET radiotracer [11C] UCB-J [180] may offer the possibility of imaging synaptic density in vivo, and potentially inform biomarker science not just for AD but for numerous other conditions involving synapse loss [181].
A recent study by Chen et al. [182] used [11C] UCB-J to quantify SV2A binding in a small sample of AD patients (amyloid positive) and healthy controls (amyloid negative). The authors found a significant reduction in SV2A binding in AD patients compared to healthy controls in addition to a relationship between overall SV2A binding and episodic memory scores. For decades, the only information that could be gleaned about synaptic integrity was indirectly through FDG-PET scans which are thought to be an indirect correlate of synapse loss given the relationship between glucose metabolism and synaptic markers. However, with this new advance, the field has the opportunity to directly examine synapses [183]. While still in the early stages, this work ushers promise in understanding the nature of synaptic alterations in AD and a host of other neurological illnesses.
Summary and Conclusions
In vivo neuroimaging in humans provides a richer understanding of the pathophysiology of AD. We discussed a number of methods that have already provided useful information in terms of diagnosing the disease during the preclinical stage, tracking its progression, and testing the efficacy of disease modifying therapeutics. For these methods to allow us to develop appropriate biomarkers that can serve as meaningful outcomes or surrogate endpoints they have to meet numerous criteria.
At a minimum, we suggest that across all these biomarkers, investigators should think carefully about test-retest reliability (see review by Henriques et al.[184]), histological validation, specificity to the disease process, sensitivity to detect abnormalities when they are subtle, practical feasibility in a clinical research setting, and relationship to cognitive/clinical outcomes. At this time, there is not a single imaging modality that meets all of the above criteria and singularly provides a rich enough understanding of pathological processes. Not only do different imaging modalities offer complementary information, but the spatial distribution of the measurements can also offer rich information that can be used for tracking and staging within individuals and groups.
Thus, we suggest that there is a need for composite neuroimaging biomarkers that combine information about glial inflammation, epigenomic alterations, amyloid and tau aggregation, structural and functional alterations, and synaptic and cellular degeneration. With the growing number of large-scale multimodal datasets (e.g. ADNI), there is a growing need for developing precision medicine approaches to better characterize, stage, and classify subtypes of dementias and discriminate AD from age-related changes.
Enabling precision medicine research in AD was identified as a key recommendation resulting from the National Institute on Aging (NIA)’s Alzheimer’s Disease Research Summit 2018: Path to Treatment and Prevention. The use of robust artificial intelligence and the integration of neuroimaging data with other -omics data will be critical to advance in the field of Alzheimer’s disease therapeutics.
Modality | Major Finding | References |
|---|---|---|
Amyloid-PET | Amyloid deposition is linked to aberrant entorhinal activity among cognitively normal older adults | [22] |
Subthreshold amyloid deposition predicts tau deposition in aging | [26] | |
Increased Aβ is associated with cortical thinning in frontoparietal regions | [24] | |
Tau-PET | Tau deposition predicts atrophy measures | |
Higher tracer uptake in the parahippocampal gyrus strongly relates to episodic memory | [185] | |
Older age is associated with binding in the medial temporal lobe (MTL), the extent of which is associated with memory deficits | [43] | |
Memory scores are strongly correlated with medial temporal tau tracer uptake, whereas whole-brain measures showed weak associations with memory and MTL atrophy | [42] | |
Task-Activated fMRI | Increased hippocampal activity during learning in individuals with MCI compared to normal controls and individuals with AD. | [66] |
Less impaired MCI patients showed this increase, while more impaired MCI patients showed a decrease in activity similar to mild AD cases | [67] | |
More impaired MCI patients showed a decrease in activity similar to mild AD cases | [68] | |
The extent of hippocampal hyperactivation at baseline predicted cognitive decline as measured by the CDR-SB scores over four years after scanning. | [69] | |
High-resolution fMRI studies have shown that this hippocampal hyperactivity is specific to the DG/CA3 subregions of the hippocampus | ||
Reduced signaling in the LEC coupled with increased signaling in DG/CA3 in the absence of structural thinning of the regions. | [73] | |
Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline | [74] | |
Resting-State fMRI | Widespread changes in DMN connectivity in MCI and AD | |
Hyperconnectivity in the anterior DMN and hypoconnectivity in the posterior DMN in AD | ||
Aß+ and tau-PET signal specific profiles | ||
Age-related decrease in connectivity between the entorhinal cortex and the dentate and CA3 regions of the hippocampus, the extent of which was correlated with memory deficits. | [64] | |
Diffusion MRI | Widespread changes in white matter in MCI and AD | |
DTI fiber tracking studies show white matter microstructural changes in the fornix and cingulum in MCI and mild AD cases | ||
Parahippocampal white matter changes in aging and MCI using structural MRI and diffusion tensor imaging (DTI) | ||
Perforant path degradation in non-demented older adults | ||
Structural MRI | Volume and shape changes in the hippocampus with healthy aging and preclinical AD | |
Volumetric loss of CA1 and DG/CA3 in APOE4 carriers, preclinical AD, MCI and AD in high resolution scans | ||
ERC thickness predicts hippocampal atrophy (including CA1-SRLM size) and is a sensitive measure of structural change in MCI and AD | ||
TSPO-PET | In the healthy brain, TSPO is only expressed at low levels and its expression is upregulated in activated and proliferating microglia and astrocytes following brain injury and neuroinflammation | |
AD patients show an increased global and regional uptake | ||
Microglial activation can correlate with both tau aggregation and amyloid deposition. | [169] | |
Epigenetic modifications | In healthy adults, HDAC expression was lowest in the hippocampus and amygdala among gray matter regions | [178] |
Imaging Synapses | Significant reduction in SV2A binding in AD patients compared to healthy controls in addition to a relationship between overall SV2A binding and episodic memory scores. | [182] |
Availability of data and materials
Not applicable
References
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 Census. Neurology. 2013;80(19):1778–83.
Petersen RC. Clinical subtypes of Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9(Suppl 3):16–24.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl). 1991;82:239–59.
Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak’s stages in the general population: can we know? Neurobiol Aging. 1997;18:362–9 discussion 389-392.
Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol (Berl). 2011;121:171–81.
Braak H, Tredici KD. Neuroanatomy and Pathology of Sporadic Alzheimer’s Disease: Springer International Publishing; 2015. Available from: //www.springer.com/us/book/9783319126784. [cited 2018 Aug 21]
Braak H, Braak E. Temporal Sequence of Alzheimer’s Disease-Related Pathology. In: Peters A, Morrison JH, editors. Cereb Cortex Neurodegener Age-Relat Chang Struct Funct Cereb Cortex. Boston: Springer US; 1999. p. 475–512. Available from: https://doi.org/10.1007/978-1-4615-4885-0_14. [cited 2018 Aug 21].
Schöll M, Schonhaut D, Lockhart S, Vogel JW, Baker S, Schwimmer H, et al. In vivo braak staging using 18F-AV1451 Tau PET imaging. Alzheimers Dement J Alzheimers Assoc. 2015;11:P4.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92.
Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011;34:430–42.
Leal SL, Yassa MA. Perturbations of neural circuitry in aging, mild cognitive impairment, and Alzheimer’s disease. Ageing Res Rev. 2013;12:823–31.
Jack CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2018;14:535–62.
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539–47.
Evans NR, Tarkin JM, Buscombe JR, Markus HS, Rudd JHF, Warburton EA. PET imaging of the neurovascular interface in cerebrovascular disease. Nat Rev Neurol. 2017;13:676–88.
Mathis CA, Wang Y, Holt DP, Huang G-F, Debnath ML, Klunk WE. Synthesis and Evaluation of 11C-Labeled 6-Substituted 2-Arylbenzothiazoles as Amyloid Imaging Agents. J Med Chem. 2003;46:2740–54.
Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19:497–510.
Ranganath C, Ritchey M. Two cortical systems for memory-guided behaviour. Nat Rev Neurosci. 2012;13:713–26.
Fotiadis P, van Rooden S, van der Grond J, Schultz A, Martinez-Ramirez S, Auriel E, et al. Cortical atrophy in patients with cerebral amyloid angiopathy: a case-control study. Lancet Neurol. 2016;15:811–9.
Hecht M, Krämer LM, von Arnim CAF, Otto M, Thal DR. Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol (Berl). 2018;135:681–94.
Huijbers W, Mormino EC, Wigman SE, Ward AM, Vannini P, McLaren DG, et al. Amyloid Deposition Is Linked to Aberrant Entorhinal Activity among Cognitively Normal Older Adults. J Neurosci. 2014;34:5200–10.
Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82:1760–7.
Villeneuve S, Reed BR, Wirth M, Haase CM, Madison CM, Ayakta N, et al. Cortical thickness mediates the effect of β-amyloid on episodic memory. Neurology. 2014;82:761–7.
Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer’s disease neurodegenerative biomarkers are associated with decreased cognitive function but not β-amyloid in cognitively normal older individuals. J Neurosci Off J Soc Neurosci. 2013;33:5553–63.
Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ. Subthreshold Amyloid Predicts Tau Deposition in Aging. J Neurosci Off J Soc Neurosci. 2018;38:4482–9.
Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs13.
Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem Off J Histochem Soc. 1989;37:209–15.
Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–72.
von Bergen M, Barghorn S, Biernat J, Mandelkow E-M, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005;1739:158–66.
Kovacech B, Skrabana R, Novak M. Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener Dis. 2010;7:24–7.
DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front Neurosci. 2018;12:267.
Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci. 2014;17:304–11.
Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife. 2018;7.
Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14:114–24.
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol (Berl). 2014;128:755–66.
Duyckaerts C, Brion JP, Hauw JJ, Flament-Durand J. Quantitative assessment of the density of neurofibrillary tangles and senile plaques in senile dementia of the Alzheimer type. Comparison of immunocytochemistry with a specific antibody and Bodian’s protargol method. Acta Neuropathol (Berl). 1987;73:167–70.
Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–7.
Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80:247–58.
Sepulcre J, Schultz AP, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, et al. In Vivo Tau, Amyloid, and Gray Matter Profiles in the Aging Brain. J Neurosci Off J Soc Neurosci. 2016;36:7364–74.
Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, et al. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol. 2016;73:1070–7.
Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. NeuroImage. 2017;157:448–63.
Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain J Neurol. 2017;140:3286–300.
Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–68.
Devous MD, Joshi AD, Navitsky M, Southekal S, Pontecorvo MJ, Shen H, et al. Test-Retest Reproducibility for the Tau PET Imaging Agent Flortaucipir F 18. J Nucl Med Off Publ Soc Nucl Med. 2018;59:937–43.
Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain J Neurol. 2017;140:764–80.
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4:58.
Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78:787–800.
Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C, et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J Nucl Med Off Publ Soc Nucl Med. 2016;57:1599–606.
Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89:971–82.
Biswal B, Yetkin FZ, Haughton VM, Hyde JS. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med Off J Soc Magn Reson Med Soc Magn Reson Med. 1995;34:537–41.
Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8:700–11.
Sheline YI, Raichle ME. Resting state functional connectivity in preclinical Alzheimer’s disease. Biol Psychiatry. 2013;74:340–7.
Gusnard DA, Raichle ME. Searching for a baseline: Functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2:685–94.
Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci. 2001;98:676–82.
Greicius MD, Menon V. Default-mode activity during a passive sensory task: uncoupled from deactivation but impacting activation. J Cogn Neurosci. 2004;16:1484–92.
Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38.
Petrella JR, Sheldon FC, Prince SE, Calhoun VD, Doraiswamy PM. Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology. 2011;76:511–7.
Sanz-Arigita EJ, Schoonheim MM, Damoiseaux JS, Rombouts SARB, Maris E, Barkhof F, et al. Loss of “small-world” networks in Alzheimer’s disease: graph analysis of FMRI resting-state functional connectivity. PloS One. 2010;5:e13788.
Supekar K, Menon V, Rubin D, Musen M, Greicius MD. Network Analysis of Intrinsic Functional Brain Connectivity in Alzheimer’s Disease. PLOS Comput Biol. 2008;4:e1000100.
Zhang Z, Lu G, Zhong Y, Tan Q, Liao W, Wang Z, et al. Altered spontaneous neuronal activity of the default-mode network in mesial temporal lobe epilepsy. Brain Res. 2010;1323:152–60.
Schultz AP, Chhatwal JP, Hedden T, Mormino EC, Hanseeuw BJ, Sepulcre J, et al. Phases of Hyperconnectivity and Hypoconnectivity in the Default Mode and Salience Networks Track with Amyloid and Tau in Clinically Normal Individuals. J Neurosci. 2017;37:4323–31.
Jones DT, Knopman DS, Gunter JL, Graff-Radford J, Vemuri P, Boeve BF, et al. Cascading network failure across the Alzheimer’s disease spectrum. Brain J Neurol. 2016;139:547–62.
Yassa MA, Mattfeld AT, Stark SM, Stark CEL. Age-related memory deficits linked to circuit-specific disruptions in the hippocampus. Proc Natl Acad Sci U S A. 2011;108:8873–8.
Fredericks CA, Sturm VE, Brown JA, Hua AY, Bilgel M, Wong DF, et al. Early affective changes and increased connectivity in preclinical Alzheimer’s disease. Alzheimers Dement Diagn Assess Dis Monit. 2018;10:471–9.
Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–11.
Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: an independent component analysis. J Neurosci Off J Soc Neurosci. 2006;26:10222–31.
Sperling R. Functional MRI Studies of Associative Encoding in Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. Ann N Y Acad Sci. 2007;1097:146–55.
Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry. 2008;79:630–5.
Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CEL. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage. 2010;51:1242–52.
Sinha N, Berg CN, Tustison NJ, Shaw A, Hill D, Yassa MA, et al. APOE ε4 status in healthy older African Americans is associated with deficits in pattern separation and hippocampal hyperactivation. Neurobiol Aging. 2018;69:221–9.
Spiegel AM, Koh MT, Vogt NM, Rapp PR, Gallagher M. Hilar interneuron vulnerability distinguishes aged rats with memory impairment. J Comp Neurol. 2013;521:3508–23.
Reagh ZM, Noche JA, Tustison NJ, Delisle D, Murray EA, Yassa MA. Functional Imbalance of Anterolateral Entorhinal Cortex and Hippocampal Dentate/CA3 Underlies Age-Related Object Pattern Separation Deficits. Neuron. 2018;97:1187–98 e4.
Leal SL, Landau SM, Bell RK, Jagust WJ. Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. eLife. 2017;6.
Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of Hippocampal Hyperactivity Improves Cognition in Amnestic Mild Cognitive Impairment. Neuron. 2012;74:467–74.
Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. 2015;7:688–98.
Taylor WD, Hsu E, Krishnan KRR, MacFall JR. Diffusion tensor imaging: background, potential, and utility in psychiatric research. Biol Psychiatry. 2004;55:201–7.
Chua TC, Wen W, Slavin MJ, Sachdev PS. Diffusion tensor imaging in mild cognitive impairment and Alzheimer’s disease: a review. Curr Opin Neurol. 2008;21:83–92.
Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol. 2003;462:144–52.
Meier-Ruge W, Ulrich J, Brühlmann M, Meier E. Age-related white matter atrophy in the human brain. Ann N Y Acad Sci. 1992;673:260–9.
Sandell JH, Peters A. Disrupted myelin and axon loss in the anterior commissure of the aged rhesus monkey. J Comp Neurol. 2003;466:14–30.
Tang Y, Nyengaard JR, Pakkenberg B, Gundersen HJ. Age-induced white matter changes in the human brain: a stereological investigation. Neurobiol Aging. 1997;18:609–15.
Bozzali M, Falini A, Franceschi M, Cercignani M, Zuffi M, Scotti G, et al. White matter damage in Alzheimer’s disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2002;72:742–6.
Huang J, Friedland RP, Auchus AP. Diffusion tensor imaging of normal-appearing white matter in mild cognitive impairment and early Alzheimer disease: preliminary evidence of axonal degeneration in the temporal lobe. AJNR Am J Neuroradiol. 2007;28:1943–8.
Naggara O, Oppenheim C, Rieu D, Raoux N, Rodrigo S, Dalla Barba G, et al. Diffusion tensor imaging in early Alzheimer’s disease. Psychiatry Res. 2006;146:243–9.
Xie S, Xiao JX, Gong GL, Zang YF, Wang YH, Wu HK, et al. Voxel-based detection of white matter abnormalities in mild Alzheimer disease. Neurology. 2006;66:1845–9.
McDonald RJ, White NM. A triple dissociation of memory systems: hippocampus, amygdala, and dorsal striatum. Behav Neurosci. 1993;107:3–22.
Sutherland RJ, Kolb B, Whishaw IQ. Spatial mapping: definitive disruption by hippocampal or medial frontal cortical damage in the rat. Neurosci Lett. 1982;31:271–6.
Gaffan D. Scene-specific memory for objects: a model of episodic memory impairment in monkeys with fornix transection. J Cogn Neurosci. 1994;6:305–20.
Gaffan D. Amnesia for complex naturalistic scenes and for objects following fornix transection in the rhesus monkey. Eur J Neurosci. 1992;4:381–8.
Gaffan D, Saunders RC, Gaffan EA, Harrison S, Shields C, Owen MJ. Effects of fornix transection upon associative memory in monkeys: Role of the hippocampus in learned action. Q J Exp Psychol Sect B. 1984;36:173–221.
Teipel SJ, Stahl R, Dietrich O, Schoenberg SO, Perneczky R, Bokde AL, et al. Multivariate network analysis of fiber tract integrity in Alzheimer’s disease. NeuroImage. 2007;34:985–95.
Bennett IJ, Huffman DJ, Stark CEL. Limbic Tract Integrity Contributes to Pattern Separation Performance Across the Lifespan. Cereb Cortex. 2015;25:2988–99.
Choo IH, Lee DY, Oh JS, Lee JS, Lee DS, Song IC, et al. Posterior cingulate cortex atrophy and regional cingulum disruption in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2010;31:772–9.
Firbank MJ, Blamire AM, Krishnan MS, Teodorczuk A, English P, Gholkar A, et al. Atrophy is associated with posterior cingulate white matter disruption in dementia with Lewy bodies and Alzheimer’s disease. NeuroImage. 2007;36:1–7.
Villain N, Desgranges B, Viader F, de la Sayette V, Mézenge F, Landeau B, et al. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer’s disease. J Neurosci Off J Soc Neurosci. 2008;28:6174–81.
Witter MP. The perforant path: projections from the entorhinal cortex to the dentate gyrus. Progress Brain Res. 2007;163:43–61. https://doi.org/10.1016/S0079-6123(07)63003-9.
Hyman BT, Van Hoesen GW, Kromer LJ, Damasio a R. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol. 1986;20:472–81.
Geinisman Y, deToledo-Morrell L, Morrell F, Persina IS, Rossi M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus. 1992;2:437–44.
Smith TD, Adams MM, Gallagher M, Morrison JH, Rapp PR. Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci Off J Soc Neurosci. 2000;20:6587–93.
Peterson DA, C a L-P, Eagle KL, Gage FH. Perforant path damage results in progressive neuronal death and somal atrophy in layer II of entorhinal cortex and functional impairment with increasing postdamage age. J Neurosci Off J Soc Neurosci. 1994;14:6872–85.
Kalus P, Slotboom J, Gallinat J, Mahlberg R, Cattapan-Ludewig K, Wiest R, et al. Examining the gateway to the limbic system with diffusion tensor imaging: The perforant pathway in dementia. NeuroImage. 2006;30:713–20.
Stoub TR, deToledo-Morrell L, Stebbins GT, Leurgans S, D a B, Shah RC. Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:10041–5.
Rogalski EJ, Murphy CM, deToledo-Morrell L, Shah RC, Moseley ME, Bammer R, et al. Changes in parahippocampal white matter integrity in amnestic mild cognitive impairment: a diffusion tensor imaging study. Behav Neurol. 2009;21:51–61.
Wang C, Stebbins GT, D a M, Shah RC, Bammer R, Moseley ME, et al. Atrophy and dysfunction of parahippocampal white matter in mild Alzheimer’s disease. Neurobiol Aging. 2012;33:43–52.
Yassa MA, Muftuler LT, Stark CEL. Ultrahigh-resolution microstructural diffusion tensor imaging reveals perforant path degradation in aged humans in vivo. Proc Natl Acad Sci U S A. 2010;107:12687–91.
Bennett IJ, Greenia DE, Maillard P, Sajjadi SA, DeCarli C, Corrada MM, et al. Age-Related White Matter Integrity Differences in Oldest-Old Without Dementia. Neurobiol Aging. 2017;56:108–14.
Augustinack JC, Helmer K, Huber KE, Kakunoori S, Zöllei L, Fischl B. Direct visualization of the perforant pathway in the human brain with ex vivo diffusion tensor imaging. Front Hum Neurosci. 2010;4:42.
Jones DK, Leemans A. Diffusion tensor imaging. Methods Mol Biol. 2011;711:127–44.
Lenglet C, Campbell JSW, Descoteaux M, Haro G, Savadjiev P, Wassermann D, et al. Mathematical Methods for Diffusion MRI Processing. NeuroImage. 2009;45:S111.
Fox NC, Freeborough PA. Brain atrophy progression measured from registered serial MRI: validation and application to Alzheimer’s disease. J Magn Reson Imaging. 1997;7:1069–75.
Sabuncu MR, Desikan RS, Sepulcre J, Yeo BTT, Liu H, Schmansky NJ, et al. The Dynamics of Cortical and Hippocampal Atrophy in Alzheimer Disease. Arch Neurol. 2011;68:1040–8.
Uylings HBM, de Brabander JM. Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 2002;49:268–76.
Jack CR, Petersen RC, Xu Y, O’Brien PC, Smith GE, Ivnik RJ, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55:484–9.
Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, et al. Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb Cortex. 2005;15:1676–89.
Jernigan TL, Archibald SL, Fennema-Notestine C, Gamst AC, Stout JC, Bonner J, et al. Effects of age on tissues and regions of the cerebrum and cerebellum. Neurobiol Aging. 2001;22:581–94.
Pruessner JC, Collins DL, Pruessner M, Evans AC. Age and gender predict volume decline in the anterior and posterior hippocampus in early adulthood. J Neurosci Off J Soc Neurosci. 2001;21:194–200.
Raz N, Rodrigue KM, Head D, Kennedy KM, Acker JD. Differential aging of the medial temporal lobe: a study of a five-year change. Neurology. 2004;62:433–8.
Rodrigue KM, Raz N. Shrinkage of the entorhinal cortex over five years predicts memory performance in healthy adults. J Neurosci Off J Soc Neurosci. 2004;24:956–63.
Rosen AC, Prull MW, Gabrieli JDE, Stoub T, O’Hara R, Friedman L, et al. Differential associations between entorhinal and hippocampal volumes and memory performance in older adults. Behav Neurosci. 2003;117:1150–60.
Rasmussen T, Schliemann T, Sørensen JC, Zimmer J, West MJ. Memory impaired aged rats: no loss of principal hippocampal and subicular neurons. Neurobiol Aging. 1996;17:143–7.
Rapp PR, Gallagher M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci U S A. 1996;93:9926–30.
Rapp PR, Deroche PS, Mao Y, Burwell RD. Neuron number in the parahippocampal region is preserved in aged rats with spatial learning deficits. Cereb Cortex. 2002;12:1171–9.
Peters A, Leahu D, Moss MB, McNally KJ. The effects of aging on area 46 of the frontal cortex of the rhesus monkey. Cereb Cortex. 1994;4:621–35.
Smith DE, Rapp PR, McKay HM, Roberts JA, Tuszynski MH. Memory Impairment in Aged Primates Is Associated with Focal Death of Cortical Neurons and Atrophy of Subcortical Neurons. J Neurosci. 2004;24:4373–81.
Stranahan AM, Jiam NT, Spiegel AM, Gallagher M. Aging reduces total neuron number in the dorsal component of the rodent prefrontal cortex. J Comp Neurol. 2012;520:1318–26.
Convit A, de Leon MJ, Golomb J, George AE, Tarshish CY, Bobinski M, et al. Hippocampal atrophy in early Alzheimer’s disease: anatomic specificity and validation. Psychiatr Q. 1993;64:371–87.
Convit A, de Leon MJ, Tarshish C, De Santi S, Kluger A, Rusinek H, et al. Hippocampal volume losses in minimally impaired elderly. Lancet Lond Engl. 1995;345:266.
Killiany RJ, Moss MB, Albert MS, Sandor T, Tieman J, Jolesz F. Temporal Lobe Regions on Magnetic Resonance Imaging Identify Patients With Early Alzheimer’s Disease. Arch Neurol. 1993;50:949–54.
Csernansky JG, Wang L, Swank J, Miller JP, Gado M, McKeel D, et al. Preclinical detection of Alzheimer’s disease: hippocampal shape and volume predict dementia onset in the elderly. NeuroImage. 2005;25:783–92.
Csernansky JG, Wang L, Joshi S, Miller JP, Gado M, Kido D, et al. Early DAT is distinguished from aging by high-dimensional mapping of the hippocampus. Dementia of the Alzheimer type. Neurology. 2000;55:1636–43.
Qiu A, Miller MI. Multi-structure network shape analysis via normal surface momentum maps. NeuroImage. 2008;42:1430–8.
Mueller SG, Weiner MW. Selective effect of age, Apo e4, and Alzheimer’s disease on hippocampal subfields. Hippocampus. 2009;19:558–64.
Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal Atrophy Patterns in Mild Cognitive Impairment and Alzheimer’s Disease. Hum Brain Mapp. 2010;31:1339–47.
Wisse LEM, Daugherty AM, Olsen RK, Berron D, Carr VA, Stark CEL, et al. A harmonized segmentation protocol for hippocampal and parahippocampal subregions: Why do we need one and what are the key goals? Hippocampus. 2017;27:3–11.
Yushkevich PA, Amaral RSC, Augustinack JC, Bender AR, Bernstein JD, Boccardi M, et al. Quantitative comparison of 21 protocols for labeling hippocampal subfields and parahippocampal subregions in in vivo MRI: Towards a harmonized segmentation protocol. NeuroImage. 2015;111:526–41.
Kerchner GA, Deutsch GK, Zeineh M, Dougherty RF, Saranathan M, Rutt BK. Hippocampal CA1 apical neuropil atrophy and memory performance in Alzheimer’s disease. NeuroImage. 2012;63:194–202.
Carr VA, Bernstein JD, Favila SE, Rutt BK, Kerchner GA, Wagner AD. Individual differences in associative memory among older adults explained by hippocampal subfield structure and function. Proc Natl Acad Sci U S A. 2017;114:12075–80.
Kerchner GA, Bernstein JD, Fenesy MC, Deutsch GK, Saranathan M, Zeineh MM, et al. Shared vulnerability of two synaptically-connected medial temporal lobe areas to age and cognitive decline: a seven tesla magnetic resonance imaging study. J Neurosci Off J Soc Neurosci. 2013;33:16666–72.
Kerchner GA, Berdnik D, Shen JC, Bernstein JD, Fenesy MC, Deutsch GK, et al. APOE ε4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology. 2014;82:691–7.
Holland D, McEvoy LK, Dale AM. Unbiased comparison of sample size estimates from longitudinal structural measures in ADNI. Hum Brain Mapp. 2012;33:2586–602.
Desikan RS, Sabuncu MR, Schmansky NJ, Reuter M, Cabral HJ, Hess CP, et al. Selective disruption of the cerebral neocortex in Alzheimer’s disease. PLoS One. 2010;5:e12853.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Roddey JC, Blennow K, et al. Amyloid-β associated volume loss occurs only in the presence of phospho-tau. Ann Neurol. 2011;70:657–61.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, et al. Amyloid-β–Associated Clinical Decline Occurs Only in the Presence of Elevated P-tau. Arch Neurol. 2012;69:709–13.
Eskildsen SF, Coupé P, García-Lorenzo D, Fonov V, Pruessner JC, Collins DL. Prediction of Alzheimer’s disease in subjects with mild cognitive impairment from the ADNI cohort using patterns of cortical thinning. NeuroImage. 2013;65:511–21.
Holland D, McEvoy LK, Desikan RS, Dale AM. Enrichment and Stratification for Predementia Alzheimer Disease Clinical Trials. PLoS ONE. 2012;7:e47739.
Ewers M, Walsh C, Trojanowski JQ, Shaw LM, Petersen RC, Jack CR, et al. Prediction of conversion from mild cognitive impairment to Alzheimer’s disease dementia based upon biomarkers and neuropsychological test performance. Neurobiol Aging. 2012;33:1203–14.
de Leon MJ, Ferris SH, George AE, Christman DR, Fowler JS, Gentes C, et al. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol. 1983;4:568–71.
Mosconi L, Tsui W-H, Santi SD, Li J, Rusinek H, Convit A, et al. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology. 2005;64:1860–7.
Herholz K, Carter SF, Jones M. Positron emission tomography imaging in dementia. Br J Radiol. 2007;80 Spec No 2:S160-S167.
Kuhl DE, Metter EJ, Riege WH, Phelps ME. Effects of human aging on patterns of local cerebral glucose utilization determined by the [18F] fluorodeoxyglucose method. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 1982;2:163–71.
Mosconi L, Tsui WH, Herholz K, Pupi A, Drzezga A, Lucignani G, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J Nucl Med Off Publ Soc Nucl Med. 2008;49:390–8.
Tripathi M, Tripathi M, Damle N, Kushwaha S, Jaimini A, D’Souza MM, et al. Differential diagnosis of neurodegenerative dementias using metabolic phenotypes on F-18 FDG PET/CT. Neuroradiol J. 2014;27:13–21.
Scheltens NME, van der Weijden K, Adriaanse SM, van Assema D, Oomen PP, Krudop WA, et al. Hypometabolism of the posterior cingulate cortex is not restricted to Alzheimer’s disease. NeuroImage Clin. 2018;19:625–32.
Benvenutto A, Giusiano B, Koric L, Gueriot C, Didic M, Felician O, et al. Imaging Biomarkers of Neurodegeneration in Alzheimer’s Disease: Distinct Contributions of Cortical MRI Atrophy and FDG-PET Hypometabolism. J Alzheimers Dis JAD. 2018;65(4):1147–57.
Yuan Y, Gu Z-X, Wei W-S. Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: a meta-analysis. AJNR Am J Neuroradiol. 2009;30:404–10.
Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72:578–86.
Jagust WJ, Landau SM, Koeppe RA, Reiman EM, Chen K, Mathis CA, et al. The Alzheimer’s Disease Neuroimaging Initiative 2 PET Core: 2015. Alzheimers Dement J Alzheimers Assoc. 2015;11:757–71.
Papadopoulos V, Lecanu L, Brown RC, Han Z, Yao Z-X. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience. 2006;138:749–56.
Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain J Neurol. 2000;123(Pt 11):2321–37.
Chen M-K, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1–17.
Su Z, Herholz K, Gerhard A, Roncaroli F, Du Plessis D, Jackson A, et al. [11C]-(R)PK11195 tracer kinetics in the brain of glioma patients and a comparison of two referencing approaches. Eur J Nucl Med Mol Imaging. 2013;40:1406–19.
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, et al. In-vivo measurement of activated microglia in dementia. Lancet Lond Engl. 2001;358:461–7.
Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF, et al. Microglial activation in Alzheimer’s disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol Aging. 2013;34:128–36.
Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, et al. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66:60–7.
Yokokura M, Mori N, Yagi S, Yoshikawa E, Kikuchi M, Yoshihara Y, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2011;38:343–51.
Edison P, Donat CK, Sastre M. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front Neurol. 2018;9 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6090997/. [cited 2018 Aug 26].
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32:1–5.
Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain J Neurol. 2018;141:2740–54.
Fan Z, Calsolaro V, Atkinson RA, Femminella GD, Waldman A, Buckley C, et al. Flutriciclamide (18F-GE180) PET: First-in-Human PET Study of Novel Third-Generation In Vivo Marker of Human Translocator Protein. J Nucl Med Off Publ Soc Nucl Med. 2016;57:1753–9.
Feeney C, Scott G, Raffel J, Roberts S, Coello C, Jolly A, et al. Kinetic analysis of the translocator protein positron emission tomography ligand [(18) F]GE-180 in the human brain. Eur J Nucl Med Mol Imaging. 2016;43:2201–10.
Kreisl WC, Henter ID, Innis RB. Imaging Translocator Protein as a Biomarker of Neuroinflammation in Dementia. Adv Pharmacol San Diego Calif. 2018;82:163–85.
Millan MJ. An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology. 2013;68:2–82.
Bihaqi SW, Schumacher A, Maloney B, Lahiri DK, Zawia NH. Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): towards a new paradigm. Curr Alzheimer Res. 2012;9:574–88.
Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM, et al. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int J Mol Sci. 2013;14:17643–63.
Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011;32:1161–80.
Wey H-Y, Wang C, Schroeder FA, Logan J, Price JC, Hooker JM. Kinetic Analysis and Quantification of [11C] Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem Neurosci. 2015;6:708–15.
Wey H-Y, Gilbert TM, Zürcher NR, She A, Bhanot A, Taillon BD, et al. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci Transl Med. 2016;8:351ra106.
Strebl MG, Wang C, Schroeder FA, Placzek MS, Wey H-Y, Van de Bittner GC, et al. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem Neurosci. 2016;7:528–33.
Nabulsi NB, Mercier J, Holden D, Carré S, Najafzadeh S, Vandergeten M-C, et al. Synthesis and Preclinical Evaluation of 11C-UCB-J as a PET Tracer for Imaging the Synaptic Vesicle Glycoprotein 2A in the Brain. J Nucl Med Off Publ Soc Nucl Med. 2016;57:777–84.
Mercier J, Provins L, Valade A. Discovery and development of SV2A PET tracers: Potential for imaging synaptic density and clinical applications. Drug Discov Today Technol. 2017;25:45–52.
Chen M-K, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin S-F, et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 2018;75(10):1215–24. https://doi.org/10.1001/jamaneurol.2018.1836.
Mormino EC, Jagust WJ. A new tool for clinical neuroscience—synaptic imaging. JAMA Neurol. 2018;75(10):1181–3. https://doi.org/10.1001/jamaneurol.2018.1643.
Henriques AD, Benedet AL, Camargos EF, Rosa-Neto P, Nóbrega OT. Fluid and imaging biomarkers for Alzheimer’s disease: Where we stand and where to head to. Exp Gerontol. 2018;107:169–77.
Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, et al. Entorhinal Tau Pathology, Episodic Memory Decline, and Neurodegeneration in Aging. J Neurosci Off J Soc Neurosci. 2018;38:530–43.
Damoiseaux JS, Seeley WW, Zhou J, Shirer WR, Coppola G, Karydas A, et al. Gender modulates the APOE ε4 effect in healthy older adults: convergent evidence from functional brain connectivity and spinal fluid tau levels. J Neurosci Off J Soc Neurosci. 2012;32:8254–62.
Sepulcre J, Sabuncu MR, Li Q, El Fakhri G, Sperling R, Johnson KA. Tau and amyloid β proteins distinctively associate to functional network changes in the aging brain. Alzheimers Dement J Alzheimers Assoc. 2017;13:1261–9.
Acknowledgements
Not applicable
Funding
FM is supported by NSF GRFP DGE-1321846 and B2D 1612490. MAY is supported by NIA P50AG05146, R21AG049220 and R01AG053555.
Author information
Authors and Affiliations
Contributions
Both authors equally contributed to the conceptual framework and to writing this manuscript. Both authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Both authors consent to publication
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Márquez, F., Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol Neurodegeneration 14, 21 (2019). https://doi.org/10.1186/s13024-019-0325-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-019-0325-5