
Abstract
Persisters refer to genetically drug susceptible quiescent (non-growing or slow growing) bacteria that survive in stress environments such as antibiotic exposure, acidic and starvation conditions. These cells can regrow after stress removal and remain susceptible to the same stress. Persisters are underlying the problems of treating chronic and persistent infections and relapse infections after treatment, drug resistance development, and biofilm infections, and pose significant challenges for effective treatments. Understanding the characteristics and the exact mechanisms of persister formation, especially the key molecules that affect the formation and survival of the persisters is critical to more effective treatment of chronic and persistent infections. Currently, genes related to persister formation and survival are being discovered and confirmed, but the mechanisms by which bacteria form persisters are very complex, and there are still many unanswered questions. This article comprehensively summarizes the historical background of bacterial persisters, details their complex characteristics and their relationship with antibiotic tolerant and resistant bacteria, systematically elucidates the interplay between various bacterial biological processes and the formation of persister cells, as well as consolidates the diverse anti-persister compounds and treatments. We hope to provide theoretical background for in-depth research on mechanisms of persisters and suggest new ideas for choosing strategies for more effective treatment of persistent infections.
Similar content being viewed by others


Introduction
Bacterial infections have long been a scourge for humanity. In the past century, the discovery and widespread use of antibiotics have somewhat improved the treatment of these infections.1,2,3 However, in recent decades, the emergence and increasing antibiotic resistance has heightened concerns regarding infectious disease.4,5,6,7,8 The constantly emerging phenomenon of “superbugs” in clinical setting is a sign that we are entering an era where traditional infection treatments are becoming increasingly ineffective.9,10,11 In addition to antibiotic resistance, persistent infections pose another major challenge and problem in managing bacterial infections.12,13,14,15 Individuals with persistent infections endure the continuous presence or recurring episodes of bacterial infections, often with poor response to antibiotic therapy.16 Clinical examples of such infections include tuberculosis,17 and typhoid fever,18 Lyme disease,19 recurrent urinary tract infections20 and others. The presence of bacterial persisters during infections is the main culprit behind relapse and treatment failure of persistent infections.14,21,22,23
Persisters are non-growing or slow growing bacteria that can continue to survive under “stress” conditions such as antibiotics, reactive oxygen, acid pH, or starvation. After “stress” removal, persisters can continue to grow and remain sensitive to the same “stress”.24 Due to their tolerance ability to antibiotics and subsequent failure in antibiotic treatments, persisters are of high clinical importance for a range of microbial pathogens. In the past three decades, researchers have made significant progress in our understanding of molecular basis of bacterial persistence,25,26 and established specific methods for isolation and analysis of persisters. Therefore, it is necessary to make a comprehensive and systematic review on bacterial persisters, so as to point out the direction for further research and accelerate the path for the control of persistent infections.
Although there have been review articles on the research progress of bacterial persister in the past decade, they either have a long-time span, and do not cover recent research advancements,22,24 or they are not comprehensive enough to provide readers with a complete understanding of all aspects of persisters.14,27,28 For instance, Fisher et al. had summarized the correlation between persistent bacteria and clinical diseases, with focuses on the mechanisms of bacterial persistence in toxin-antitoxin modules, stringent response, bacterial communication, drug efflux, and others.14,27,28 However, the mechanisms discussed do not include other mechanisms of persistence such as trans-translation and protein degradation systems, metabolism of purine and amino acid, epigenetic modifications, RNA degradation, and small non-coding RNA. Additionally, the review does not cover research advancements in anti-persister drugs. Therefore, we believe that systematic review on the discovery history, characteristics, detection methods, mechanisms of persisters, and persister drugs for more effective treatment of persistent infections is necessary and important. This review will provide an update on the mechanisms and treatment of persisters, as well as put forward the potential targets for developing new drugs against persisters and challenging problems facing persister research.
Our research group has dedicated many years to investigating the mechanisms of bacterial persistence and screening drugs that target persister cells. We have proposed a “Yin-Yang” model of bacterial persistence and treatment strategy,24 elucidated many novel mechanisms of persistence in diverse pathogens such as Mycobacterium tuberculosis (M. tuberculosis),29,30,31,32,33,34,35 Escherichia coli (E. coli),36,37,38,39,40,41,42 Staphylococcus aureus (S. aureus),43,44,45,46 and Borrelia burgdorferi (B. burgdorferi),47,48 and identified several drugs or drug combinations that are more effective against persisters and persistent infections than the current standard treatments.49,50,51,52,53,54,55,56,57,58,59,60 Drawing upon our research findings and existing literature reports, we conducted this comprehensive review of the various aspects of bacterial persisters mentioned above, aiming to provide important references for future research.
Historical overview
Persisters were first identified more than 80 years ago and are closely associated with chronic persistent infections (Fig. 1). In 1942, Gladys Hobby discovered the phenomenon of bacterial persistence when she experimented with the newly developed antibiotic penicillin and found that the drug killed 99% bacteria (pneuniococci, heniolytic streptococci and staphylococci), with 1% organisms not killed.61 In 1944, Joseph Bigger, who had studied the above bacterial persistence in more detail with staphylococci, named the small numbers of non-growing dormant bacteria that survived penicillin attack as “persisters”, and suggested a scheme of treatment in which penicillin is alternately administered and withheld for the treatment of bacteria in the persister phase.62 However, this discovery at that time did not catch much attention to persisters due to lack of deep understanding.

Milestone events in persister research. Since the initial discovery of bacterial persistence phenomenon in 1942, significant findings have progressively unveiled the clinical implications of bacterial persisters in human diseases. Created with BioRender.com
In the following 30 years, researchers conducted numerous studies to better understand persister cells and their important role in clinic.63 Until 1970, another category of persistence was observed by Alexandre Tomasz: a novel type of pneumococcal mutant that grows at normal generation times, is as sensitive to growth inhibition by penicillin as the wild-type parent strain, experiences only a very slow loss of viability, and does not lyse at all during exposure to penicillin.64 To differentiate it from the persistence found earlier (named as physiological or phenotypic tolerance many years ago), the terms “antibiotic tolerance” and “genotypic tolerance” were introduced to describe this novel type of bacterial response to antibiotic treatment.64 In 1974, Gary Best identified the first genotypically tolerant clinical isolate of S. aureus (strain Evans), which had the same oxacillin MIC as the rest of the strain but survive from high concentrations of the drug.65 In 1976, Mayhall et al. described a high prevalence (55%) of persisters among clinical isolates of staphylococci.66 In 1977, tolerant response was observed in Streptococcus sanguis (S. sanguis) by Diane Horne and Alexander Tomasz.67 At that time, it came to realize that persistence, which differs from previously described forms of penicillin resistance, is common and important in clinic.68
Since then, numerous cases of treatment failures caused by over 20 species of tolerant bacteria in humans with infections were reported,69,70,71,72,73,74,75,76,77 but there was no strong direct evidence showing that antibiotic tolerance affects the treatment of human infections. In 1983, Brennan and Durack showed a clear relationship between degree of S. sanguis tolerance and efficacy of treatment in the rabbit endocarditis model, in which tolerant S. sanguis survived better than non-tolerant bacteria after 5 days of treatment.78 At the same year, by repeatedly exposing growing E. coli to ampicillin, Harris Moyed identified hipA mutant with no change in MIC but had higher persistence, which was considered to be due to mutation in a gene related to the formation of persisters.79 These initial studies have deepened our understanding of persister/tolerant bacteria and bacterial persistence at that period.
During the 1990s-2000s, as the wide application of indwelling devices (cardiac stents, urinary tract indwelling catheters, etc.) and the increase of immunocompromised patients (cancer chemotherapy or HIV infection, etc.), the number of chronic persistent infections including biofilm infections increased dramatically.80,81 In 2000, Kim Lewis established a link between bacterial persistence and biofilm infections in Pseudomonas aeruginosa (P. aeruginosa).82,83 It was then discovered that biofilms contain persisters which are subsequently recognized as the culprit for the difficulty in curing biofilm infections, relapse after treatment, and other chronic persistent infections.22,84 Since then, persisters and persistent infections have garnered the interest of an increasing number of scientists worldwide.14,28,85,86,87,88,89,90 Nevertheless, no persister drugs that kill persister bacteria and eradicate biofilm infections exist until recently. Tuberculosis serves as a prime example of the significance of bacterial persisters during infection and drugs targeting persisters, as the unique anti-persister drug pyrazinamide (PZA) plays a crucial role in shortening TB therapy and reducing relapse rates.17,91
Characteristics and detection methods of persisters
Characteristics of persister cells and their distinctions from resistant and “tolerant bacteria”
Persisters exhibit phenotypic heterogeneity, which includes metabolic diversity, variation in persistence levels, and differences in colony sizes. (1) Metabolic diversity. The bacteria in the community have different metabolic states, including metabolic quiescence, slow metabolism, etc., that is, the individual bacterial persisters have different persistence abilities. Balaban and colleagues proposed that the non-growing (metabolically stagnant) persisters induced by external environmental factors are called type I persisters, such as those produced by culturing bacteria in liquid medium to stationary phase in vitro; The slow-growing (slow-metabolizing) persisters that are spontaneously generated by non-external factors are called type II persisters, and this group of bacteria will continue to divide and proliferate slowly and can return to normal bacteria.25 In fact, the metabolic heterogeneity of persisters is much more complex than that of type I and type II persisters. For example, in the group of type I or type II persisters, bacteria do not display the same metabolic states. Additionally, the metabolic state of persisters is not invariable, and it will change with the change of environmental conditions. (2) Variation in persistence level. It has been proposed that there is a hierarchy of persistence levels within persister continuum, where some persisters have strong persistence ability, which is called deep persistence, while some other persisters have weak persistence ability, which is called shallow persistence.24,92,93 In addition, in the studies of dormant microbes such as Vibrio cholerae (V. cholerae),94,95,96 Legionella pneumophila,97,98,99,100,101,102 M. tuberculosis103,104 etc., it has been found that persisters with deeper persistence levels can be viable but non-culturable (VBNC)105 but resuscitated and grow under appropriate conditions such as conditioned medium or co-culture with host cells,106,107,108 which is not covered by the conventional persister definition.24 (3) Differences in colony size. Heterogeneity in colony size can be reflected in the emergence of small colony variants (SCVs),109 which have been observed in various bacteria including M. tuberculosis,110 S. aureus,111,112,113,114 E. coli.115,116 These variants are characterized by their significantly smaller size (approximately 5 or 10 times smaller than the most common colony type) and slower growth rate compared to the parent strain. SCVs have been linked to increased antibiotic tolerance and persistence.109,117,118 These small colonies represent a subpopulation of persister cells with an extended bacterial lag phase,119 and their frequency within a bacterial population tends to increase following exposure to stressors such as acidic pH120 or reactive oxygen species.121 Thus, the heterogeneity of persisters is complex and dependent on the particular conditions under which the persisters are studied. We previously proposed a Yin-Yang model to more accurately reflect the heterogeneity and transformation of persisters,24 in which “Yin” and “Yang” represent persisters and growing/metabolically active bacteria, respectively. These two states of bacteria are not absolutely independent of each other, but can be interconverted to each other.24
Persisters are relative and highly dependent on different factors, including the type of bacterial strain, the specific antibiotic used, and the environmental conditions,37 such as the following: (1) The growth phase of bacteria. The percentage of persister in stationary phase was higher than that in logarithmic phase.122,123,124 For example, in the study of the formation of persisters during the growth of E. coli in vitro, it was found that the proportion of persisters was very low at the early stage of log phase growth, but the proportion of persisters increased rapidly at the late stage of log growth; In the stationary phase of bacteria, the proportion of persisters reached the highest and stabilized at about 1%;125 (2) Nutrient composition, pH and gas composition in the environment during bacterial culture can also affect persister formation. For example, amino acid limitation leads to a decrease in the growth rate of bacteria and triggers stress responses, including the inhibition of drug target function mediated by ppGpp, which renders bactericidal drugs ineffective in killing the bacteria.126 However, it is important to note that the tolerance induced by amino acid-limiting conditions is typically shorter in duration compared to tolerance observed during the stationary phase of bacterial growth.126 Furthermore, it also has been demonstrated that hydrogen sulfide (H2S) and nitric oxide (NO) can induce antibiotic tolerance through anti-oxidative defense.127,128 (3) The environment of antibiotic exposure, including the type, concentration, time and other factors of antibiotic exposure.129,130 Specifically, the number of persisters in the same bacterial strain can vary depending on the type of antibiotics used.122,131,132,133,134 For instance, drugs that reduce bacterial metabolic activity and interfere with bacterial growth and replication, such as quinolones and macrolides, will lead to more bacteria entering a persister state to resist the effects of antibiotics.42 Additionally, the duration and concentration of antibiotic exposure also influence the formation of persister bacteria, affecting both the number and degree of persistence.122 It is important to note that different bacterial species, even when exposed to the same antibiotic at the same concentration, may not produce the exact same number of persisters.129 Furthermore, the mechanism of persistent bacteria formation under the action of different antibiotics is also different, such as varying importance of individual persister genes in tolerance to different antibiotics.41
Persisters have the ability to survive in the presence of antibiotics, but they differ from resistant bacteria. Persisters have strong tolerance to antibiotics, and show multi-drug tolerance (MDT).135 The tolerance of persisters to antibiotics is only expressed at the phenotypic level, and there is no mutation in resistance genes as in persister bacteria, so it is different from antibiotic-resistant bacteria due to genetic mutations or antibiotic resistance genes. The minimum inhibitory concentration (MIC) of antibiotic-resistant bacteria to antibiotics is increased, while that of persisters remained unchanged or decreased. However, persisters and antibiotic-resistant bacteria are not completely unrelated, as they may interconvert and overlap as indicated in the Yin-Yang model. For instance, persisters under specific conditions can also facilitate drug-resistance gene mutations to form antibiotic-resistant bacteria, and persisters are also found in the antibiotic-resistant bacterial population.136,137 At present, the mechanism of tolerance of persisters to multiple antibiotics is not very clear, but the current research shows that the mechanism of tolerance of persisters to antibiotics is generally different from the mechanism of bacterial resistance,125 though enhanced efflux and reduced entry may be shared.
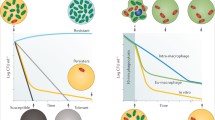
In addition to persister and resistant bacteria, another term “tolerant bacteria,” has also been coined to describe bacteria that survive antibiotic treatment. As both persisters and “tolerant bacteria” exhibit the characteristic of phenotypic drug tolerance, and mechanisms associated with tolerant bacteria (such as reduced metabolism or ATP levels) have also been identified in persister,125 there is often confusion between the two, which puzzles many researchers.12,138,139 Balaban and colleagues had attempted to distinguishing between resistance, tolerance and persistence to antibiotic treatment. She proposed that persistence characterizes a bacterial subpopulation (typically less than 1%) to survive antibiotic exposure, while tolerance describes the same ability but pertains an entire bacterial population85,137 (Fig. 2a, b). This view distinguishes tolerance and persistence only by their penetrance within a population, but it did not define and distinguish persisters and “tolerant bacteria”. Then, Helaine and colleagues differentiated persister and “tolerant bacteria” based on their individual growth ability in the absence of antibiotic: “tolerant bacteria” are either slow-growing or non-growing, while persisters are exclusively non-growing.140 However, this view is inconsistent with the phenotypic heterogeneity of persisters, which suggests that persister include both (type I persisters) and slow growing bacteria (type II persisters).25 Additionally, Bassler and colleagues stated that persistence is another form of tolerance that is not acquired through heritable mutations, but rather through phenotypic differentiation.86 This claim cannot be accepted either, because mutations in genes (such as hipA mutant79) are also associated with the formation of persisters. It can be seen that how to distinguish between tolerance and persistence has remained somewhat ambiguous so far, and no one has been able to pinpoint their exact difference. We hold the opinion that there might be no fundamental difference between persisters and “tolerant bacteria” essentially but a matter of degree, and that tolerant bacteria represent shallow persisters in the heterogeneous persister continuum as proposed in the Yin-Yang model.24 This hypothesis can actually find answers in the research history of persisters.

Distinguishing characteristics of persister cells from resistant and tolerant bacteria in in vitro models. According to the definition reported in the literature, when exposed to antibiotics, the homogeneous population of sensitive (green), persister (red), “tolerant,” (yellow) or resistant bacteria (purple) exhibit different scenarios (a) and antibiotic killing kinetics (b). Following the addition of a bactericidal antibiotic, sensitive bacteria could be completely killed and its kill curve is a decreasing straight line. Even after the antibiotic is removed, the bacteria cannot revive. Persister refers to a small fraction within the bacterial population that, when exposed to antibiotics, the bulk growing bacteria are killed rapidly, while the persisters are still alive. Once the antibiotic is removed, persisters can resuscitate and resume growth. “Tolerant bacteria” is a whole population of bacteria with persister-like tolerance, which are killed more slowly than normal growing bacteria and are capable of regrowth upon antibiotic removal. Resistant cells, unaffected by antibiotics, grow in the presence of the antibiotic and exhibit an ascending straight-line without being killed, indicating their survival and proliferation despite antibiotic exposure. Created with BioRender.com
Specifically, in 1970, since persistence was not well known, people mistakenly believed that “antibiotic tolerance”, refer in particular to “genotypic tolerance”, was unrelated to persistence and represented a new form of bacterial survival from antibiotic treatment. This misunderstanding led to the emergence and confusion of both tolerance and persistence in later reports. Actually, both “phenotypic tolerance” which was discovered in 1942 and named in 1986, and “genotypic tolerance” contribute to the clinical problem of bacterial “persistence”. Therefore, we believe that there is no essential difference between persisters and “tolerant bacteria” previously described in the literatures. This could explain why researchers have difficulty distinguishing between them and why they are often used interchangeably, including our own.
During host infections, sensitive bacteria, persisters/tolerant bacteria and resistant bacteria may not exist independently, but could coexist simultaneously and undergo dynamic interconversions (Fig. 3). This might be a manifestation of the phenotypic heterogeneity of bacterial populations in the host.141 In the proposed evolutionary connection between persistence, tolerance, and resistance to antibiotics, the anticipated evolutionary trajectory starts with the development of antibiotic tolerance from antibiotic persistence, eventually culminating in antibiotic resistance,142 which is in line with our views as expressed in the Yin-Yang model. Based on the dynamic transformation process and the complex phenotypic heterogeneity of persisters, the “tolerant bacteria” described in the literature may be considered as one hierarchy of persisters. To avoid unnecessary confusion, in our review article, we refer persisters as metabolically quiscent bacteria exhibiting phenotypic tolerance or genotypic tolerance (mutations in persistence genes) to stress conditions.

The coexistence and dynamic interconversions of sensitive bacteria, persisters/tolerant bacteria and resistant bacteria during host infections. In the host environment, the bacterial population is heterogeneous and significantly more complex than that in vitro. The metabolic activities of bacteria within the persister population are not uniform; there are bacteria with slow metabolism known as shallow persisters and those with metabolic dormancy known as deep persisters. From an evolutionary perspective, persistence under certain conditions can lead to resistance development, via mutations or transfer of resistance genes. Created with BioRender.com
Methods for detecting persister cells
(1) Time-kill assay. Bacterial persistence refers to the ability of bacteria to survive during exposure to lethal bactericidal drugs or stress environment. The stronger the survival ability, the stronger the persistence ability. The traditional method to detect persisters is the time-kill assay, which was even employed by Hobby and Bigger in their original studies on persister cells in 1942 and 1944.61,62 This assay involves exposing bacteria to a lethal dose of antibiotics or stresses, followed by the quantification of the remaining viable bacteria using the colony-forming unit (CFU) counting method at different time points of antibiotic exposure. The persistence ability of bacteria can then be inferred based on the number of viable bacteria that remain after the exposure. While the time-kill assay is a commonly used method for detecting persister cells, it does have certain limitations. For instance, although it was already known that persistence is defined by a decreased rate of killing, the time-kill assay lacks criteria to determine the bacteria that survive antibiotic or stress treatment are persister cells, and may not distinguish between shallow and deep persisters. Moreover, the time-kill assay cannot be used to detect deep persisters in VBNC state as pointed out previously.24
(2) Minimum duration for killing 99% or 99.99% of bacteria (MDK99/ MDK99.99). The academic school represented by Nathalie Balaban and colleagues believe that tolerance is different from persistence. In 2017, Nathalie Balaban introduced a metric, MDK99, for measuring tolerance.143 Subsequently, in 2019, another metric, MDK 99.99, was proposed to determine the persistence capacity of bacteria.85 This approach appears to overcome the limitation of the absence of specific criteria in the time-kill assay, rendering it a promising method for detecting persister cells. In essence, MDK99 and MDK99.99 respectively represent the time it takes to kill 99% and 99.99% of bacteria with antibiotics. From this perspective, they reflect two levels of bacterial persistence to antibiotics, indirectly suggesting that their proposed tolerance and persistence do not have a fundamental difference.
Theoretically, both MDK99 and MDK 99.99 can be extracted from a time-kill curve. However, we have encountered challenges in applying this experimental framework to real-world scenarios. Specifically, when examining the time-dependent kill curve of E. coli under levofloxacin exposure, we observed that it did not follow a smooth mathematical pattern. As a result, it became difficult to accurately extract the MDK99 or 99.99 from a bacterial time-kill curve. We could only estimate the range based on the time points on the X-axes, such as between 2 and 3 (days or hours). This issue is evident in a study that utilized MDK99 assays to measure the variation in antibiotic tolerance among M. tuberculosis clinical isolates.144 It is worth noting that there are no practical experimental examples in published articles that determine bacterial persistence by calculating MDK99.99. Thus, it can be seen that the practicality of MDK assays still requires further validation.
(3) “ScanLag” and “ColTapp”. As early as 2010, Nathalie Balaban and her colleagues developed the “ScanLag” method for E. coli, which enables for the monitoring of a two-dimensional distribution of the lag time and growth of each colony on agar plates.145,146 Subsequently, Annelies Zinkernagel modified the ScanLag for the analysis of S. aureus lag time distributions, naming it ColTapp.147 Both ScanLag and ColTapp methods determine bacterial persisters based on colony and bacterial lag times, as persister cells tend to exhibit relatively long lag times and small colonies.119 Both the time-kill assay and the MDK99.99 method mentioned earlier provide a measurement at the population level but do not allow for the observation of individual persister cells within a bacterial population. As a result, both methods have limitations in accurately reflecting the phenotypic heterogeneity of persister cells within a bacterial population. However, the ScanLag and ColTapp methods have the ability to detect single colony growth and lag time, enabling them to measure the heterogeneity of persister cells. In addition, microfluidic device has also been used to detect individual persister cells with time-lapse microscopy.25
(4) Tolerance disk test (TDtest). In vitro studies have demonstrated that bacterial persistence can be induced by external environmental factors. It is important to note that bacterial persistence may also occur in patients, potentially leading to treatment failure. Therefore, in addition to assessing antibiotic susceptibility, the detection of bacterial persistence in a clinical setting is crucial for identifying effective treatment regimens. The Kirby-Bauer disk-diffusion assay has been widely utilized in clinical settings for determining bacterial antibiotic resistance. However, it is not suitable for detecting persistent bacteria. In 2016, Balaban and colleagues modified the standard disk-diffusion assay to enable the evaluation of persister tolerance levels.148 This modified test, known as the Tolerance Disk (TD) test, specifically targets persister cells. The TD test method consists of two steps: (1) placing an antibiotic disk on top of the agar plate, where the antibiotic diffuses into the agar, resulting in the formation of an inhibition zone around the disk for antibiotic-sensitive bacteria; (2) replacing the antibiotic disk with a glucose disk, which promotes the re-growth of bacteria and enables the detection of surviving bacteria. Although the TDtest method offers several advantages for routine clinical use, such as simplicity, low cost, and the ability to detect different levels of persistence, it does have limitations. One limitation is that it cannot provide the exact frequency of persister cells. Additionally, it is limited to detecting bacterial persistence under antibiotic exposure and cannot detect persister cells induced by other stress factors. It is important to address these limitations in future research to further enhance the utility of the TDtest in clinical settings.
(5) Replica plating tolerance isolation system (REPTIS). Inspired by the TDtest, in 2019, Matsuo and colleagues developed a replica plating tolerance isolation system called “REPTIS”, which is used to not only isolate but also calculate the frequency of tolerant cells.149 The “REPTIS” method also consists of two steps. In the first step, bacteria are exposed to a master agar plate containing antibiotics for 72 h of incubation. In the second step, colonies from the master plate that did not grow due to the presence of antibiotics are transferred onto a replica plate without antibiotics for another 72 h of incubation. The number of regrown bacteria indicates the presence of persister cells. Similar to the TDtest, the “REPTIS” method also has the potential to be used in clinical settings, but it is limited in its inability to detect triggered persistence induced by specific stressors. Further research is needed to address this limitation and develop methods that can effectively detect and characterize persister cells under various stress conditions in clinical settings.
(6) For VBNC persisters in non-culturable state, non-culture method could be used to evaluate if they are viable bacteria. VBNC bacteria have an intact cell membrane and can be tested with the LIVE/DEAD™ Bacterial Viability Assay.47,150 Reverse transcription polymerase chain reaction (RT-PCR) can also be used to detect VBNC.151 To detect VBNC bacteria using RT-PCR, specific marker genes or gene regions associated with the target bacteria can be selected. These marker genes may vary depending on the bacterial species of interest. For instance, using mRNAs encoding the lipopolysaccharide gene rfbE and the H7 flagellin gene fliC as the markers to detect the VBNC state of E. coli O157:H7;152 the VCA0656 gene related to the aminoimidazole riboside kinase protein was used as a genetic marker to precisely detect the VBNC state of V. cholerae;151 two selected housekeeping genes, 16S-23S rDNA and rpoS, proved to be good viability markers for Vibrio parahemolyticus VBNC state.153 In addition to the RT-PCR, droplet digital PCR (ddPCR) method is another suitable approach for counting the numbers of single-copy genes on the chromosome. It has emerged as a new tool for quantifying VBNC cells in recent years, demonstrating higher accuracy and sensitivity compared to qPCR.154
In summary, these detection methods are designed based on certain characteristics of persister cells, and each method has its own advantages and disadvantages. Among them, the TDtest method has great potential for clinical application due to its advantages in evaluating tolerance levels. However, methods such as time-kill assay, MDK99.99, and “REPTIS” are labor-intensive, which limits their application in clinic. “ScanLag” and “ColTapp” have advantages in detecting the heterogeneity of persister cells, but require special equipment (such as automated agar plate imaging as well as single-cell microscopy) and professional analysts. Therefore, research still needs to further improve and develop more comprehensive, flexible, and easy-to-operate methods to promote routine detection of persister cells in clinic.
Persisters and chronic persistent infections
Persisters have been identified in various pathogens, including bacteria, fungi, and parasites. Among these, chronic persistent bacterial infections have been extensively studied in urinary tract infections, tuberculosis, endocarditis, refractory cystic fibrosis infections,21,84,155,156 as well as indwelling device-related infections like pacemaker-related infections and prosthetic joint infections.157,158 The presence of persister cells in these infections contributes to their chronicity and the difficulties in eradicating them with conventional antibiotic treatments. In recent years, as our understanding of persisters has grown, numerous studies have highlighted the association between persisters and treatment challenges in chronic persistent infections. These challenges include recurrent infections, bacterial resistance, and biofilm formation. The following descriptions provide further insight into these issues:
-
(1)
Recurrence of infection. Persisters play a significant role in the recurrence or relapse of infections after treatment. After bacterial infections, under the joint action of antibiotics and immune system, most bacteria die, and a small number of bacteria adjust their metabolic state and change into persisters with slow or reduced metabolism to survive. At this time, the infection symptoms may be alleviated or disappear. However, if antibiotic treatment is stopped or the immune system function of the host is impaired, the bacteria can resume growth, resulting in recurrence of infection and associated symptoms. If antibiotics are continued for a long period of time, it may still not be able to kill the persisters, while increasing the probability of drug-resistant bacteria.22,159
-
(2)
Bacterial resistance. Persisters serve as an evolutionary reservoir from which resistant bacteria can emerge.160 In the early days, researchers believed that persisters were growth arrested.25 At this stage, the persistence and drug resistance of bacteria were considered as independent mechanisms to resist drugs. Later, it was gradually realized that the apparent stability of persister numbers was actually due to a dynamic state of balanced death and division, rather than generally arrested growth.161 Furthermore, whole-genome sequencing of microbes isolated from patients who experienced recrudescent infections revealed the presence of several drug resistance mutations in antibiotic-resistant bacteria, indicating that evolution towards resistance might occur during the persistent infection.162,163,164 In 2017, Nathalie Balaban conducted in vitro evolution experiments that revealed the precedence of persister cells over resistance.165 These experiments showed that the bacterial cultures exhibited tolerance several cycles before the emergence of resistance. The reason behind this phenomenon may be attributed to the fact that stress response programs, crucial for the formation and survival of persister cells, can also expedite adaptive genome-wide mutagenesis.166,167,168,169 Another significant mechanism through which persistence contributes to antibiotic resistance is the promotion of horizontal transfer of drug resistance genes through stress responses.170,171 For example, the SOS response increase the frequency of horizontal gene transfer in E. coli and V. cholerae, specifically facilitating the exchange of resistance elements for aminoglycosides, lincosamides, and antifolate antibiotics.170 Salmonella persister cells have also been found to facilitate the dissemination of antibiotic resistance plasmids in the intestinal tract.172 Overall, persistence not only enables bacteria to survive antibiotic exposure but also serves as a pathway for the development of antibiotic resistance.
-
(3)
Biofilm infections. In the 2000s, researchers established a link between bacterial persistent infections and biofilms.82 Since then, studies on persister cells and biofilm infections have been conducted. In both P. aeruginosa and S. aureus infections, it has been observed that biofilm cells exhibit greater tolerance to antibiotics compared to planktonic cells.83,173 This increased tolerance is attributed to the higher number of persister cells present in the biofilm compared to planktonic cells.174,175 The mechanism by which bacteria within biofilms become highly tolerant to antibiotics is believed to involve the starvation-signaling stringent response,166,175 important for persister formation, but also physical barrier to antibiotic exposure posed by the biofilm structure. Within biofilms, nutrient availability can be limited, triggering the stringent response and promoting the formation of persister cells.176 The discovery that the anti-persister drug candidate ADEP4 can effectively kill persister cells by activating ClpP and subsequently eradicate chronic biofilm infections provides further confirmation of the role of persisters in biofilm-induced persistent infections.177 Kim Lewis proposed a model of recurrent biofilm infection which shows a good relationship between biofilm-persisters and biofilm infection recurrence.178 The model suggested that metabolically active bacteria and persisters coexist in biofilm infections. Antibiotic treatment can kill most of the metabolically active bacteria, and the immune system can remove part of the metabolically active bacteria and the persisters outside the biofilm, while the persisters within the biofilm continue to survive under the protection of the biofilm. When the antibiotic is removed or the concentration of the drug is reduced, the remaining persister bacteria in the biofilm continue to reproduce and re-form the biofilm infection,24 resulting in recurrence and persistence of the infection.
Molecular mechanisms of persister formation and survival
Due to the complex characteristics of persisters, such as heterogeneity, relative nature, transience, small numbers, the study of the molecular mechanisms of their formation and survival is challenging. There are mainly two models for the study of genes related to persisters:
-
(1)
Screening single gene knockout or overexpression library or transposon insertion library.179,180,181,182,183,184 Firstly, screening for bacteria with reduced or increased persistence capacities, and then further verifying the relevant gene by PCR and sequencing. Through the gene knockout library model, the genes related to the formation of persisters are mainly global regulatory factors phoU,37 dksA,181 dnaK,181 hupAB181 and ihfAB,185 energy metabolism-related genes sucB92 and ubiF,92 etc., suggesting that the genes affecting bacterial persistence are not a single gene or a pathway-related gene.
-
(2)
Sorting or isolating persisters for transcriptome sequencing analysis. One approach is to treat bacteria with bactericidal antibiotics such as beta-lactam antibiotics, killing metabolically active bacteria and subjecting the remaining persisters to transcriptome sequencing.124,135 However, this method will change the external environment of the persisters after lysis of most bacteria, resulting in gene expression of the persisters being affected. The second method is to construct fluorescent bacteria containing degradable GFP controlled by ribosome promoter, and use flow cytometry or magnetic bead sorting to sort out bacteria with weak GFP fluorescence (bacteria with low translation level are considered as persisting bacteria) for transcriptome sequencing analysis.26,186 This method also has certain disadvantages, such as the environment and density of the bacteria are changed after the sorted persisters are transferred to a new buffer or medium. In the new environment, the persister bacteria will begin to recover, resulting in a decrease in persistence capacity. Through transcriptome sequencing, the genes related to persistence bacteria were mainly related to biosynthesis (down-regulated expression), such as atpA, fdxC, etc.,124 and toxin-related genes (up-regulated expression), such as relBE, mazEF, dinJ, yafQ and ygiU.22 Additionally, laboratory evolution of persistence and whole-genome sequencing has been used in the recent studies to identify genes involved in persistence.187,188,189 Through whole-genome sequencing, some new persistence related genes such as atl which encodes a bifunctional autolysin,187 nuoAHJKLMN which encodes NADH/ubiquinone oxidoreductase that translocates protons across the membrane,188 gadC which encodes a central component of the E. coli acid resistance system, and oppB which encodes membrane topology of the integral membrane components have been identified.189
Now we know the persistence mechanisms, mainly through the study of E. coli, S. aureus, M. tuberculosis, however, it is noteworthy that the mechanisms of persister formation in different bacteria has a certain degree of conservation, that is, with convergent evolution characteristics.24 Moreover, different persistence states of the same bacteria, such as stationary phase persisters, SCVs, L-form bacteria, VBNC, biofilm persisters, etc., may have similar and overlapping related persister genes and pathways.24,190,191,192 In addition, many persister genes or mechanisms exist in the same bacterial persisters and their roles are additive and redundant. Below we describe specific genes and pathways associated with persistence (Table 1).
Toxin-antitoxin modules (TA modules)
TA modules are composed of toxin and antitoxin genes and are widely distributed in bacterial and archaeal plasmids or genomes.193,194,195 Toxins are a stable protein, which are involved in the inhibition of bacterial replication or translation. Antitoxins are unstable proteins or RNAs that inhibit the activity of toxins. When exposed to “stresses”, bacteria can make toxins work by degrading antitoxins.196 TA modules are classified based on the nature of the antitoxin and its mechanism of action. In the context of persister formation, the widely studied TA modules are type I and type II.197,198,199,200
hipA was the earliest persistence-related gene found in ref. 79 which encodes HipA toxin protein and combines with antitoxin encoded by hipB to form HipBA module, but its role in persistence is still inconclusive. It has been proposed that the HipA toxin works by phosphorylating the active site of glutamyl-tRNA synthetase (GltX), which leads to its inactivation.201 This, in turn, causes an increase in the concentration of uncharged tRNAs at the ribosomal A site, triggering RelA-dependent (p)ppGpp synthesis and inducing a high level of persistence.201 Some other studies have also suggested that HipA-induced persistence depends not only on (p)ppGpp but also on the 10 mRNase-encoding TA modules, polyphosphate and Lon protease.202 However, four years after publishing the article, the authors became aware that the previously described contribution of polyphosphate to bacterial persistence was an artifact resulting from inadvertent lysogenization with a bacteriophage.203 Therefore, the connection of polyphosphate and HipBA modules in persister formation is no longer supported. Recently, it has been found that HipA could mediate persistence through not only GltX but also several other targets including another aminoacyl-tRNA synthetase TrpS,204 the negative modulator of replication initiation SeqA, transcriptional factor RcsB, and 30S ribosomal protein RpsI.205 In addition to hipBA, several other TA modules have been associated with persister formation. These include relBE (toxin RelE),206,207 mazEF (toxin MazF),208 dinJ-yafQ (toxin YafQ),209,210 mqsRA (toxin MqsR),211,212 ccdAB (toxin CcdB),213 tisAB (toxin TisB),214,215,216 yefM-yoeB (toxin YoeB),217 tacAT (toxin TacT)218 and others.219,220,221,222 Among them, the majority belong to type II TA modules, including hipBA, relBE, mazEF, dinJ-yafQ, tacAT and yefM-yoeB;222,223,224 the remaining ones belong to type I TA modules.
The activation of TA modules often leads to the accumulation of free toxins, which can result in bacteriostatic growth inhibition and antibiotic tolerance.225,226 However, the activation and mechanisms of the type II and type I TA modules differ. In general, the anti-toxins of type II TA modules are proteins which are usually degraded by the protease ClpP or Lon in response to (p)ppGpp signaling.208,223,224,227,228 The anti-toxins of type I TA modules are antisense RNAs which are activated by the “SOS” response and (p)ppGpp signaling.215,225 Type II toxins mediate bacteria to enter a persistence state through various mechanisms,229,230,231,232 as illustrated in Fig. 4. Primarily, these toxins primarily act as inhibitors of replication and translation.233 They interfere with DNA gyrase, leading to the inhibition of DNA replication (e.g., HipA). Moreover, they function as mRNA endonucleases, either ribosome-dependent (e.g., RelE family) or ribosome-independent (e.g., MazF family),197,199 consequently disrupting the translation process (Fig. 4). Additionally, certain type II toxins, such as HipA201 and TacT,218,234,235 have been found to inactivate glutamyl-tRNA synthetase (GltX) or acetylate tRNA respectively. Type I toxins are usually small proteins, such as TisB and HokB, that insert and form pores in bacterial membranes, causing loss of proton motive force (PMF) and ATP production236 (Fig. 4). Thus, the activation of TA modules and the accumulation of toxins have been implicated in persister formation,124,237,238 a state in which a subpopulation of bacteria becomes dormant and exhibits increased tolerance to antibiotics or other stresses.26

Mechanisms of persister formation via TA modules. The anti-toxins of type II TA modules are proteins that are usually degraded by the protease ClpP or Lon in response to (p)ppGpp signaling. These toxins mediate bacteria to enter a persistence state by disrupting replication and translation processes, such as interfering with DNA gyrase, acting as mRNA endonucleases, inactivating glutamyl-tRNA synthetase (GltX) and acetylate-tRNA. The anti-toxins of type I TA modules are antisense RNAs which are activated by the “SOS” response and (p)ppGpp signaling. These toxins are usually small proteins that insert and form pores in bacterial membranes, causing loss of proton motive force (PMF) and ATP production. Created with BioRender.com
Research findings have demonstrated that over-expression of toxin genes can indeed enhance bacterial persistence.135,239,240,241 However, the knockout of cumulative deletion of 10 TA modules or more decreases tolerance to antibiotics, but single deletions had no effect on persister levels,226,242 which may be due to the abundance of TA elements in bacteria. For example, E. coli has at least 15 TA modules and M. tuberculosis has at least 80 TA modules, which are highly expressed in bacteria exposed to stress factors. Knockout of one of these genes is compensated by other genes, and the persistence of bacteria is reduced when multiple genes are knocked out simultaneously.243 Due to their redundancy, though multiple TA systems have been found to be involved in bacterial persistence, developing novel anti-persister drugs targeting these systems may be challenging. However, recent research has shown that inhibiting toxin HipA can interfere with persister formation.244 This finding suggests that interfering with toxin-antitoxin modules may have therapeutic implications for treating bacterial infections. Finally, it is worth mentioning that bacteria such as B. burgdorferi and S. aureus had persisters but B. burgdorferi has no TA modules and S. aureus has TA modules but do not play a role in persistence,48 suggesting that TA modules are not the main determinant of bacterial persistence, despite TA was first identified to be involved in persistence phenomenon.79 And further studies are still needed to uncover the precise roles of different TA modules in different bacterial persistence.
Metabolism and metabolic regulation
Energy metabolism
Persisters are dormant and the metabolic process is obviously slowed down or stopped,245 and thus genes that alter cell metabolism are involved in the formation of persisters, such as sucB encoding the E2 subunit of the α-ketoglutarate dehydrogenase complex,92 sucC and sucD encoding succinyl coenzyme A (succinyl-CoA) synthetase242 in the tricarboxylic acid (TCA) cycle (Fig. 5). In the TCA cycle, both the α-ketoglutarate dehydrogenase complex and succinyl-CoA synthetase play critical roles in cellular metabolism, particularly in the generation of energy. Specifically, α-ketoglutarate dehydrogenase catalyzes the dehydrogenase of α-ketoglutarate to succinyl-CoA (2-oxoglutarate + NAD+ + CoA-SH → Succinyl-CoA + NADH + H+ + CO2); succinyl-CoA synthetase breaks down succinyl-CoA into succinate and CoA (Succinyl-CoA+ GDP+ Pi → Succinate + GTP + CoA). We and others have previously found that bacterial persistence levels decreased when genes (sucB, sucC, and sucD) encoding the relevant enzymes in the catalytic α-ketoglutarate to succinate reaction pathway are disrupted.92,242 In contrast, disruption of acnB gene encoding aconitase, which catalyzes the conversion of citrate to isocitrate, improved the bacterial persistence ability.242

Mechanisms of persister formation via energy metabolism. Persisters have lower metabolic activity than non-persisters and this reduced energy metabolic activity could be mediated through (a) G3P metabolism, (b) aerobic respiration, (c) tricarboxylic acid (TCA) cycle and (d) methylcitrate cycle (MCC), which may allow persisters to enter a dormant state and survive in adverse conditions. For example, reduced ATP levels decrease the activity of ATP-dependent antibiotic targets, while decreased proton motive force (PMF) restricts the entrance of antibiotics such as aminoglycosides and thus enhances the tolerance ability. Created with BioRender.com
Changes in the respiratory chain activity can also cause alterations in bacterial persistence levels (Fig. 5). In the respiratory chain, ubiquinone forms hydroquinone upon acceptance of 2e- and 2H+ from the cytosol, which plays a key role in ATP production and maintenance of membrane potential. ubiF is the key gene for ubiquinone formation in the aerobic biosynthetic pathway.246 Compared with the wild strain, the knockout strain of ubiF is more sensitive to antibiotics and the persistence level is correspondingly reduced.92,247 Aerobic respiration in bacteria is mediated by NADH. The inactivation of NADH dehydrogenase I (nuoI) and NADH dehydrogenase II (ndh) reduces oxidized NAD (NAD + ), proton motive force (PMF), and positively charged aminoglycosides are difficult to enter cells, thus inhibiting the killing effect of aminoglycosides on bacteria, that is, improving the persistence capacity of bacteria.248 After S. aureus randomly enters the stationary phase, the activity of ATP-dependent antibiotic targets (DNA gyrase, DNA topoisomerase, RNA polymerase, etc.) decreases with the decrease of ATP, and the bacteria can only maintain limited physiological activities, and the persistence ability is enhanced.249 Therefore, the intracellular ATP level also affects the formation of persisters.
During the process of glycerol-3-phosphate metabolism in E. coli, glycerol kinase GlpK catalyzes the conversion of glycerol to glycerol-3-phosphate (G3P), the G3P acetyltransferase PlsB catalyzes the first step of phospholipid synthesis (G3P → 1 acyl G3P), and G3P dehydrogenase GlpD converts G3P to DHAP under aerobic conditions (Fig. 5). It has been reported that the overexpression of GlpD in E. coli increased the bacterial tolerance to ampicillin and ofloxacin, while a glpD deletion mutant had a decreased level of persisters in the stationary phase.250 The persistence ability of plsB mutant was 100–1000 times lower than that of wild strain, but the mRNA of plsB gene in persisters was not changed compared with that of metabolically active bacteria in the wild-type strain,250 suggesting that plsB may be a gene related to the entry of bacteria into persistence state, but not related to maintenance of bacteria in the persistence state. Glycerol kinase (GlpK) catalyzes the conversion of glycerol to glycerol-3-phosphate, allowing it to enter the process of glucose metabolism. In our previous studies, we found that glpK was involved in persister, L-form and biofilm formation in S. aureus.43 A recent study has found that the glpK of M. tuberculosis is associated with the tolerance of the bacterium to anti-tuberculosis drugs,251 which further suggests the relationship between GlpK and the formation of persisters and stress survival.
Aspartate decarboxylase (encoded by panD) catalyzes L-aspartate to β-alanine, which is one of the key enzymes in pantothenate metabolism and acetyl-CoA synthesis, and is essential for energy metabolism and fatty acid metabolism (Fig. 5). Our previous studies found that aspartate decarboxylase PanD is a target of pyrazinamide (PZA), which can explain the mechanism of PZA in killing persisters by inhibiting PanD to affect acetyl-CoA synthesis and energy metabolism of persisters.31,252,253 In addition, in the study of M. tuberculosis, it was also found that the transcription factor prpR of the prpDC operon, which is involved in energy metabolism, is also involved in persistence.254 The proteins encoded by prpD and prpC catalyze the first two steps of the methylcitrate cycle (MCC), converting propionyl-CoA to pyruvate, while prpR is a transcription factor of the operon, so prpR mutations affect the energy metabolism and persistence capacity of M. tuberculosis.
Trans-translation and protein degradation systems
Trans-translation exists in almost all bacteria, and plays an important role in rescuing stranded ribosomes and degrading toxic proteins during protein translation.255,256,257,258 It is required by diverse bacteria, especially in response to stresses.259,260 In our previous studies, we discovered that one of the mechanisms of action of PZA, an anti-persister drug, is to inhibit trans-translation and protein degradation by binding to bacterial ribosomal protein S1 (RpsA), suggesting that trans-translation and protein degradation is involved in persistence.29 Indeed, subsequent studies with mutant strains defective in trans-translation component ssrA and smpB in E. coli, showed that the persistence ability of the mutants to diverse antibiotics and stresses decreased significantly, which further confirms the roles of trans-translation and protein degradation in bacterial persistence.36
The finely tuned Clp protease system plays a crucial role in bacterial survival under stress conditions.261,262,263 During this process, damaged proteins are identified as substrates and a variety of major stress regulators are controlled by Clp-mediated proteolysis.264 A functional Clp protease complex consists of an ATP-consuming hexameric Clp-ATPase and the barrel-shaped proteolytic core ClpP. Usually, bacteria have various Clp-ATPases, including ClpC, ClpX, ClpA, or ClpE, that interact with the same proteolytic core.264,265 It has been found that mutations in the gene clpC or clpP are associated with PZA resistance in M. tuberculosis266,267 and persister formation in S. aureus,268,269 which verify the role of protein degradation in bacterial persistence. Furthermore, ATP depletion is the main force driving the formation of protein aggregation, and protein aggregation could be used as an indicator of dormancy depth.93 DnaK-ClpB can promote the degradation of protein aggregates, and interfering with the function of this gene will affect the recovery of persisters.93 These results further indicate the correlation between protein degradation and bacterial persistence.
Acyldepsipeptide antibiotic (ADEP4) can activate the ClpP protease, which can result in non-specific protease activity and subsequent degradation of over 400 proteins. These degraded proteins included ribosomal proteins, such as proteins S21, L9, S1 and ribosomal recycling factor, as well as FtsZ and proteins from various functional types such as purine metabolism, glycolysis, and aminoacyl-tRNA biosynthesis.177 Furthermore, ClpP is crucial for the proteolytic regulation of cellular levels of the MazE and RelB in type II toxin-antitoxin systems in Streptococcus mutans,228,269,270 suggesting that it is involved in the degradation of antitoxins. This may be the mechanisms through which ClpP is involved in bacterial persistence. Combining ADEP4 with rifampicin was shown to produce complete eradication of S. aureus biofilm persisters both in vitro and in vivo.177 The anti-persister effects of PZA targeting ClpC1 as well as ADEP4 targeting ClpP suggest that protein degradation pathway may serve as excellent therapeutic targets of anti-persister drug development.271
Metabolism of purines and amino acids
Purines are essential components of DNA, RNA, and ATP, and their metabolisms play crucial roles in various cellular processes. Disruption of the purine metabolism in S. aureus,272 B. subtilis,273 A. veronii,274and V. splendidus,275 influened persister cell formation. Moreover, it has been found that purine is crucial for persistence of E. faecalis in vivo, such as urinary tract and wound infections.276 Thus, it can be seen that the metabolism of purine has been linked to bacterial persister formation. Through screening a gene mutation library, we discovered that purine metabolism-related genes purB, purM and purN are involved in S. aureus persistence to antibiotics and stresses.277,278 Additionally, microarray analyses of persistent methicillin-resistant S. aureus (MRSA) bacteria showed that the transcription of multiple purine metabolism genes, including purF, purM, and purN, were significantly higher compared to non-persistent bacteria.272
The mechanism of purine metabolism in bacterial persistence encompasses various aspects, including energy metabolism, protein aggresome formation and intracellular efflux mechanisms.275 For instance, a decrease in purine metabolism has been linked to reduced energy production, such as ATP,275 which has been suggested to affect bacterial persistence to antibiotics by lowering the activity of ATP-dependent antibiotic targets.186,249,279 Additionally, the decrease in purine metabolism has been associated with protein aggresome formation,275 which has been reported to be linked to persister cells, as described earlier. Furthermore, inhibition of purine metabolism has been found to decrease membrane potential,275 which results in lower intracellular antibiotic concentrations compared to active cells, leading to enhanced bacterial survival under antibiotic treatment.280 Purine nucleosides, such as adenosine, guanosine, and inosine, have also been demonstrated to play significant roles in bacterial persistence.281,282 Adenosine and guanosine have been shown to restore susceptibility of V. splendidus persister cells to tetracycline.281 Inosine, on the other hand, has been found to modulate membrane permeability by upregulating the expression of OmpF.283 Inosine activates CpxA, which dephosphorylates CpxR-P and subsequently promotes the transcription of ompF,284 thereby increasing the uptake of antimicrobial molecule. All of these findings are consistent with the notion that purine metabolism is inversely correlated with bacterial persistence.
Amino acid metabolism plays a crucial role in the growth and survival of bacteria, including persistent bacteria. In persistent bacteria, amino acid metabolism is often altered to adapt to the stressful conditions they encounter. For example, persistent bacteria can exhibit changes in amino acid transport systems, allowing them to acquire amino acids more efficiently from their surroundings.285,286,287 In this discussion, we specifically focus on the role and mechanism of glutamate and arginine in bacterial persistence. Glutamate plays a crucial role in various metabolic processes in bacterial cells. Among the L-glutamate transport systems, GadC was initially identified as a regulator of acid tolerance in E. coli.285 In our previous research, we discovered that genes related to glutamate metabolism, such as gltT, as well as glutamate transporter genes gadC, gltS, gltP and gltI are all involved in bacterial persistence.286,287 Our findings demonstrated that the deletion gltS, gltP and gltI, resulted in decreased tolerance to various stresses, including antibiotics, acidic pH, hyperosmosis, and heat, in both E. coli and uropathogenic E. coli strains.286 Additionally, it also has been reported that global regulator Fis was beneficial for persister formation in S. typhi by repressing the gene expression associated with glutamate transport.288 Antibiotic tolerant E. coli cells upregulates glutamate decarboxylases (GadA) to counteract its intracellular acidification.289 These further supports the important role of glutamate metabolism in bacterial persistence.278,286,287
Glutamate may mediate tolerance to antibiotics and stresses through several potential mechanisms. Firstly, the glutamate-dependent acid resistance system is considered the most effective defense mechanism in safeguarding cells against low pH environments, providing a crucial protective role.290,291,292 Secondly, glutamate and its metabolite GABA serve as prominent compatible solutes in bacteria, enhancing enzyme functionality and protecting cells from various stresses such as high temperatures, freeze-thaw treatments, and drying.293 Thirdly, glutamate dehydrogenase catalyzes the conversion of glutamate to α-ketoglutaric acid (α-KG) through oxidation and deamination, which enables glutamate to enter the energy metabolism pathway via the TCA cycle, thereby facilitating bacterial tolerance.287
Besides glutamate, arginine has also been implicated in bacterial persistence. Through a comprehensive genetic screen targeting the clinically relevant strain USA300, we have identified the importance of the arginine metabolism gene argJ in persister formation, survival, and virulence in mice and C. elegans.46,278,287 Notably, mutations in the active site of the ArgJ protein resulted in a persistence defect, which was restored by genetic complementation and arginine supplementation in the argJ mutant.46 Furthermore, quantitative RT-PCR analysis revealed the upregulation of genes within the arg operon under drug-stressed conditions and in stationary phase cultures. Despite the identified role of the arginine biosynthesis pathway in persistence, the underlying molecular mechanisms remain unclear. One proposed mechanism is that the downstream products of arginine production, such as ammonia, mitigate hydroxyl radicals generated during antibiotic action that promote cell death and neutralize the acidic environment.294 Additionally, ornithine and polyamines have been shown to enhance cell fitness and survival against reactive oxygen species.295
Metabolic regulators
Cell metabolism is closely related to the formation and survival of persisters, so metabolic regulators are also associated with bacterial persistence, such as phoU. phoU is located on the pstSCAB operon of phosphate transport system and has negative regulation effect on phosphate metabolism in E. coli.296 Through screening the transposon mutant library of E. coli, we found that the phoU mutant had significant defect in persistence to various drugs and stress conditions (nutritional deficiency, high temperature, acidity, etc.), while the metabolic activity of the phoU mutant was higher than that of the parent strain (high expression of energy metabolism-related genes, phosphate metabolism-related genes and flagellum synthesis-related genes).37 These results suggest that PhoU may be a negative global regulator, mediating the persistence of bacteria by down-regulating their metabolic activities. The effect of PhoU on bacterial persistence was further validated in different bacterial species such as M. tuberculosis, S. aureus and P. aeruginosa. In the study of M. tuberculosis, we and others found that the persistence of M. tuberculosis in vitro and in vivo was significantly reduced after mutation of phoY proteins, a phoU homolog.33,297 In S. aureus, phoU mutation caused up-regulation of genes related to carbon metabolism and pyruvate metabolism, down-regulation of virulence genes and virulence regulatory genes,298 which further verified the mechanism of PhoU as a global regulator mediating bacterial persistence.
In addition, other global regulators have also been found to be involved in bacterial persistence. Lewis et al. evaluated the correlation between global regulator gene knockout strains and their persistence ability in E. coli and found that ATP levels increased and persistence levels decreased after integration host factor (ihf) gene knockout.185 Brynildsen et al. evaluated the role of seven global transcriptional regulators, ArcA, Cra, cAMP receptor protein[CRP], DksA, FNR, Lrp, and RpoS, in bacterial persistence, and found that cAMP/CRP played a central role.299 Recently, the RNA-binding protein ProQ has been found to be another global regulator of gene expression and contributes to persister formation in Salmonella by activating energy-consuming cellular processes.300
Stress responses mediated by SOS and sigma factor RpoS
The SOS response is triggered by DNA damage, allowing repair of genetic material to enhance cell survival.301 The proteins that make up the SOS system include a transcriptional repressor called LexA and a DNA-binding activating protein, RecA. After recA gene knockout, the persistence ability of E. coli to ciprofloxacin was significantly decreased, but it had no effect on the persistence to penicillin and gentamicin. The main reason is that quinolones cause bacterial DNA breaks,134 which activate the SOS initiation repair mechanism and related genes. This phenomenon showed that the same bacteria, under the action of different antibiotics, the genes related to the formation of persisters will have certain differences. By screening the single-gene knockout library of E. coli, we found that the mutations in other genes related to SOS response, recC, ruvA and uvrD, also cause defective persistence under the induction of rifampicin and tetracycline.42 Moreover, activation of the SOS response increases the frequency of SCVs, a special type of persisters, in S. aureus.302 These findings indicate that SOS response and DNA repair participate in the formation and survival of persisters (Fig. 6). In addition to DNA repair, the SOS response contributes to activating the type I TA module TisB/IstR as well.215 This leads to strong transcription of tisB, which forms ion channels in the plasma membrane, reducing the proton motive force and ATP formation, ultimately resulting in the formation of persisters.236,303 The induction of SOS response can also enhance the expression of fibronectin-binding protein, promote the attachment to host cells and accelerate the formation of biofilm, and improve the viability of bacteria in extreme conditions.304 Moreover, the SOS system (RecA), which is crucial for the development and survival of persisters, has been discovered to promote horizontal gene transfer between V. cholerae, thereby enhancing antibiotic resistance.170

Mechanisms of persister formation via SOS response, stringent response and quorum sensing. The SOS response is triggered by DNA damage, facilitating genetic repair to enhance cell survival under stress. It also contributes to the activation of type I TA module TisB/IstR as well. The stringent response is a global response to nutrition deprivation or limitation (such as carbon, amino acid nitrogen and phosphate), where (p)ppGpp is an important messenger molecule. This response induces bacterial dormancy through downstream pathways involving TA modules and ribosomes. Quorum sensing is a bacterial communication process that coordinates behavior based on population density. Bacteria release signaling molecules (AHL, CSP, indole etc.), which can enhance efflux pumps or disrupt metabolism. Created with BioRender.com
RpoS (σS)-mediated response can be induced by several general stresses,305 including nutrient deprivation, extreme pH, temperature and oxidative stress, which is associated with bacterial survival in stressful conditions as in stationary phase.174,306 However, its specific functions and regulatory mechanisms of RpoS in the process of persister formation are still uncertain. Some studies have found that SOS response increases tolerance to antibiotics in P. aeruginosa306 and E. coli.307 On the contrary, certain studies have demonstrated that the deletion of rpoS significantly enhances persistence through the upregulation of the MqsR toxin, indicating that bacteria with impaired general stress response are more prone to generating persister cells.308 Moreover, it has been reported that the antitoxins DinJ of the YafQ/DinJ TA module and MqsA of the MqsR/MqsA TA module have the ability to repress rpoS transcription and translation.309,310 These findings suggest that RpoS response and TA systems interact in the process of bacterial persistence. However, a recent study has revealed that RpoS had minimal impact on antibiotic persistence in E. coli.311 Therefore, further studies are required to address these discrepancies.
Cell communication
Quorum sensing and signaling molecules
Bacteria can communicate with each other through chemical signals to produce phenotypic heterogeneity, which is called quorum sensing (QS).312,313 Many significant bacterial pathogens, such as P. aeruginosa, S. aureus, S. typhimurium and V. cholerae, employ QS cell communication to regulate the expression of numerous virulence factors and related behaviors.314 In 2010, Nina Möker first reported the connection between the intraspecies quorum-sensing regulatory pathway and the formation of persisters.315 She discovered that the quorum-sensing-related signaling molecules, phenazine pyocyanin and the acyl-homoserine lactone (AHL) 3-OC12-HSL, notably elevated the number of persisters in logarithmic P. aeruginosa. In 2012, Leung and Lévesque reported that another chemical signal, competence-stimulating peptide (CSP), can induce the formation of Streptococcus mutans persisters.316 This finding further supports the hypothesis that quorum sensing is implicated in bacterial persistence. Following that, the involvement of quorum sensing in bacterial persistence has been confirmed in a range of other pathogens, including E. coli,317,318 V. cholerae,319 S. aureus320 and others.314,321 It is possible that the impact of quorum sensing on the formation of persister cells is linked to its capacity to regulate the secretion systems (T3SS and T6SS)322,323,324 and toxin like pyocyanin.325 Nevertheless, additional research is needed to elucidate these connections.
The QS systems identified in E. coli consist of the LuxR homolog, LuxS, autoinducer-2 (AI-2) and autoinducer-3 (AI-3) systems, as well as an indole-mediated signaling system.314 Indole-mediated signaling is widely distributed between cells and has been shown to be involved in the formation of E. coli persisters.317 It has been reported that indole can be sensed by a population of cells in a heterogeneous manner, activate oxidative stress and phage shock pathways through periplasmic or membrane components, induce high expression of oxyR, pspBC genes, and thus contribute to the generation of persister subpopulation.317 This result has been confirmed in another study.326 Furthermore, the intestinal pathogen Salmonella typhimurium has also been observed to increase its antibiotic tolerance in response to indole.327 These findings highlight the complex interactions between indole and bacterial antibiotic tolerance. Additionally, in E. coli, indole still has been found to induce the expression of several multidrug efflux pumps.328,329 These research findings have established the relationship between quorum sensing, reactive oxygen species (ROS), and efflux pumps (Fig. 6), suggesting that quorum sensing does not act alone in mediating bacterial persistence.
However, it is important to note that previous studies have reported contrasting effects of indole on antibiotic persistence. For example, indole has been shown to decrease antibiotic tolerance of E. coli.209,330 Additionally, indole derivatives have demonstrated the ability to enhance the efficacy of aminoglycosides against stationary-phase Gram-positive bacteria under hypotonic conditions. However, these derivatives have also been found to suppress the effects of aminoglycosides against Gram-positive bacteria in the exponential-phase stage and both stages of Gram-negative bacteria.331 Furthermore, indole has been observed to exhibit opposite effects on antibiotic resistance, as it has been shown to reverse antibiotic resistance in Lysobacter enzymogenes.332,333 These diverse effects of indole on antibiotic persistence and resistance highlight the complexity of its interactions with bacteria. It is important to further investigate the underlying mechanisms behind these opposing effects of indole on antibiotic persistence and resistance.
Stringent response and guanosine pentaphosphate/tetraphosphate (pppGpp/ppGpp)
Stringent response in bacteria is a global response to amino acid deprivation or carbon, nitrogen and phosphate limitation,334,335 where (p)ppGpp is an important messenger molecule in this process, responsible for sensing environmental stress and inducing downstream pathways to drive bacteria into dormancy.28,336 The (p)ppGpp network comprises Rel/SpoT homolog (RSH) proteins with a nucleotidyl-transferase domain.337 In E. coli, the (p)ppGpp synthetase RelA is activated by amino acid starvation and heat shock, while the synthetase/hydrolase enzyme SpoT is activated by carbon, nitrogen, phosphate, iron, and fatty acid starvation.338 When bacteria are exposed to these stresses, the concentration of (p)ppGpp increases and acts as an alarm to coordinate many concentration-dependent process reprogramming, such as replication, transcription, and translation339 (Fig. 6). Interestingly, diverse bacteria deficient in (p)ppGpp production usually display massive defects in persister formation and survival.126,166,340,341,342,343,344,345 Also, clinical S. aureus mutations that partially activate the stringent response confer multidrug tolerance.346
The ppGpp ribosome dimerization has been proposed as a model of persister formation where ppGpp may regulate bacterial persistence levels by affecting ribosome function347,348 (Fig. 6). This model suggests that stress factors such as nutritional deficiency, hyperosmolality and acidity can induce the production of ppGpp and cAMP through RelA/SpoT, and ppGpp and cAMP further induce the expression of ribosome-associated inhibitor A (raiA) (which binds to the 70S ribosomes to inactivate it) and ribosome modulation factor (rmf) (which converts active 70S ribosomes into inactive 100S ribosomes through inactive 90S dimer complex). Both can cause protein synthesis disorders by inactivating ribosomes.347 Wood et al. previously found that after ribosome function inactivation, persister cell population in the exponential growth stage increased nearly 10,000 times.279 This result further confirmed the mechanism of ppGpp involved in the formation of persisters by regulating ribosome function. Additionally, since both type I and type II TA modules are activated by the (p)ppGpp signaling, it suggests that the mechanism of ppGpp involved in the formation of persisters might be through TA modules. However, this mechanism, following the retraction of a closely related previously published article titled “(p)ppGpp Controls Bacterial Persistence by Stochastic Induction of Toxin-Antitoxin Activity”,227 still requires further validation.
Efflux pumps/transport systems
In addition to “passive defense” through dormancy, persisters can also excrete drugs to resist antibiotic attack by enhancing efflux activity, a form of “active defense”.349,350,351 Efflux pumps are a type of membrane protein that can actively pump drugs out of bacterial cells, reducing the concentration of antibiotics inside the cells and thus reducing their effectiveness.352,353,354 In 2016, Bai and colleagues from Peking University in China discovered that several multi-drug efflux genes, with a focus on the central component tolC, exhibited elevated expression levels in E. coli persisters.355 Moreover, through time-lapse imaging and mutagenesis studies, they further confirmed a direct positive correlation between tolC expression and bacterial persistence.355 This study garnered significant attention at that time,349,350,351,356 because it represented the first instance of an active mechanism contributing to stochastically induced multiple-drug tolerance. Subsequent studies further demonstrated that enhanced efflux activity contributes to the formation of persister cells.357,358 For example, when penicillin-exposed persisters of S. pyogenes (Group A streptococcus-GAS) were compared with susceptible strains, a significant number of efflux pump-related genes were transcriptionally upregulated, including at least a 4-fold increase in Resistance-Nodulation-cell Division (RND) family efflux pump-related operon genes and a 2.2-fold increase in gene expression of a homolog of the polysaccharide transporting ATP-binding protein MsmK.359 In addition, loss of the AcrAB efflux pump in Aeromonas veronii (A. veronii) decreased the formation of persisters when treated with chloramphenicol, tetracycline, fluoroquinolone and β-lactam antibiotics.360 Moreover, AcrR, the repressor of the AcrAB efflux pump, was also found to play a role in regulating persister formation by repressing the activity of the AcrAB efflux pump in A. veronii.360 The involvement of efflux pumps in bacterial persistence had been validated by proteomic analysis361 and in ex vivo models as well.362 The mechanism might be that efflux pumps with high activity can reduce the concentration of antibiotics in persisters, which is of great significance to the survival of the persisters.
As is well known, the efflux system is also a crucial control element for antibiotic-resistance.352,363,364,365,366 However, the triggers and outcomes of efflux pump changes might differ between resistant and persistent bacteria. In the case of resistant bacteria, the efflux pumps are often triggered by low concentrations of antibiotics. When antibiotics are present at levels below their effective killing threshold, as can occur with the unregulated use of antibiotics in clinical settings, bacteria can detect and respond to these low levels by increasing the expression of efflux transporters and other mechanisms through genetic alterations.364 This leads to the development of a more robust defense mechanism that enables bacteria to withstand higher concentrations of antibiotics that would typically be lethal to them. On the other hand, by tracking bacteria using the FlAsH-labeled TolC, researchers found that persister cells could emerge from a subpopulation that had increased levels of TolC even before treatment with the antibiotic.355 This suggested that in persistent bacteria, the efflux pumps are triggered by not only antibiotics but also by other factors in environment, like metabolic toxic compounds. Although persister cells enter a dormant or slow-growing state, they are not completely metabolically inactive and may produce toxic compounds that also need to be expelled.364 This important result also raises the question of whether the mechanism of bacterial persistence mediated by efflux pumps is independent of other mechanisms that have already been discovered.350 Further detailed and mechanistic investigations are essential to answer this question.
Moreover, as a recent study demonstrated the role of the AcrAB-TolC multidrug efflux pump on drug resistance acquisition by plasmid transferring,363 suggesting that the mechanism of bacterial resistance mediated by efflux pumps is probably not primarily through antibiotic efflux. It appears that when bacteria rely on efflux pumps as their primary defense against antibiotics and only pump out intracellular levels of antibiotics, persistence is formed. In the proposed evolutionary connection between persistence and resistance to antibiotics, the expected evolutionary trajectory begins with the emergence of antibiotic persistence, ultimately leading to antibiotic resistance.142 From this perspective of the mechanism involving efflux pumps in bacterial persistence and resistance, such an evolutionary trajectory seems reasonable.
Epigenetic modifications, RNA degradation, and small non-coding RNA
Strain with deletion in adenine methyltransferase-encoding gene dam (Δdam) in uropathogenic E. coli (UPEC) had a significant defect in the formation of persisters, while strain with deletion in cytosine methyltransferase-encoding gene dcm (Δdcm) did not affect the formation of persisters, suggesting that adenine methylation but not cytosine methylation is involved in persister formation or survival.367 Dam could mediate the formation of persisters by regulating the transcription of related genes, cell movement, DNA repair and other processes.367 In addition to this, under stress, cellular DNA methylation status regulates global gene expression patterns to promote bacterial dormancy, when persisters recover from dormancy, some of the epigenetic traits that promote persistence can be retained in a few cells as heritable “memories” that drive evolutionary pathways over time, thereby increasing the frequency of persister formation or survival.368
RNA methylation has also been found to play a role in the formation of persisters. For example, methylation of the G37 site of tRNA m1 (at the 37th guanosine N1site at the 3’ end of the tRNA anticodon) in Gram-negative bacteria can maintain a unique double-membrane structure in its envelope, which is closely related to drug resistance and persistence in Gram-negative bacteria.369 In addition, the double membrane structure is an essential component of the recovery of Gram-negative bacteria, suggesting that tRNA methylation modification is not only involved in the formation but also in the recovery of persisters. It has also been found that an acetyltransferase, TacT, blocks formation of peptide bonds between nascent peptide and tRNA by modifying the primary amine group of amino acids on tRNA molecules, thereby inhibiting protein translation and promoting the formation of persisters;218 the accumulated deacylated tRNA trigger bacterial persistence independent of stringent response,370 suggesting that tRNA acetylation also plays a role in persister formation. In addition, polynucleotide phosphorylase (PNPase), a component of RNA degradosome (composed of RNase E, helicase RhlB, and enolase together with PNPase), involved in RNA degradation in E. coli, has been found to affect the formation of persisters by regulating cAMP/CRP, known to be involved in bacterial metabolism.38 Interestingly, we also found that small non-coding RNAs (sRNAs), such as ryhB, are also involved in persister formation.40 The discovery of these new mechanisms has not only further improved our understanding of the mechanisms of persister formation but also provides new targets for developing anti-persister drugs.
Compounds or drugs that target and eliminate persister cells
As mentioned above, bacterial persisters not only hinder our ability to effectively treat infections but also act as reservoirs for antibiotic resistant strains. Therefore, killing persisters is crucial for improving treatment outcomes in face of challenges posed by chronic persistent infections, including recurrent infections, bacterial resistance, and biofilm formation. However, the antibiotics commonly used in clinic mainly kill growing bacteria and usually have poor activity against persisters.371 As a result, the treatment of chronic persistent infections and biofilm infections becomes exceptionally challenging, resulting in unsatisfactory treatment outcomes.84,125 A notable illustration of this challenge is evident in biofilm infections caused by P. aeruginosa and M. abscessus among individuals with cystic fibrosis. Encouragingly, with the advancement of our understanding of mechanisms and characteristics of persisters, several anti-persister compounds have been discovered89,372,373 (Table 2). These compounds can be categorized into three main types: direct killing of persisters, sensitizing persisters to conventional antibiotics, and inhibition of persister formation.
Killing of persisters directly
Compounds targeting the molecular pathway related to bacterial persistence
As described earlier, multiple processes such as toxin-antitoxin systems, metabolism and metabolic regulatory factors, DNA repair, cell communication, efflux pumps, and epigenetic modifications are involved in the formation and survival of persisters. In these critical processes, targeting the metabolism of trans-translation and protein degradation has emerged as the most successful approach for directly eliminating persisters with anti-persister drugs, including pyrazinamide (PZA), bedaquiline, ADEP4, lassomycin and kitamycobactin.
PZA is a first-line tuberculosis drug that plays a unique role in shortening the duration of tuberculosis chemotherapy. As a well-established prototype anti-persister drug, it has notably decreased the treatment duration for tuberculosis from 9 to 12 months to just 6 months,91 highlighting the clinical importance of anti-persister drugs.17 It has been found that PZA is a prodrug that is hydrolyzed intracellularly to pyrazinoic acid (POA) by pyrazinamidase (PZase) encoded by pncA,374 then POA bound to the ribosomal protein S1 (RpsA), a vital protein involved in protein translation and the ribosome-sparing process of trans-translation.29 PanD, an aspartate decarboxylase, is another target of PZA and that POA binding to PanD could inhibit pantothenate and CoA biosynthesis, which is essential for the central metabolism necessary for energy production and fatty acid metabolism in M. tuberculosis.31,252,375 In brief, PZA kills M. tuberculosis persisters by targeting metabolic processes including trans-translation and protein degradation, as well as energy metabolism.376 Bedaquiline, also known as TMC207, is a new antimycobacterial agent recently approved by the FDA for treating pulmonary MDR-TB by inhibiting ATP synthase.377 Research has shown that when combined with other agents such as pretomanid and moxifloxacin, bedaquiline effectively eliminates M. tuberculosis in non-replicating persister state.378,379 This indicates that bedaquiline may serve as an anti-persister drug against M. tuberculosis, similar to PZA.
ADEP4 is a compound against S. aureus persisters targeting ClpP ATPase, which was isolated from the fermentation broth of Streptococcus hawaiiensis NRRL 15010 177,195,380,381. The combination of ADEP4 with rifampicin has been demonstrated to achieve complete eradication of S. aureus biofilm persisters in both in vitro and in vivo studies.177 Though, this was not confirmed in a recent study using a more persistent infection model.56 Nevertheless, it is worth highlighting that the effectiveness of an antimicrobial drug does not solely depend on targeting essential components for bacterial viability. Antimicrobial drugs that target previously unexplored pathways, such as activating the ClpP protease, have shown no cross-resistance to any antibiotic classes currently available or in development. This unique characteristic makes them an ideal candidate for combination therapy in the treatment of infections caused by drug-resistant or drug-tolerant bacteria. Lassomycin382 and its analog kitamycobactin,383 which were screened from extracts of uncultured soil microbes, have also shown activity against M. tuberculosis persisters. Their targets have been identified as the Clp ATPase complex as well. Specifically, lassomycin binds to a highly acidic region of the ClpC1 ATPase, leading to ATPase activation and changes in protein degradation profile.384 These findings suggest that exploring anti-persister drugs based on the molecular mechanisms of bacterial persistence, especially metabolism, holds promise for future research.
In summary, in addition to the above compounds, there are still compounds targeting other molecular mechanisms of bacterial persistence. For instance, mavintramycin A was found to be active against persistent infection of M. avium complex (MAC) by binding to 23S ribosomal RNA and inhibiting protein synthesis.385 However, although many molecular mechanisms of bacterial persistence have been identified and validated over time, these persister drugs mainly target metabolism. Therefore, in future studies, it is crucial to explore the development of more anti-persister drugs targeting other essential molecular mechanisms involved in the formation and survival of persisters.
Membrane active small molecules
Another prominent target for directly attacking persisters is the bacterial membrane.386 HT61 is a novel quinolone-derived compound against non-multiplying persisters, including methicillin sensitive and resistant S. aureus.387,388 HT61 has also been proposed as an adjunct to other antimicrobials to extend their usefulness. For example, it has been shown to enhance the effect of tobramycin against P. aeruginosa both in vitro and in vivo.389 Additionally, HT61 has been found to enhance the activity of neomycin, gentamicin, mupirocin, and chlorhexidine against both methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA) in vitro and in vivo.390 It is worth mentioning that HT61 nasal gel has also shown promising results in Phase II clinical trials (EudraCT number, 2009-017398-39) and Phase III trials (EudraCT number, 2010-021193-11) by enhancing the effectiveness of conventional antibiotics in eradicating persistent nasal colonization of S. aureus.372 The mechanism of action of HT61 involves non-specific targeting of anionic lipids that are abundant in bacterial membranes.391,392 By interacting with these lipids, HT61 disrupts the integrity of the bacterial cell membrane, leading to cell lysis and release of ATP from the bacterial cells.
XF-73 is another membrane-active agent, porphyrin compound, with rapid bactericidal activity, which is active against both slow-growing and non-dividing cultures of S. aureus including biofilms.393,394,395,396 In a randomized, open-label, Phase I clinical trial, nasal formulations of XF-73 demonstrated good safety and local tolerability.397 In a Phase II clinical trial, the intranasal administration of XF-73 24 h prior to surgery significantly reduced S. aureus nasal carriage in patients undergoing cardiac surgery.398 These findings suggest the potential of XF-73 as an effective intervention to reduce S. aureus colonization and associated infections in surgical settings. Further research has revealed that XF-73 also exhibits antimicrobial effects against Candida albicans (C. albicans) and its biofilms.399,400 This expands the potential applications of XF-73 beyond its activity against S. aureus, highlighting its broad-spectrum antimicrobial properties. Continued investigation is necessary to fully understand the extent of XF-73’s antimicrobial activity and its potential clinical applications. XF-70, a compound structurally similar to XF-73, has also demonstrated the ability to combat slow-growing and non-dividing cultures of S. aureus including biofilms.394 Furthermore, XF-70 has exhibited additional activity against small-colony variant hemB mutants of MSSA and MRSA.401
NCK10 is an aryl-alkyl-lysine compound with a decyl chain appendage that has been shown to effectively lyse persister cells of E. coli and biofilms of A. baumannii (MTCC 1425), E. coli (MTCC 443), K. pneumoniae (ATCC 700603) and P. aeruginosa (MTCC 424) in murine model of burn infection.402 Furthermore, NCK10 has exhibited activities against planktonic cells, persister cells, biofilms of MRSA, and has shown the potential to protect mice from skin infections.403 Additionally, NCK10 has displayed broad-spectrum antibacterial activity, demonstrating potency against both immature and mature biofilms of C. albicans.404 This compound achieves its antimicrobial effects by depolarizing and permeabilizing the bacterial outer membrane, leading to cell lysis. These findings suggest that NCK10 holds potential as a broad-spectrum antimicrobial agent against persistent bacterial infections and biofilms. Further research is needed to fully understand the mechanism of action and potential clinical applications of NCK10.
Another class of membrane-active agent against persisters is synthetic retinoid derivatives. Two notable representatives of this class are CD437 and CD1530, which were identified through a screening of approximately 82,000 compounds using a C. elegans-MRSA infection model.405 Both CD437 and CD1530 have demonstrated the ability to eliminate persisters formed in MRSA biofilms by disrupting the bacterial membrane bilayer and inducing membrane permeabilization.406 Furthermore, an analogue of CD437 has shown antimicrobial activity against MRSA persisters while exhibiting improved cytotoxicity compared to CD437.406 In a MRSA mouse deep-seated thigh infection model, CD437 or its analogue, either alone or in combination with gentamicin, exhibited promising efficacy.406 These findings highlight the potential of synthetic retinoid compounds as effective agents against persisters and their potential for combination therapy with conventional antibiotics.
NH125 (1-hexadecyl-2-methyl-3-[phenylmethyl]-1H-imidazolium iodide), a bacterial histidine kinase inhibitor, was identified using SYTOX Green screening assay. It was found to possess strong bactericidal properties against MRSA persisters by inducing cell membrane permeabilization.407,408 Furthermore, NH125 has demonstrated either no toxicity or low toxicity to C. elegans.407 In addition, several analogues of NH125 have been identified to possess the ability to kill MRSA, methicillin-resistant Staphylococcus epidermidis (MRSE), and vancomycin-resistant Enterococcus faecium (VRE) persisters and biofilms and eradicate fungal biofilms.409,410 These findings suggest that NH125 and its analogues hold potential as promising drug candidates for combating persistent bacterial and fungal infections. In addition to NH125, several other membrane-active small molecules, such as PQ401411 and NTZDpa,412 have shown promising activity against MRSA persisters in in vitro studies or in models like C. elegans and Galleria mellonella. These compounds have demonstrated potential as effective agents against persistent MRSA infections. Further research is needed to evaluate their efficacy, safety, and potential for clinical use.
From the above information, it is evident that the currently discovered membrane-active small molecules have primarily been studied for their activity against S. aureus, including drug-resistant strains and biofilms. However, among these compounds, NCK10 stands out with its relative broad-spectrum antibacterial activity. NCK10 has demonstrated efficacy not only against Gram-positive bacteria like MRSA but also against Gram-negative bacteria such as A. baumannii, E. coli, K. pneumoniae and P. aeruginosa. Except for NCK10, esculentin (1–21), an amphibian skin membrane-active peptide, has also shown activity against both planktonic and biofilm cells of P. aeruginosa and prolong survival of animals in models of sepsis and pulmonary infection.413 Derivatives of esculentin peptides have exhibited efficacy against infections caused by E. coli strains such as O157:H7.414,415 Furthermore, numerous other peptides have been identified as anti-persister drugs. These include 2D-24416 and piscidin417 targeting P. aeruginosa, Trp/Arg-containing peptides effective against E. coli,418 Art-175 (fusion of the sheep myeloid 29-amino acid peptide and the KZ144 endolysin) effective against A. baumannii419 and P. aeruginosa,420 quaternary ammonium cations (QACs) effective against MRSA and E. faecalis,421 P14KanS effective against S. aureus and S. epidermidis, as well as P. aeruginosa and A. baumannii,422 and pentobra effective against E. coli and S. aureus.423 These broad-spectrum activities highlight the potential of peptides as versatile antimicrobial agents capable of targeting a range of bacterial pathogens. Additionally, a propanol-amine derivative (1-((2,4-dichlorophenethyl) amino)-3-phenoxypropan-2-ol), known as SPI009, also showed broad-spectrum activities against persister cells of various Gram-positive and Gram-negative pathogens by inducing significant membrane permeabilization.424,425
Polycationic glycopolymer, such as poly (acetyl, arginyl) glucosamine (PAAG), represent another type of membrane-active compound against mycobacteria. Studies have shown that PAAG treatment can effectively eradicate antibiotic-induced persister cells in planktonic cultures of non-tuberculosis mycobacteria (NTM).426 Additionally, PAAG has demonstrated the ability to disperse NTM biofilms.426 These findings suggest that PAAG holds promise as a potential therapeutic agent for combating persistent NTM infections. However, its efficacy has only been demonstrated in in vitro experiments. Furthermore, there are many other membrane active molecules exhibiting effective anti-persister activity are currently under evaluation in vitro. For example, 3,6-Di[4-(diethylamino)-butoxy]-1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-enyl)-9H-xanthen-9-one (AM-0016)427 and boromycin428 kills mycobacterial persisters; carvacrol (2-methyl-5-(1-methylethyl) fenol) showed activity against B. burgdorferi persisters;429 lipidated lysine 9, was capable of lysing persister cells of E. coli and S. aureus.430 Further research is needed to evaluate the activity of these agents.
Overall, disrupting the bacterial membrane bilayer has emerged as a promising strategy for killing persisters. While many membrane-active compounds have demonstrated good activity against persisters, and some even show promising clinical application prospects, there are several obstacles that need to be considered during the discovery and assessment of these agents. For instance, potential candidates have been found to effectively kill pathogens, but they may also disrupt mammalian membranes and have potential toxicity.431,432 Therefore, structural transformation and optimization are necessary to develop species-specific molecules. NH125 serves as an example of this, as it exhibits low cytotoxicity due to careful design and optimization.
Phage-derived therapy
Phage therapy is an ancient anti-infective tool that predates even penicillin.433 However, the challenges of phage therapy include insufficient pharmacokinetic and pharmacodynamic studies, the need for further evaluation of clinical efficacy, and the management of foreign proteins, nucleic acids, and virulence factors introduced by phages, all of which pose significant obstacles to its widespread implementation.433,434 In recent years, phage therapy has been sporadically approved for compassionate use, which refers to emergency use when no other approved therapy is available.435,436 Furthermore, phage therapy has shown promising therapeutic outcomes in the majority of cases.437,438,439 Therefore, studies on phage therapy for anti-infective treatments, especially drug-resistant infections, have been increasing.439,440,441,442
Research on phage therapy for the treatment of persistent infections has also made some progress. The most advanced development is the recombinant bacteriophage lysin CF-301, known as PlySs2 or exebacase, which is the first agent in this category to complete late-stage clinical trials (phase 3, NCT04160468) for the treatment of S. aureus bacteremia, including right-sided endocarditis.443 CF-301 was first cloned by the team of Vincent Fischetti, with the gene derived from a Streptococcus suis (S. equi) phage sequence. It was found that the chromatographically purified CF-301 exhibited broad lytic activity against various bacteria, including MRSA, vancomycin-intermediate S. aureus (VISA), Streptococcus suis, Listeria, Staphylococcus simulans, Staphylococcus epidermidis, S. equi, Streptococcus agalactiae, Streptococcus pyogenes, Streptococcus sanguinis, group G streptococci (GGS), group E streptococci (GES), and Streptococcus pneumoniae.444 Subsequently, the antibacterial activity of CF-301 against S. aureus persister cells in exponential-phase and stationary-phase populations and biofilm was further demonstrated.445,446 Additionally, the endolysin LysH5 derived from the phage vB_SauS-phiIPLA88 induces cell lysis in both actively growing and non-growing S. aureus and S. epidermidis, enabling the elimination of persister cells.447 Both of these therapies, derived from phages, act through the mechanism of cell wall hydrolases. Moreover, engineered bacteriophages expressing the outer membrane protein OmpF enhanced the eradication of persister cells by fluoroquinolones compared to monotherapy.448 Another type of bacteriophage, expressing the SOS-response repressor LexA3, also successfully reduced E. coli persister levels.448
Repurposed existing anti-cancer drugs or antibiotics
Moreover, existing anti-cancer drugs or antibiotics were re-purposed to kill bacterial persisters, which have the advantage of saving the cost and time of new drug research and development. For example, mitomycin C (MMC), an FDA-approved anti-cancer drug, has been found to effectively eradicate E. coli, S. aureus and P. aeruginosa persisters both in vitro and in vivo,449 as well as A. baumannii450 and B. burgdorferi451 persisters. Likewise, another FDA-approved anti-cancer drug, cisplatin, has demonstrated the ability to eradicate persister cells of E. coli, S. aureus, and P. aeruginosa as well.452 The antimicrobial mechanisms of these two drugs involve causing growth-independent crosslinking of DNA, thereby effectively eliminating persister cells.449 In addition, anti-cancer drug anthracyclines were also found to be capable of killing B. burgdorferi persisters,453 although the exact mechanism of action remains to be determined they may act by DNA damage. Nevertheless, it should be pointed out that the high toxicity of anti-cancer drugs may affect their applications in clinic as anti-persister therapies. In addition, there are also known antibiotics that have been repurposed for persister killing, such as tosufloxacin,52 colistin,49 clinafloxacin,56 daptomycin60 etc., which have the advantage of not requiring additional clinical safety evaluations.
Sensitizing persisters to conventional antibiotics
Due to the inactive metabolic state of persisters, conventional antibiotics are rarely effective against persisters. However, researchers have discovered that strategies such as reverting or awakening persisters, enhancing antibiotic uptake or increasing bactericidal action of antibiotics can render them susceptible to conventional antibiotics.
Reverting or awakening persisters
Reversing or awakening persisters to their normal bacterial states, which respond to antibiotic treatment, is an ideal approach for studying anti-persister drugs. While resuscitation factors have been identified for bacterial resuscitation, the practical implementation of reverting or awakening persisters remains limited. It has been observed that the addition of spent medium containing protein and peptide factors resuscitated S. aureus persister cells, enhancing their susceptibility to antibiotic-induced eradication.454
Recently, the fatty acid signaling molecule cis-2-decenoic acid has been shown to induce persisters of both P. aeruginosa and E. coli to transition into ciprofloxacin-susceptible states, but the mechanism involved is still unclear.455 Another compound, BF8, was also found to revert antibiotic tolerance of E. coli and P. aeruginosa persister cells, although the underlying mechanism remains unknown.456,457 In addition, 3-[4-(4-methoxyphenyl)piperazin-1-yl]piperidin-4-ylbiphenyl-4-carboxylate (C10), identified from a chemical library screen, demonstrated the ability to reverse the persister phenotype of both P. aeruginosa and E. coli to antibiotic-sensitive cells by enhancing their metabolism.458 This action rendered these tolerant cells susceptible to various classes of antibiotics.
Enhancing drug uptake
Carbon metabolites were first discovered to promote the uptake of aminoglycoside antibiotics in persisters. Allison et al. demonstrated that carbon metabolites like glucose, mannitol, fructose and pyruvate can increase the generation of proton-motive force (PMF), thereby promoting the uptake of aminoglycosides in persisters.248 This mechanism facilitates the killing of persisters by aminoglycosides in both E. coli and S. aureus.248 This work establishes a metabolism reprogramming strategy for eradicating bacterial persisters and emphasizes the significance of the metabolic environment in antibiotic treatment. Furthermore, other studies have also found that mannitol (10–40 mM) can increase the sensitivity of P. aeruginosa persister cells to tobramycin by up to 1000-fold.459 Similarly, fumarate has been shown to have a similar effect.460 Rhamnolipids, which are biosurfactant molecules produced by P. aeruginosa, have been found to enhance the uptake and activity of aminoglycosides against S. aureus persisters including phenotype of SCVs.461 It is worth mentioning that rhamnolipids promoting the uptake of aminoglycosides in persisters is PMF-independent, unlike carbon metabolites.
Moreover, specific environmental factors have also been found to sensitize persistent bacteria to antibiotics by increasing drug uptake. For instance, a two-minute treatment under hypoionic shock conditions (e.g., in pure water) significantly enhances the bactericidal effects of aminoglycosides against both spontaneous and triggered E. coli persisters.462 This enhancement may be attributed to the involvement of mechanosensitive channels (MS). Additionally, freezing has been observed to dramatically increase the bacterial uptake of aminoglycosides independent of PMF.463 This effect may be attributed to freezing-induced cell membrane damage, such as the activation of the mechanosensitive ion channel MscL. These findings provide valuable insights into the development of new strategies against bacterial persisters by combining existing antibiotics with MS or MscL agonists. Such combination approaches have the potential to enhance the efficacy of antibiotics and overcome the challenges posed by persistent bacterial infections.
Potentiating antibiotic activity
Amino acids have also been found to enhance antibiotic-mediated killing through several pathways, including inhibiting amino acid synthesis, boosting endogenous reactive oxygen species (ROS) production, and altering membrane pH gradient. For instance, supplying amino acids (such as serine, threonine, glutamine, and tryptophan) to wild-type E. coli sensitizes stationary-phase cells to gentamicin, potentially by inhibiting amino acid synthesis.242 Additionally, the addition of serine can enhance the effectiveness of ofloxacin or moxifloxacin against E. coli by increasing NADH production.464 This strategy works by elevating the NAD (+)/NADH ratio, disrupting Fe-S clusters, and boosting the generation of endogenous ROS. Furthermore, L-arginine enhanced gentamicin activity against S. aureus, E. coli and P. aeruginosa persisters by modifying the membrane pH gradient.465 Moreover, glutamine has been found to promote antibiotic uptake, leading to killing of multidrug-resistant uropathogenic bacteria.283 Our previous studies have demonstrated the significant role of glutamate in the formation and survival of bacterial persistence.286 Therefore, facilitation of these amino acids may hold promise for future development of effective approaches to manage chronic bacterial persistent infections.
Besides amino acids, metal ions could also potentiate antibiotic activity against persisters by enhancing ROS production. It has been found that combinations of silver and antibiotics eradicate Gram-negative bacterial persisters both in vitro and in vivo.466 Additionally, iron has been shown to enhance the activity of the persister drug PZA against M. tuberculosis. With recent progress in nanotechnology and surface chemistry, it is conceivable to create silver nanoparticles with antibiotic-functionalized surfaces embedded in medical materials that regulates the release of active antimicrobial agents at the infection site.
Inhibiting persister formation
Unlike strategies that specifically aim to eliminate persisters to cure persistent infections, an alternative strategy for addressing persistent infections involves preventing the formation of persisters in the first place. By interfering with the mechanisms involved in persister formation, it may be possible to prevent the development of persistent infections. Specifically, targeting quorum sensing (QS) and (p)ppGpp, as mentioned earlier, can be effective as they play crucial regulatory roles in persister formation.
Researches have demonstrated that inhibiting the transcriptional regulator MfvR in QS system with a synthetic benzamide-benzimidazole derivative (referred to as M64) is effective against persistent P. aeruginosa infections both in vitro and in vivo.467,468 This compound represents the first identified to decrease the formation of antibiotic-tolerant persister cells. In addition, another compound, relacin, which targets the (p)ppGpp signaling pathway, has been shown to reduce persister levels in E. coli, B. subtilis, B. anthracis, Group A streptococci and E. faecalis.469,470 Relacin works by preventing the accumulation of the alarmone (p)ppGpp through the inhibition of RelA, thereby disrupting the bacterial stringent response.469 The stringent response is a key pathway involved in bacterial persistence which is mediated by the alarmone (p)ppGpp. In M. tuberculosis, it has been found that targeting various components of the stringent response could lead to altered antibiotic susceptibility and potentially shorten tuberculosis treatment.471 These suggest that intervening in QS and (p)ppGpp pathways can potentially disrupt the formation of bacterial persisters, thereby assisting in the treatment of infectious diseases. Although these compounds have not yet entered clinical application, these research findings bring hope for the treatment of persistent bacterial infections.
In addition to QS and (p)ppGpp, other pathways could also be used in controlling persister formation. For example, enhancing respiration and thus metabolism using N-acetylcysteine reduced the formation of M. tuberculosis persisters;472 inhibiting the accumulation of protein aggregates by MOPS (3-(N-morpholino) propanesulfonic acid) or osmolytes (such as betaine, trehalose, glucose, and glycerol) reduced the frequency of persister formation;473 increasing bacterial sensitivity to oxidative stress by 5-aminosalicylic acid (also known as mesalamine) is also capable of reducing persister formation;474 or interfering with the bacterial response to oxidative stress by using bacteriophages expressing the SOS-response repressor LexA3 increased persister killing as well.448 Therefore, the growing understanding of the mechanisms underlying bacterial persistence has resulted in the discovery of additional strategies that disrupt processes involved in persister formation, consequently reducing the number of persisters.
Proposal of persister drug combination approach for more effective treatment of persistent infections
The earliest treatment strategy for persistent infections can be traced back to the 1940s, coinciding with the first description of the phenomenon of bacterial persisters.62 Joseph Bigger suggested that intermittent or pulse antibiotic use would enable surviving persister cells to resuscitate during non-treatment periods, followed by their rapid elimination during subsequent treatment. However, this approach is difficult to use due to unpredictable resuscitation of the persisters in vivo and has not been used clinically. In the following clinical treatment of challenging persistent infections like tuberculosis17,378 and acquired immune deficiency syndrome (AIDS),475,476 combination therapies with multiple drugs are employed, which may also be the most effective approach for treating other persistent infections. This proposal aligns with the findings and recommendations of other scientists,88,477,478,479 and its validity has been further supported by recent animal studies. For example, chronic persistent infections caused by B. burgdorferi, S. aureus, P. aeruginosa and E. coli can be effectively eradicated using this approach by applying different drug combinations (cefoperazone + doxycycline + daptomycin;480 clinafloxacin + meropenem + daptomycin;56 clinafloxacin + cefuroxime + gentamicin;481 colistin + ofloxacin + nitrofurantoin49) in respective animal models. Further clinical evaluations are needed to see if these antibiotic combinations can eradicate the corresponding persistent infections in patients. However, it is important to note that different antibiotics have distinct mechanisms of action and can sometimes exhibit antagonistic effects on each other482 or promote the emergence of tolerance.483 Therefore, it is necessary to continue to search for rational drug combinations that can effectively kill persisters and cure persistent infections.
Firstly, combination of drugs killing both metabolically active bacteria and persister cells (Fig. 7). In theory, due to the complex heterogeneous bacterial population consisting of growing and non-growing persister cells,24,484,485 a drug combination approach targeting these two bacterial populations would be important to cure persistent infections, according to the Yin-Yang model.24,477 The clinical experience with the persister drug PZA to kill persisters and shorten the treatment of tuberculosis without relapse, indicates that using a combination of drugs that not only kill growing bacteria but more importantly kill the persisters would more effectively cure persistent infections.24 Additionally, the anti-persister drug daptomycin486 and colistin,49,487 when used in combination with other conventional antibiotics, has demonstrated effective eradication of the aforementioned persistent infections,49,56,480 further underscoring the success of the persister drug combination approach.24 Moreover, in combination with conventional antibiotic rifampin, ADEP4 was effective against a deep-seated murine S. aureus biofilm infection,177 although there is still debate over whether it could completely clear persistent infections.481

Proposed drug combination strategies for treating persistent infections. (1) Combination of drugs killing both metabolically active bacteria and persister cells per Yin-Yang model 24. (2) Compounds sensitizing persisters or inhibiting persister formation combined with traditional antibiotics. (3) Utilizing mechanistically diverse drugs to potentially eliminate the persister population. (4) Combining antibacterial agents and immunomodulators to modulate host immunity. Created with BioRender.com
Secondly, combination of compounds that sensitize persisters or inhibit persister formation with traditional antibiotics488 (Fig. 7). This approach takes into account the complex interactions between antibiotics and persister cells, aiming to enhance the effectiveness of treatment by traditional antibiotics. Currently, there are several compounds that sensitize persister cells, such as cis-2-decenoic acid,455 carbon metabolites,248,459,460 amino acids,242,283,464,465 metal ions,466,489 and others. However, their effectiveness in clearing persistent infections still needs further validation in vivo.
Additionally, the combination of mechanistically different drugs also has the potential to eradicate the persister population (Fig. 7). When bacteria are exposed to stressful environments, they employ various strategies to ensure their survival, such as forming metabolically slow or dormant persister cells. Through the elucidation of the bacterial persister mechanisms mentioned above, it becomes evident that persister formation is a result of multiple factors working in concert, rather than being caused by a single factor. Therefore, it is speculated that the use of anti-persister drugs targeting a single target may not fully control persister cells.
Finally, combination of antibacterial agents and immunomodulators targeting the host immunity (Fig. 7). The formation of persisters is influenced not only by environmental stresses such as antibiotic, temperature, pH, and nutrients in vitro, but also influenced by host factors in vivo.490,491 For example, it has been found that the internalization of Salmonella typhimurium by macrophages triggers the formation of non-replicating persister cells.491,492 The observations with S. aureus493,494 and uropathogenic E. coli persisters495,496 in host cells further illustrate the impact of host factors. These findings suggest that modulating host immunity could be effective in combating persistent infections. This idea was confirmed by the finding that inactivated vaccine, such as the Salmonella cells killed by peracetic acid, reduces the formation of reservoirs of persisters after oral infection with S. typhimurium.172 Moreover, DNA vaccine497 and cytokine granulocyte macrophage colony-stimulating factor (GM-CSF)498 also showed activities against persistent infections. Under these conditions, the use of immunotherapy alongside traditional chemotherapeutic antibacterial drugs could potentially lead to a quicker or more definitive resolution of persistent infections in humans.
Conclusion and future perspective
In this review, we comprehensively summarized the historical background of bacterial persisters, including its discovery in vitro, validation through clinical isolation and in animal models. We detailed the complex characteristics of persister bacteria and argue their relationship with tolerant and resistant bacteria. Additionally, we systematically addressed the interplay between various bacterial biological processes and the formation of persister cells. Moreover, we consolidate the diverse anti-persister compounds and therapies documented in the literature to date. We conclude that the widespread recognition of the clinical significance of persistent infections has driven research on persister cells, resulting in a deeper understanding of their mechanisms and potential treatment strategies. Nevertheless, there remain numerous unanswered questions regarding the molecular mechanism and treatment strategy of persisters that necessitate further exploration and discussion.
For example, while in vitro screening of genes associated with persisters is primarily carried out under the action of antibiotics, there are many other factors (such as oxygen, pH, nutrient starvation and other environmental factors such as those encountered in the host) that play a role in the formation of persisters but have not been well studied. Moreover, the relative importance and interaction of different persistence genes and pathways under different conditions are very complex, and many details in regulating the formation and survival of persisters are still unclear189,190 and need further study. The molecular basis for the heterogeneous nature of persisters from shallow to deep persistence to VBNC in the continuing spectrum of persisters also still remains to be investigated.24 Additionally, different host-specific niches can promote persistence in the context of infection, but our understanding of this mechanism is far from complete. Further elucidation of these unclear mechanisms of bacterial persistence should help to provide new targets for development of new anti-persister drugs. Upon reviewing the mechanisms of persistence, it becomes evident that persister bacteria do not arise from a single factor or gene regulation. Therefore, it is imperative to identify key synthetic lethal gene combinations that impact persister formation or survival as potential drug targets for future intervention studies.
Regarding drug development for persisters, there still remain significant challenges and a long road ahead. For instance, although researchers have previously developed compounds or drug candidates that directly target and kill persisters, such as ADEP4, providing hope for the treatment of persistent bacteria, the experimental treatments using ADEP4 + rifampin have failed to effectively clear persistent infections.481 Perhaps the most effective strategy is to employ persister drug combinations per the Ying-Yang model, such as drugs targeting growing cells + drugs targeting persister cells, or drugs sensitizing persister cells or preventing persister formation + traditional antibiotics. Furthermore, it has been suggested that during long-term persistent infections, bacterial persisters emerge and adapt within a complex host environment,490 indicating potential involvement of the immune system in this process. Therefore, modulating the host immune system and host environment could hold importance for more efficacious treatment of chronic persistent infections. For instance, therapeutic vaccines could be developed based on antigenic key proteins of persisters. Additionally, Traditional Chinese Medicines or extracts with immunomodulatory effects could be employed to regulate the host immune microenvironment, enhancing the immune response against persistent bacteria.
From the mechanism in Table 1 to the drugs in Table 2, we found that the drug mechanisms of killing persisters mainly focus on altering metabolism, disrupting membranes, and DNA cross-linking, while the drug mechanisms of sensitizing persisters to conventional antibiotics or inhibiting persister formation primarily concentrate on stress response and cell communication. There are still many persister mechanisms that have not been utilized for drug development, indicating significant potential and opportunities for the development of anti-persister drugs. It is encouraging that the persister drug combination approach, developed per the Yin-Yang model based on the persister drug PZA, has produced promising results to more effectively kill persisters and biofilm bacteria not only in vitro but also cure the persistent infections in animal models in vivo.56,480,481 Further studies are warranted to validate this approach for more effective treatment of persistent infections in clinical studies.
References
Cook, M. A. & Wright, G. D. The past, present, and future of antibiotics. Sci. Transl. Med. 14, eabo7793 (2022).
Hutchings, M. I., Truman, A. W. & Wilkinson, B. Antibiotics: past, present and future. Curr. Opin. Microbiol. 51, 72–80 (2019).
Khardori, N. Antibiotics–past, present, and future. Med. Clin. North Am. 90, 1049–1076 (2006).
Ferrara, F. et al. Antibacterial agents and the fight against antibiotic resistance: a real-world evidence analysis of consumption and spending by an Italian healthcare company. Ann. Pharm. Fr. 82, 545–552 (2024).
Klingeberg, A. et al. The percentage of antibiotic resistance in uncomplicated community-acquired urinary tract infections-findings of the RedAres project. Dtsch Arztebl Int 121, 175–181 (2024).
Zhou, Y., Zhang, Y. & Du, S. Antibiotic resistance in Helicobacter pylori among children and adolescents in East Asia: a systematic review and meta-analysis. Chin. Med. J. (ahead of print) 10–1097 (2024).
Geyi, D. et al. Salmonella enterica serovars linked with poultry in India: antibiotic resistance profiles and carriage of virulence genes. Braz. J. Microbiol. 55, 969–979 (2024).
Aminov, R. I. A brief history of the antibiotic era: lessons learned and challenges for the future. Front. Microbiol. 1, 134 (2010).
Livermore, D. M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64, i29–i36 (2009).
Painuli, S., Semwal, P., Sharma, R. & Akash, S. Superbugs or multidrug resistant microbes: a new threat to the society. Health Sci. Rep. 6, e1480 (2023).
Chahine, E. B., Dougherty, J. A., Thornby, K. A. & Guirguis, E. H. Antibiotic approvals in the last decade: are we keeping up with resistance? Ann. Pharmacother. 56, 441–462 (2022).
Balaban, N. Q., Gerdes, K., Lewis, K. & McKinney, J. D. A problem of persistence: still more questions than answers? Nat. Rev. Microbiol. 11, 587–591, (2013).
Levin, B. R. & Rozen, D. E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 4, 556–562, (2006).
Fisher, R. A., Gollan, B. & Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15, 453–464 (2017).
Gollan, B., Grabe, G., Michaux, C. & Helaine, S. Bacterial persisters and infection: past, present, and progressing. Annu. Rev. Microbiol. 73, 359–385 (2019).
Levin, B. R., Concepcion-Acevedo, J. & Udekwu, K. I. Persistence: a copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr. Opin. Microbiol. 21, 18–21 (2014).
Zhang, Y., Yew, W. W. & Barer, M. R. Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 56, 2223–2230 (2012).
Gunn, J. S. et al. Salmonella chronic carriage: epidemiology, diagnosis, and gallbladder persistence. Trends Microbiol. 22, 648–655 (2014).
Stricker, R. B. & Johnson, L. The pain of chronic Lyme disease: moving the discourse backward? FASEB J. 25, 4085–4087 (2011).
Durrani, B., Mohammad, A., Ljubetic, B. M. & Dobberfuhl, A. D. The potential role of persister cells in urinary tract infections. Curr. Urol. Rep. 24, 541–551 (2023).
Fauvart, M., De Groote, V. N. & Michiels, J. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 60, 699–709 (2011).
Lewis, K. Persister cells. Annu. Rev. Microbiol. 64, 357–372 (2010).
Rhen, M. et al. The basis of persistent bacterial infections. Trends Microbiol. 11, 80–86 (2003).
Zhang, Y. Persisters, persistent infections and the Yin-Yang model. Emerg. Microbes Infect. 3, e3 (2014).
Balaban, N. Q. et al. Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004).
Shah, D. et al. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6, 53 (2006).
Maisonneuve, E. & Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 157, 539–548, (2014).
Harms, A., Maisonneuve, E. & Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 354, aaf4268, (2016).
Shi, W. et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333, 1630–1632 (2011).
Zhang, Y. et al. Identification of novel efflux proteins Rv0191, Rv3756c, Rv3008, and Rv1667c involved in pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 61, 10–1128, (2017).
Shi, W. et al. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 3, e58 (2014).
Liu, J. et al. Mutations in efflux pump Rv1258c (Tap) cause resistance to pyrazinamide, isoniazid, and streptomycin in M. tuberculosis. Front. Microbiol. 10, 216 (2019).
Shi, W. & Zhang, Y. PhoY2 but not PhoY1 is the PhoU homologue involved in persisters in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 65, 1237–1242 (2010).
Shi, W. et al. Introducing RpsA point mutations Delta438A and D123A into the chromosome of Mycobacterium tuberculosis confirms their role in causing resistance to pyrazinamide. Antimicrob. Agents Chemother. 63, 10–1128 (2019).
Shi, W. et al. Identification of novel mutations in LprG (rv1411c), rv0521, rv3630, rv0010c, ppsC, and cyp128 associated with pyrazinoic acid/pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 62, 10−1128, (2018).
Li, J. et al. Trans-translation mediates tolerance to multiple antibiotics and stresses in Escherichia coli. J. Antimicrob. Chemother. 68, 2477–2481 (2013).
Li, Y. & Zhang, Y. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51, 2092–2099 (2007).
Wu, N. et al. Polynucleotide phosphorylase mediates a new mechanism of persister formation in escherichia coli. Microbiol. Spectr. 11, e0154622 (2023).
Liu, S. et al. Variable persister gene interactions with (p)ppGpp for persister formation in escherichia coli. Front. Microbiol. 8, 1795 (2017).
Zhang, S. et al. Small non-coding RNA RyhB mediates persistence to multiple antibiotics and stresses in uropathogenic escherichia coli by reducing cellular metabolism. Front. Microbiol. 9, 136 (2018).
Wu, N. et al. Ranking of persister genes in the same Escherichia coli genetic background demonstrates varying importance of individual persister genes in tolerance to different antibiotics. Front. Microbiol. 6, 1003 (2015).
Cui, P. et al. Identification of genes involved in bacteriostatic antibiotic-induced persister formation. Front. Microbiol. 9, 413 (2018).
Han, J. et al. Conditions and mutations affecting Staphylococcus aureus L-form formation. Microbiology 161, 57–66 (2015).
Wang, W. et al. Transposon mutagenesis identifies novel genes associated with staphylococcus aureus persister formation. Front. Microbiol. 6, 1437 (2015).
Xu, T. et al. Absence of protoheme IX farnesyltransferase CtaB causes virulence attenuation but enhances pigment production and persister survival in MRSA. Front. Microbiol. 7, 1625 (2016).
Yee, R. et al. Identification of a novel gene argJ involved in arginine biosynthesis critical for persister formation in Staphylococcus aureus. Discov. Med. 29, 65–77 (2020).
Oliver, J. D. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol. Rev. 34, 415–425 (2010).
Feng, J., Shi, W., Zhang, S. & Zhang, Y. Persister mechanisms in Borrelia burgdorferi: implications for improved intervention. Emerg. Microbes Infect. 4, e51 (2015).
Cui, P. et al. Disruption of membrane by Colistin Kills uropathogenic escherichia coli persisters and enhances killing of other antibiotics. Antimicrob. Agents Chemother. 60, 6867–6871 (2016).
Wade, M. M. & Zhang, Y. Effects of weak acids, UV and proton motive force inhibitors on pyrazinamide activity against Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 58, 936–941, (2006).
Niu, H. et al. Identification of anti-persister activity against uropathogenic escherichia coli from a clinical drug library. Antibiotics 4, 179–187 (2015).
Niu, H. et al. A clinical drug library screen identifies tosufloxacin as being highly active against staphylococcus aureus persisters. Antibiotics 4, 329–336 (2015).
Feng, J., Leone, J., Schweig, S. & Zhang, Y. Evaluation of natural and botanical medicines for activity against growing and non-growing forms of B. burgdorferi. Front. Med. 7, 6 (2020).
Feng, J. et al. Identification of novel activity against Borrelia burgdorferi persisters using an FDA approved drug library. Emerg. Microbes Infect. 3, e49 (2014).
Feng, J. et al. A drug combination screen identifies drugs active against amoxicillin-induced round bodies of in vitro borrelia burgdorferi persisters from an FDA drug library. Front. Microbiol. 7, 743 (2016).
Yee, R. et al. Eradication of staphylococcus aureus biofilm infection by persister drug combination. Antibiotics. 11, 1278, (2022).
Feng, J., Zhang, S., Shi, W. & Zhang, Y. Ceftriaxone pulse dosing fails to eradicate biofilm-like Microcolony B. burgdorferi persisters which are sterilized by daptomycin/ doxycycline/cefuroxime without pulse dosing. Front. Microbiol. 7, 1744 (2016).
Feng, J., Zhang, S., Shi, W. L. & Zhang, Y. Activity of sulfa drugs and their combinations against stationary phase in vitro. Antibiotics 6, 10 (2017).
Feng, J., Auwaerter, P. G. & Zhang, Y. Drug combinations against Borrelia burgdorferi persisters in vitro: eradication achieved by using daptomycin, cefoperazone and doxycycline. PLoS One 10, e0117207 (2015).
Feng, J. et al. Eradication of biofilm-like microcolony structures of borrelia burgdorferi by daunomycin and daptomycin but not Mitomycin C in combination with doxycycline and cefuroxime. Front. Microbiol. 7, 62 (2016).
HOBBY, G. L., MEYER, K. & CHAFFEE, E. Observations on the mechanism of action of penicillin. Exp. Biol. Med. 50, 5 (1942).
Bigger, J. W. Treatment of staphylococcal infections with penicillin. Lancet 244, 497–500 (1944).
Tuomanen, E., Durack, D. T. & Tomasz, A. Antibiotic tolerance among clinical isolates of bacteria. Antimicrob. Agents Chemother. 30, 7 (1986).
Tomasz, A., Albino, A. & Zanati, E. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227, 138–140 (1970).
Best, G. K., Best, N. H. & Koval, A. V. Evidence for participation of autolysins in bactericidal action of oxacillin on Staphylococcus aureus. Antimicrob. Agents Chemother. 6, 825–830, (1974).
Mayhall, C. G., Medoff, G. & Marr, J. J. Variation in the susceptibility of strains of Staphylococcus aureus to oxacillin, cephalothin, and gentamicin. Antimicrob. Agents Chemother. 10, 707–712, (1976).
Horne, D. & Tomasz, A. Tolerant response of Streptococcus sanguis to beta-lactams and other cell wall inhibitors. Antimicrob. Agents Chemother. 11, 888–896, (1977).
Sabath, L. D. et al. A new type of penicillin resistance of Staphylococcus aureus. Lancet 1, 443–447 (1977).
Traub, W. H. Variable tolerance of a clinical isolate of Staphylococcus epidermidis from an infected hydrocephalus shunt for several beta-lactam antibiotics, vancomycin and fosfomycin. Chemotherapy 27, 432–443 (1981).
Holloway, Y., Dankert, J. & Hess, J. Penicillin tolerance and bacterial endocarditis. Lancet 1, 589 (1980).
Glauser, M. P., Bernard, J. P., Moreillon, P. & Francioli, P. Successful single-dose amoxicillin prophylaxis against experimental streptococcal endocarditis: evidence for two mechanisms of protection. J. Infect. Dis. 147, 568–575, (1983).
Holloway, Y. & Dankert, J. Penicillin tolerance in nutritionally variant streptococci. Antimicrob. Agents Chemother. 22, 1073–1075, (1982).
Krogstad, D. J. & Pargwette, A. R. Defective killing of enterococci: a common property of antimicrobial agents acting on the cell wall. Antimicrob. Agents Chemother. 17, 965–968, (1980).
Kaye, D. Enterococci. Biologic and epidemiologic characteristics and in vitro susceptibility. Arch. Intern Med. 142, 2006–2009 (1982).
Savitch, C. B., Barry, A. L. & Hoeprich, P. D. Infective endocarditis caused by Streptococcus bovis resistant to the lethal effect of penicillin G. Arch. Intern Med. 138, 931–934 (1978).
Moellering, R. C. Jr et al. Antibiotic synergism against Listeria monocytogenes. Antimicrob. Agents Chemother. 1, 30–34 (1972).
Gordon, R. C., Barrett, F. F. & Clark, D. J. Influence of several antibiotics, singly and in combination, on the growth of Listeria monocytogenes. J. Pediatr. 80, 667–670, (1972).
Brennan, R. O. & Durack, D. T. Therapeutic significance of penicillin tolerance in experimental streptococcal endocarditis. Antimicrob. Agents Chemother. 23, 273–277, (1983).
Moyed, H. S. & Bertrand, K. P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155, 768–775, (1983).
Davidson, D. J., Spratt, D. & Liddle, A. D. Implant materials and prosthetic joint infection: the battle with the biofilm. EFORT Open Rev. 4, 633–639 (2019).
Allon, M. Saving infected catheters: why and how. Blood Purif. 23, 23–28, (2005).
Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999).
Spoering, A. L. & Lewis, K. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183, 6746–6751 (2001).
Hoiby, N. et al. ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clin. Microbiol. Infect. 21, S1–S25 (2015).
Balaban, N. Q. et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448 (2019).
Yan, J. & Bassler, B. L. Surviving as a community: antibiotic tolerance and persistence in bacterial biofilms. Cell Host Microbe 26, 15–21 (2019).
Wilmaerts, D., Windels, E. M., Verstraeten, N. & Michiels, J. General mechanisms leading to persister formation and awakening. Trends Genet. 35, 401–411 (2019).
Wood, T. K. Combatting bacterial persister cells. Biotechnol. Bioeng. 113, 476–483 (2016).
Huemer, M., Mairpady Shambat, S., Brugger, S. D. & Zinkernagel, A. S. Antibiotic resistance and persistence-implications for human health and treatment perspectives. EMBO Rep. 21, e51034 (2020).
Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry 70, 267–274 (2005).
Zhang, Y. & Mitchison, D. The curious characteristics of pyrazinamide: a review. Int. J. Tuberc. Lung Dis. 7, 6–21 (2003).
Ma, C. et al. Energy production genes sucB and ubiF are involved in persister survival and tolerance to multiple antibiotics and stresses in Escherichia coli. FEMS Microbiol. Lett. 303, 33–40 (2010).
Pu, Y. et al. ATP-dependent dynamic protein aggregation regulates bacterial dormancy depth critical for antibiotic tolerance. Mol. Cell 73, 143–156.e144 (2019).
Xu, H. S. et al. Survival and viability of nonculturableEscherichia coli andVibrio cholerae in the estuarine and marine environment. Micro. Ecol. 8, 313–323 (1982).
Debnath, A. & Miyoshi, S. I. The impact of protease during recovery from viable but non-culturable (VBNC) state in vibrio cholerae. Microorganisms. 9, 2628, (2021).
Ayibieke, A., Nishiyama, A., Senoh, M. & Hamabata, T. Gene expression analysis during the conversion from a viable but nonculturable to culturable state in Vibrio cholerae. Gene 863, 147289 (2023).
Cervero-Aragó, S. et al. Viability and infectivity of viable but nonculturable Legionella pneumophila strains induced at high temperatures. Water Res. 158, 268–279 (2019).
Ducret, A., Chabalier, M. & Dukan, S. Characterization and resuscitation of ‘non-culturable’ cells of Legionella pneumophila. BMC Microbiol. 14, 3 (2014).
Dietersdorfer, E. et al. Starved viable but non-culturable (VBNC) Legionella strains can infect and replicate in amoebae and human macrophages. Water Res. 141, 428–438 (2018).
Whiley, H., Bentham, R. & Brown, M. H. Legionella persistence in manufactured water systems: pasteurization potentially selecting for thermal tolerance. Front. Microbiol. 8, 1330 (2017).
Al-Bana, B. H., Haddad, M. T. & Garduno, R. A. Stationary phase and mature infectious forms of Legionella pneumophila produce distinct viable but non-culturable cells. Environ. Microbiol. 16, 382–395, (2014).
Epalle, T. et al. Viable but not culturable forms of Legionella pneumophila generated after heat shock treatment are infectious for macrophage-like and alveolar epithelial cells after resuscitation on Acanthamoeba polyphaga. Micro. Ecol. 69, 215–224 (2015).
Sun, Z. & Zhang, Y. Spent culture supernatant of Mycobacterium tuberculosis H37Ra improves viability of aged cultures of this strain and allows small inocula to initiate growth. J. Bacteriol. 181, 7626–7628 (1999).
Pai, S. R. et al. Identification of viable and non-viable Mycobacterium tuberculosis in mouse organs by directed RT-PCR for antigen 85B mRNA. Micro. Pathog. 28, 335–342 (2000).
Ayrapetyan, M., Williams, T. & Oliver, J. D. Relationship between the viable but nonculturable state and antibiotic persister cells. J. Bacteriol. 200, 10–1128 (2018).
Senoh, M. et al. Conversion of viable but nonculturable Vibrio cholerae to the culturable state by co-culture with eukaryotic cells. Microbiol. Immunol. 54, 502–507 (2010).
Kim, J. S., Chowdhury, N., Yamasaki, R. & Wood, T. K. Viable but non-culturable and persistence describe the same bacterial stress state. Environ. Microbiol. 20, 2038–2048 (2018).
Mishra, A., Taneja, N. & Sharma, M. Demonstration of viable but nonculturable Vibrio cholerae O1 in fresh water environment of India using ciprofloxacin DFA-DVC method. Lett. Appl. Microbiol. 53, 124–126 (2011).
Kahl, B. C., Becker, K. & Loffler, B. Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clin. Microbiol. Rev. 29, 401–427 (2016).
Park, H. E. et al. Bigger problems from smaller colonies: emergence of antibiotic-tolerant small colony variants of Mycobacterium avium complex in MAC-pulmonary disease patients. Ann. Clin. Microbiol. Antimicrob. 23, 25 (2024).
Zhou, S. et al. Staphylococcus aureus small-colony variants: formation, infection, and treatment. Microbiol. Res. 260, 127040 (2022).
Liu, S. et al. Unravelling staphylococcal small-colony variants in cardiac implantable electronic device infections: clinical characteristics, management, and genomic insights. Front. Cell Infect. Microbiol. 13, 1321626 (2023).
Tsujino, Y., Ogawa, E. & Ito, K. Thymidine-dependent small-colony variants of Staphylococcus aureus isolated from infective endocarditis in a postlung transplant patient. Transpl. Infect. Dis. 26, e14176 (2024).
Lee, J. et al. The bacteriology of diabetic foot ulcers and infections and incidence of staphylococcus aureus small colony variants. J. Med. Microbiol. 72, 001716 (2023).
Gil-Gil, T. et al. The evolution of heteroresistance via small colony variants in escherichia coli following long term exposure to bacteriostatic antibiotics. bioRxiv, (2023).
Noaman, K. A. et al. The transmutation of Escherichia coli ATCC 25922 to small colony variants (SCVs) E. coli strain as a result of exposure to gentamicin. J. Infect. Public Health 16, 1821–1829 (2023).
Proctor, R. A. et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305 (2006).
Loss, G. et al. Staphylococcus aureus small colony variants (SCVs): news from a chronic prosthetic joint infection. Front. Cell Infect. Microbiol. 9, 363 (2019).
Vulin, C. et al. Prolonged bacterial lag time results in small colony variants that represent a sub-population of persisters. Nat. Commun. 9, 4074 (2018).
Leimer, N. et al. Nonstable staphylococcus aureus small-colony variants are induced by low pH and sensitized to antimicrobial therapy by phagolysosomal alkalinization. J. Infect. Dis. 213, 305–313 (2016).
Painter, K. L. et al. Staphylococcus aureus adapts to oxidative stress by producing H2O2-resistant small-colony variants via the SOS response. Infect. Immun. 83, 1830–1844 (2015).
Li, Y. et al. Survival of bactericidal antibiotic treatment by tolerant persister cells of Klebsiella pneumoniae. J. Med. Microbiol. 67, 273–281 (2018).
Keren, I. et al. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18 (2004).
Keren, I., Minami, S., Rubin, E. & Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2, e00100–e00111 (2011).
Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56 (2007).
Fung, D. K., Chan, E. W., Chin, M. L. & Chan, R. C. Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother. 54, 1082–1093, (2010).
Shatalin, K., Shatalina, E., Mironov, A. & Nudler, E. H2S: a universal defense against antibiotics in bacteria. Science 334, 986–990, (2011).
Gusarov, I., Shatalin, K., Starodubtseva, M. & Nudler, E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325, 1380–1384, (2009).
Nierman, W. C., Yu, Y. & Losada, L. The in vitro antibiotic tolerant persister population in Burkholderia pseudomallei is altered by environmental factors. Front. Microbiol. 6, 1338 (2015).
Kawai, Y. et al. AldB controls persister formation in Escherichia coli depending on environmental stress. Microbiol. Immunol. 62, 299–309 (2018).
Kaplan, Y. et al. Observation of universal ageing dynamics in antibiotic persistence. Nature 600, 290–294 (2021).
Patel, H., Buchad, H. & Gajjar, D. Pseudomonas aeruginosa persister cell formation upon antibiotic exposure in planktonic and biofilm state. Sci. Rep. 12, 16151 (2022).
Manandhar, S. et al. High level of persister frequency in clinical staphylococcal isolates. BMC Microbiol. 22, 109 (2022).
Drlica, K., Malik, M., Kerns, R. J. & Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52, 385–392, (2008).
Keren, I. et al. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 (2004).
Wiegand, I., Hilpert, K. & Hancock, R. E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175, (2008).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330, (2016).
Kester, J. C. & Fortune, S. M. Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 49, 91–101 (2014).
Meylan, S., Andrews, I. W. & Collins, J. J. Targeting antibiotic tolerance, pathogen by pathogen. Cell 172, 1228–1238 (2018).
Ronneau, S., Hill, P. W. & Helaine, S. Antibiotic persistence and tolerance: not just one and the same. Curr. Opin. Microbiol. 64, 76–81 (2021).
Weigel, W. A. & Dersch, P. Phenotypic heterogeneity: a bacterial virulence strategy. Microbes Infect. 20, 570–577 (2018).
Zhou, Y., Liao, H., Pei, L. & Pu, Y. Combatting persister cells: the daunting task in post-antibiotics era. Cell Insight 2, 100104 (2023).
Brauner, A., Shoresh, N., Fridman, O. & Balaban, N. Q. An experimental framework for quantifying bacterial tolerance. Biophys. J. 112, 2664–2671 (2017).
Vijay, S. et al. Most-probable-number-based minimum duration of killing assay for determining the spectrum of rifampicin susceptibility in clinical Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 65, 10−1128 (2021).
Levin-Reisman, I. et al. Automated imaging with ScanLag reveals previously undetectable bacterial growth phenotypes. Nat. Methods 7, 737–739 (2010).
Levin-Reisman, I., Fridman, O. & Balaban, N. Q. ScanLag: high-throughput quantification of colony growth and lag time. J. Vis. Exp. 89, e51456 (2014).
Bar, J. et al. Efficient microbial colony growth dynamics quantification with ColTapp, an automated image analysis application. Sci. Rep. 10, 16084 (2020).
Gefen, O., Chekol, B., Strahilevitz, J. & Balaban, N. Q. TDtest: easy detection of bacterial tolerance and persistence in clinical isolates by a modified disk-diffusion assay. Sci. Rep. 7, 41284 (2017).
Matsuo, M. et al. Genetic and transcriptomic analyses of ciprofloxacin-tolerant staphylococcus aureus isolated by the replica plating tolerance isolation system (REPTIS). Antimicrob Agents Chemother. 63, e02019-18 (2019).
Wideman, N. E., Oliver, J. D., Crandall, P. G. & Jarvis, N. A. Detection and potential virulence of viable but non-culturable (VBNC) listeria monocytogenes: a review. Microorganisms. 9, 194 (2021).
Casasola-Rodriguez, B. et al. Detection of VBNC vibrio cholerae by RT-real time PCR based on differential gene expression analysis. FEMS Microbiol. Lett. 365, (2018).
Yaron, S. & Matthews, K. R. A reverse transcriptase-polymerase chain reaction assay for detection of viable Escherichia coli O157:H7: investigation of specific target genes. J. Appl. Microbiol. 92, 633–640 (2002).
Coutard, F., Pommepuy, M., Loaec, S. & Hervio-Heath, D. mRNA detection by reverse transcription-PCR for monitoring viability and potential virulence in a pathogenic strain of Vibrio parahaemolyticus in viable but nonculturable state. J. Appl. Microbiol. 98, 951–961 (2005).
Zhao, S. et al. Enumeration of viable non-culturable vibrio cholerae using droplet digital PCR combined with propidium monoazide treatment. Front. Cell Infect. Microbiol. 11, 753078 (2021).
Zhang, Y. Persistent and dormant tubercle bacilli and latent tuberculosis. Front. Biosci. 9, 1136–1156 (2004).
Mulcahy, L. R., Burns, J. L., Lory, S. & Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192, 6191–6199, (2010).
Dengler Haunreiter, V. et al. In-host evolution of Staphylococcus epidermidis in a pacemaker-associated endocarditis resulting in increased antibiotic tolerance. Nat. Commun. 10, 1149 (2019).
Jansen, B. & Peters, G. Foreign body associated infection. J. Antimicrob. Chemother. 32, 69–75 (1993).
Windels, E. M. et al. Antibiotics: combatting tolerance to stop resistance. mBio. 10, e02095-19 (2019).
Cohen, N. R., Lobritz, M. A. & Collins, J. J. Microbial persistence and the road to drug resistance. Cell Host Microbe 13, 632–642, (2013).
Wakamoto, Y. et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013).
Mwangi, M. M. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl Acad. Sci. USA 104, 9451–9456 (2007).
Lieberman, T. D. et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat. Genet. 43, 1275–1280 (2011).
Hayden, H. S. et al. Evolution of Burkholderia pseudomallei in recurrent melioidosis. PLoS One 7, e36507 (2012).
Levin-Reisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Nguyen, D. et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 (2011).
Al Mamun, A. A. et al. Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344–1348 (2012).
Cirz, R. T. et al. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J. Bacteriol. 189, 531–539 (2007).
Cirz, R. T. et al. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J. Bacteriol. 188, 7101–7110 (2006).
Beaber, J. W., Hochhut, B. & Waldor, M. K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427, 72–74, (2004).
Stecher, B. et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc. Natl Acad. Sci. USA 109, 1269–1274 (2012).
Bakkeren, E. et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 573, 276–280 (2019).
Kamble, E. & Pardesi, K. Antibiotic tolerance in biofilm and stationary-phase planktonic cells of staphylococcus aureus. Micro. Drug Resist. 27, 3–12 (2021).
Stewart, P. S. et al. Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 59, 3838–3847 (2015).
Bernier, S. P. et al. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 9, e1003144 (2013).
Conlon, B. P., Rowe, S. E. & Lewis, K. Persister cells in biofilm associated infections. Adv. Exp. Med. Biol. 831, 1–9 (2015).
Conlon, B. P. et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503, 365–370 (2013).
Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 45, 999–1007 (2001).
Hu, Y. & Coates, A. R. Transposon mutagenesis identifies genes which control antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiol. Lett. 243, 117–124, (2005).
Molina-Quiroz, R. C., Lazinski, D. W., Camilli, A. & Levy, S. B. Transposon-sequencing analysis unveils novel genes involved in the generation of persister cells in uropathogenic escherichia coli. Antimicrob. Agents Chemother. 60, 6907–6910 (2016).
Hansen, S., Lewis, K. & Vulic, M. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52, 2718–2726, (2008).
Girgis, H. S., Harris, K. & Tavazoie, S. Large mutational target size for rapid emergence of bacterial persistence. Proc. Natl Acad. Sci. USA 109, 12740–12745, (2012).
Cameron, D. R. et al. A genetic determinant of persister cell formation in bacterial pathogens. J. Bacteriol. 200, (2018).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006–0008, (2006).
Nicolau, S. E. & Lewis, K. The role of integration host factor in escherichia coli persister formation. mBio 13, e0342021 (2022).
Shan, Y. et al. ATP-dependent persister formation in Escherichia coli. mBio. 8, e02267-16 (2017).
Chung, M. et al. Phenotypic signatures and genetic determinants of oxacillin tolerance in a laboratory mutant of Staphylococcus aureus. PLoS One 13, e0199707 (2018).
Van den Bergh, B. et al. Mutations in respiratory complex I promote antibiotic persistence through alterations in intracellular acidity and protein synthesis. Nat. Commun. 13, 546 (2022).
Van den Bergh, B. et al. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 1, 16020 (2016).
Glover, W. A., Yang, Y. & Zhang, Y. Insights into the molecular basis of L-form formation and survival in Escherichia coli. PLoS One 4, e7316 (2009).
Ayrapetyan, M., Williams, T. C., Baxter, R. & Oliver, J. D. Viable but nonculturable and persister cells coexist stochastically and are induced by human serum. Infect. Immun. 83, 4194–4203, (2015).
Bollen, C., Dewachter, L. & Michiels, J. Protein aggregation as a bacterial strategy to survive antibiotic treatment. Front. Mol. Biosci. 8, 669664 (2021).
Hayes, F. & Van Melderen, L. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 46, 386–408 (2011).
Unterholzner, S. J., Poppenberger, B. & Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 3, e26219 (2013).
Kedzierska, B. & Hayes, F. Emerging roles of toxin-antitoxin modules in bacterial pathogenesis. Molecules. 21, 790 (2016).
Harms, A., Brodersen, D. E., Mitarai, N. & Gerdes, K. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784 (2018).
Page, R. & Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214, (2016).
Singh, G., Yadav, M., Ghosh, C. & Rathore, J. S. Bacterial toxin-antitoxin modules: classification, functions, and association with persistence. Curr. Res. Micro. Sci. 2, 100047 (2021).
Gerdes, K. & Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66, 103–123, (2012).
Ronneau, S. & Helaine, S. Clarifying the link between toxin-antitoxin modules and bacterial persistence. J. Mol. Biol. 431, 3462–3471 (2019).
Germain, E., Castro-Roa, D., Zenkin, N. & Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 52, 248–254 (2013).
Germain, E., Roghanian, M., Gerdes, K. & Maisonneuve, E. Stochastic induction of persister cells by HipA through (p)ppGpp-mediated activation of mRNA endonucleases. Proc. Natl Acad. Sci. USA 112, 5171–5176 (2015).
Germain, E., Roghanian, M., Gerdes, K. & Maisonneuve, E. Stochastic induction of persister cells by HipA through (p) ppGpp-mediated activation of mRNA endonucleases (Retraction of Vol 112, Pg 5171, 2015). Proc. Natl Acad. Sci. USA 116, 11077–11077 (2019).
Huang, C. Y. et al. hipBA toxin-antitoxin systems mediate persistence in Caulobacter crescentus. Sci. Rep. 10, 2865 (2020).
Semanjski, M. et al. The kinases HipA and HipA7 phosphorylate different substrate pools in Escherichia coli to promote multidrug tolerance. Sci. Signal. 11, eaat5750 (2018).
Singh, R., Barry, C. E. 3rd & Boshoff, H. I. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J. Bacteriol. 192, 1279–1291 (2010).
Korch, S. B., Malhotra, V., Contreras, H. & Clark-Curtiss, J. E. The Mycobacterium tuberculosis relBE toxin:antitoxin genes are stress-responsive modules that regulate growth through translation inhibition. J. Microbiol. 53, 783–795, (2015).
Tripathi, A., Dewan, P. C., Siddique, S. A. & Varadarajan, R. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 289, 4191–4205, (2014).
Hu, Y. et al. Toxin YafQ increases persister cell formation by reducing indole signalling. Environ. Microbiol. 17, 1275–1285 (2015).
Harrison, J. J. et al. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 53, 2253–2258 (2009).
Sun, C. et al. MqsR/MqsA toxin/antitoxin system regulates persistence and biofilm formation in pseudomonas putida KT2440. Front. Microbiol. 8, 840 (2017).
Kwan, B. W. et al. The MqsR/MqsA toxin/antitoxin system protects Escherichia coli during bile acid stress. Environ. Microbiol. 17, 3168–3181 (2015).
Tripathi, A., Dewan, P. C., Barua, B. & Varadarajan, R. Additional role for the ccd operon of F-plasmid as a transmissible persistence factor. Proc. Natl Acad. Sci. USA 109, 12497–12502 (2012).
Wagner, E. G. & Unoson, C. The toxin-antitoxin system tisB-istR1: expression, regulation, and biological role in persister phenotypes. RNA Biol. 9, 1513–1519 (2012).
Dorr, T., Vulic, M. & Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8, e1000317 (2010).
Su, W. L. et al. TisB protein protects escherichia coli cells suffering massive DNA damage from environmental toxic compounds. mBio 13, e0038522 (2022).
Ma, D. et al. Edwardsiella piscicida YefM-YoeB: a Type II toxin-antitoxin system that is related to antibiotic resistance, biofilm formation, serum survival, and host infection. Front. Microbiol. 12, 646299 (2021).
Cheverton, A. M. et al. A salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell 63, 86–96 (2016).
Fasani, R. A. & Savageau, M. A. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl Acad. Sci. USA 110, E2528–E2537 (2013).
Paul, P., Sahu, B. R. & Suar, M. Plausible role of bacterial toxin-antitoxin system in persister cell formation and elimination. Mol. Oral. Microbiol. 34, 97–107 (2019).
Wiradiputra, M. R. D., Khuntayaporn, P., Thirapanmethee, K. & Chomnawang, M. T. Toxin-antitoxin systems: a key role on persister formation in salmonella enterica serovar typhimurium. Infect. Drug Resist. 15, 5813–5829 (2022).
Curtis, T. D., Takeuchi, I., Gram, L. & Knudsen, G. M. The influence of the toxin/antitoxin mazEF on growth and survival of listeria monocytogenes under stress. Toxins 9, 31 (2017).
Holden, D. W. & Errington, J. Type II toxin-antitoxin systems and persister cells. mBio. 9, e01574-18 (2018).
Goormaghtigh, F. et al. Reassessing the role of Type II toxin-antitoxin systems in formation of Escherichia coli Type II persister cells. mBio. 9, e00640-18 (2018).
Verstraeten, N. et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 59, 9–21 (2015).
Maisonneuve, E., Shakespeare, L. J., Jorgensen, M. G. & Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl Acad. Sci. USA 108, 13206–13211 (2011).
Maisonneuve, E., Castro-Camargo, M. & Gerdes, K. p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150 (2013).
Donegan, N. P., Thompson, E. T., Fu, Z. & Cheung, A. L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 192, 1416–1422 (2010).
Fernández De Henestrosa, A. R. et al. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 35, 1560–1572 (2000).
McKenzie, G. J., Magner, D. B., Lee, P. L. & Rosenberg, S. M. The dinB operon and spontaneous mutation in Escherichia coli. J. Bacteriol. 185, 3972–3977 (2003).
Courcelle, J. et al. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158, 41–64 (2001).
Motiejūnaite, R., Armalyte, J., Markuckas, A. & Suziedeliene, E. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol. Lett. 268, 112–119 (2007).
Winther, K., Tree, J. J., Tollervey, D. & Gerdes, K. VapCs of Mycobacterium tuberculosis cleave RNAs essential for translation. Nucleic Acids Res. 44, 9860–9871 (2016).
Rycroft, J. A. et al. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat. Commun. 9, 1993 (2018).
Yashiro, Y. et al. Molecular basis of glycyl-tRNA(Gly) acetylation by TacT from Salmonella Typhimurium. Cell Rep. 37, 110130 (2021).
Berghoff, B. A., Hoekzema, M., Aulbach, L. & Wagner, E. G. Two regulatory RNA elements affect TisB-dependent depolarization and persister formation. Mol. Microbiol. 103, 1020–1033 (2017).
Vazquez-Laslop, N., Lee, H. & Neyfakh, A. A. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 188, 3494–3497 (2006).
Walter, N. D. et al. Transcriptional adaptation of drug-tolerant mycobacterium tuberculosis during treatment of human tuberculosis. J. Infect. Dis. 212, 990–998 (2015).
Correia, F. F. et al. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 188, 8360–8367 (2006).
Kaspy, I. et al. HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun. 4, 3001 (2013).
Schumacher, M. A. et al. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323, 396–401 (2009).
Shan, Y. et al. Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. mBio. 6, e00078-15 (2015).
Tsilibaris, V., Maenhaut-Michel, G., Mine, N. & Van Melderen, L. What is the benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome? J. Bacteriol. 189, 6101–6108 (2007).
Li, T. et al. Novel inhibitors of toxin HipA reduce multidrug tolerant persisters. ACS Med. Chem. Lett. 7, 449–453 (2016).
Di Pietro, M., Filardo, S., De Santis, F. & Sessa, R. New insights into Chlamydiae persistence: an energy metabolism strategy? Int. J. Immunopathol. Pharm. 26, 525–528 (2013).
Kwon, O., Kotsakis, A. & Meganathan, R. Ubiquinone (coenzyme Q) biosynthesis in Escherichia coli: identification of the ubiF gene. FEMS Microbiol. Lett. 186, 157–161 (2000).
Martini, M. C. et al. ubiF is involved in acid stress tolerance and symbiotic competitiveness in Rhizobium favelukesii LPU83. Braz. J. Microbiol. 53, 1633–1643 (2022).
Allison, K. R., Brynildsen, M. P. & Collins, J. J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473, 216–220 (2011).
Conlon, B. P. et al. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 1, 16051 (2016).
Spoering, A. L., Vulic, M. & Lewis, K. GlpD and PlsB participate in persister cell formation in Escherichia coli. J. Bacteriol. 188, 5136–5144 (2006).
Safi, H. et al. Phase variation in Mycobacterium tuberculosis glpK produces transiently heritable drug tolerance. Proc. Natl Acad. Sci. USA 116, 19665–19674 (2019).
Zhang, S. et al. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2, e34 (2013).
Sun, Q. et al. The molecular basis of pyrazinamide activity on Mycobacterium tuberculosis PanD. Nat. Commun. 11, 339 (2020).
Hicks, N. D. et al. Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol. 3, 1032–1042 (2018).
Keiler, K. C. Biology of trans-translation. Annu Rev. Microbiol. 62, 133–151 (2008).
Kurita, D. et al. Molecular mechanism of trans-translation. Nucleic Acids Symp. Ser. 51, 43–44 (2007).
Shpanchenko, O. V., Bugaeva, E., Golovin, A. V. & Dontsova, O. A. Trans-translation: facts and hypothesis. Mol. Biol. (Mosk.) 44, 563–572 (2010).
Campos-Silva, R. et al. Trans-translation is an appealing target for the development of new antimicrobial compounds. Microorganisms 10, 3 (2021).
Thibonnier, M., Thiberge, J. M. & De Reuse, H. Trans-translation in Helicobacter pylori: essentiality of ribosome rescue and requirement of protein tagging for stress resistance and competence. PLoS One 3, e3810 (2008).
Muto, A. et al. Requirement of transfer-messenger RNA for the growth of Bacillus subtilis under stresses. Genes Cells 5, 627–635 (2000).
Meyer, A. S. & Baker, T. A. Proteolysis in the Escherichia coli heat shock response: a player at many levels. Curr. Opin. Microbiol 14, 194–199 (2011).
Illigmann, A. et al. Contribution of the Clp protease to bacterial survival and mitochondrial homoeostasis. Micro. Physiol. 31, 260–279 (2021).
Ju, Y. et al. Recent advances in Clp protease modulation to address virulence, resistance and persistence of MRSA infection. Drug Discov. Today 26, 2190–2197 (2021).
Frees, D., Savijoki, K., Varmanen, P. & Ingmer, H. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63, 1285–1295 (2007).
Olivares, A. O., Baker, T. A. & Sauer, R. T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 14, 33–44 (2016).
Zhang, S. et al. Mutation in clpC1 encoding an ATP-dependent ATPase involved in protein degradation is associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 6, e8 (2017).
Modlin, S. J. et al. Atypical genetic basis of pyrazinamide resistance in monoresistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 65, e01916-20 (2021).
Springer, M. T. et al. Effect of clpP and clpC deletion on persister cell number in Staphylococcus aureus. J. Med. Microbiol. 65, 848–857 (2016).
Tian, X. L. et al. ClpP is required for proteolytic regulation of type II toxin-antitoxin systems and persister cell formation in Streptococcus mutans. Access Microbiol. 1, e000054 (2019).
Karimaei, S., Aghamir, S. M. K. & Pourmand, M. R. Comparative analysis of genes expression involved in type II toxin-antitoxin system in Staphylococcus aureus following persister cell formation. Mol. Biol. Rep. 51, 324 (2024).
Moreno-Cinos, C. et al. ClpP protease, a promising antimicrobial target. Int. J. Mol. Sci. 20, 2232 (2019).
Li, L. et al. Role of purine biosynthesis in persistent methicillin-resistant staphylococcus aureus infection. J. Infect. Dis. 218, 1367–1377 (2018).
Stepanek, J. J. et al. Purine biosynthesis is the bottleneck in trimethoprim-treated Bacillus subtilis. Proteom. Clin. Appl. 10, 1036–1048 (2016).
Wang, D. et al. SmpB and tmRNA orchestrate purine pathway for the trimethoprim resistance in aeromonas veronii. Front. Cell Infect. Microbiol. 10, 239 (2020).
Li, Y., Wood, T. K., Zhang, W. & Li, C. Purine metabolism regulates Vibrio splendidus persistence associated with protein aggresome formation and intracellular tetracycline efflux. Front. Microbiol. 14, 1127018 (2023).
Tan, C. A. Z. et al. Purine and carbohydrate availability drive Enterococcus faecalis fitness during wound and urinary tract infections. mBio 15, e0238423 (2023).
Peng, Q. et al. PurN is involved in antibiotic tolerance and virulence in staphylococcus aureus. Antibiotics 11, 1702 (2022).
Yee, R. et al. Genetic screen reveals the role of purine metabolism in staphylococcus aureus persistence to Rifampicin. Antibiotics 4, 627–642 (2015).
Kwan, B. W., Valenta, J. A., Benedik, M. J. & Wood, T. K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 57, 1468–1473 (2013).
Le, D. et al. Active efflux leads to heterogeneous dissipation of proton motive force by protonophores in bacteria. mBio 12, e0067621 (2021).
Li, Y., Liang, W. & Li, C. Exogenous adenosine and/or guanosine enhances tetracycline sensitivity of persister cells. Microbiol. Res. 270, 127321 (2023).
Li, X. et al. Metabolomics method in understanding and sensitizing carbapenem-resistant acinetobacter baumannii to meropenem. ACS Infect Dis. 10, 184–195 (2023).
Zhao, X. L. et al. Glutamine promotes antibiotic uptake to kill multidrug-resistant uropathogenic bacteria. Sci. Transl. Med. 13, eabj0716 (2021).
Raivio, T. L. & Silhavy, T. J. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179, 7724–7733 (1997).
Richard, H. & Foster, J. W. Sodium regulates Escherichia coli acid resistance, and influences GadX- and GadW-dependent activation of gadE. Microbiology 153, 3154–3161 (2007).
Niu, H. et al. Glutamate transporters GltS, GltP and GltI are involved in escherichia coli tolerance in vitro and pathogenicity in mouse urinary tract infections. Microorganisms. 11, 1173 (2023).
Yee, R. et al. Identification of genes regulating cell death in staphylococcus aureus. Front. Microbiol 10, 2199 (2019).
Yan, D. et al. Disruption of Fis reduces bacterial persister formation by regulating glutamate metabolism in Salmonella. Micro. Pathog. 152, 104651 (2021).
Goode, O. et al. Persister Escherichia coli cells have a lower intracellular pH than susceptible cells but maintain their pH in response to antibiotic treatment. Mbio. 12, e0090921 (2021).
Biase, D. & Pennacchietti, E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: function, distribution and biomedical implications of the gadBC operon. Mol. Microbiol. 86, 770–86 (2012).
Foster, J. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2, 898–907 (2004).
Biase, D. D., Tramonti, A., Bossa, F. & Visca, P. The response to stationary-phase stress conditions in Escherichia coli: role and regulation of the glutamic acid decarboxylase system. Mol. Microbiol. 32, 1198–1211 (1999).
Welsh, D. T. Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. Fems Microbiol. Rev. 24, 263–290 (2000).
Lindgren, J. K. et al. Arginine deiminase in Staphylococcus epidermidis functions to augment biofilm maturation through pH homeostasis. J. Bacteriol. 196, 2277–2289 (2014).
Wortham, B. W., Patel, C. N. & Oliveira, M. A. Polyamines in bacteria: pleiotropic effects yet specific mechanisms. Adv. Exp. Med Biol. 603, 106–115 (2007).
Steed, P. M. & Wanner, B. L. Use of the rep technique for allele replacement to construct mutants with deletions of the pstSCAB-phoU operon: evidence of a new role for the PhoU protein in the phosphate regulon. J. Bacteriol. 175, 6797–6809 (1993).
Namugenyi, S. B., Aagesen, A. M., Elliott, S. R. & Tischler, A. D. Mycobacterium tuberculosis PhoY proteins promote persister formation by mediating Pst/SenX3-RegX3 phosphate sensing. mBio. 8, e00494-17 (2017).
Shang, Y. et al. Staphylococcus aureus PhoU homologs regulate persister formation and virulence. Front. Microbiol. 11, 865 (2020).
Mok, W. W., Orman, M. A. & Brynildsen, M. P. Impacts of global transcriptional regulators on persister metabolism. Antimicrob. Agents Chemother. 59, 2713–2719 (2015).
Rizvanovic, A. et al. The RNA-binding protein ProQ promotes antibiotic persistence in salmonella. mBio 13, e0289122 (2022).
Baharoglu, Z. & Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 38, 1126–1145 (2014).
Vestergaard, M., Paulander, W. & Ingmer, H. Activation of the SOS response increases the frequency of small colony variants. BMC Res. Notes 8, 749 (2015).
Podlesek, Z. & Žgur Bertok, D. The DNA damage inducible SOS response is a key player in the generation of bacterial persister cells and population wide tolerance. Front. Microbiol. 11, 1785 (2020).
Dörr, T., Lewis, K. & Vulić, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5, e1000760 (2009).
Hengge, R. Stationary-phase gene regulation in Escherichia coli. EcoSal Plus. 4 (2011).
Murakami, K. et al. Role for rpoS gene of Pseudomonas aeruginosa in antibiotic tolerance. FEMS Microbiol. Lett. 242, 161–167 (2005).
Pribis, J. P. et al. Gamblers: an antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol. Cell 74, 785–800.e787 (2019).
Hong, S. H. et al. Bacterial persistence increases as environmental fitness decreases. Micro. Biotechnol. 5, 509–522 (2012).
Wang, X. et al. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7, 359–366 (2011).
Hu, Y., Benedik, M. J. & Wood, T. K. Antitoxin DinJ influences the general stress response through transcript stabilizer CspE. Environ. Microbiol. 14, 669–679 (2012).
Valencia, E. Y., de Moraes Gomes, F., Ospino, K. & Spira, B. RpoS role in antibiotic resistance, tolerance and persistence in E. coli natural isolates. BMC Microbiol. 24, 72 (2024).
Bassler, B. L. & Losick, R. Bacterially speaking. Cell 125, 237–246 (2006).
Papenfort, K. & Bassler, B. L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016).
Castillo-Juarez, I. et al. Role of quorum sensing in bacterial infections. World J. Clin. Cases 3, 575–598 (2015).
Moker, N., Dean, C. R. & Tao, J. Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol. 192, 1946–1955 (2010).
Leung, V. & Levesque, C. M. A stress-inducible quorum-sensing peptide mediates the formation of persister cells with noninherited multidrug tolerance. J. Bacteriol. 194, 2265–2274 (2012).
Vega, N. M., Allison, K. R., Khalil, A. S. & Collins, J. J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 8, 431–433 (2012).
Lee, J. H. & Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 34, 426–444 (2010).
Jubair, M., Morris, J. G. Jr & Ali, A. Survival of Vibrio cholerae in nutrient-poor environments is associated with a novel “persister” phenotype. PLoS One 7, e45187 (2012).
Xu, T. et al. The Agr quorum sensing system represses persister formation through regulation of phenol soluble modulins in staphylococcus aureus. Front. Microbiol. 8, 2189 (2017).
Trastoy, R. et al. Mechanisms of bacterial tolerance and persistence in the gastrointestinal and respiratory environments. Clin. Microbiol. Rev. 31, e00023-18 (2018).
Ruwandeepika, H. A., Karunasagar, I., Bossier, P. & Defoirdt, T. Expression and quorum sensing regulation of Type III secretion system genes of Vibrio harveyi during infection of gnotobiotic brine shrimp. PLoS One 10, e0143935 (2015).
Li, M. et al. HigB of pseudomonas aeruginosa enhances killing of phagocytes by up-regulating the Type III secretion system in ciprofloxacin induced persister cells. Front. Cell Infect. Microbiol. 6, 125 (2016).
Zheng, J., Shin, O. S., Cameron, D. E. & Mekalanos, J. J. Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc. Natl Acad. Sci. USA 107, 21128–21133 (2010).
Ali, L. et al. Molecular mechanism of Quorum-Sensing in Enterococcus faecalis: its role in virulence and therapeutic approaches. Int. J. Mol. Sci. 18, 960 (2017).
Han, T. H. et al. Environmental factors affecting indole production in Escherichia coli. Res. Microbiol. 162, 108–116 (2011).
Vega, N. M. et al. Salmonella typhimurium intercepts Escherichia coli signaling to enhance antibiotic tolerance. Proc. Natl Acad. Sci. USA 110, 14420–14425 (2013).
Hirakawa, H. et al. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol. Microbiol. 55, 1113–1126 (2005).
Lee, H. H., Molla, M. N., Cantor, C. R. & Collins, J. J. Bacterial charity work leads to population-wide resistance. Nature 467, 82–85 (2010).
Kwan, B. W. et al. Phosphodiesterase DosP increases persistence by reducing cAMP which reduces the signal indole. Biotechnol. Bioeng. 112, 588–600 (2015).
Sun, F. et al. 5-Methylindole potentiates aminoglycoside against gram-positive bacteria including staphylococcus aureus persisters under hypoionic conditions. Front. Cell Infect. Microbiol. 10, 84 (2020).
Wang, Y. et al. Indole reverses intrinsic antibiotic resistance by activating a novel dual-function importer. mBio. 10, e00676-19 (2019).
Han, Y. et al. Indole-induced reversion of intrinsic multiantibiotic resistance in lysobacter enzymogenes. Appl. Environ. Microbiol. 83, e00995-17 (2017).
Potrykus, K. & Cashel, M. (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51 (2008).
Jain, V., Kumar, M. & Chatterji, D. ppGpp: stringent response and survival. J. Microbiol. 44, 1–10 (2006).
Cabello, F. C., Godfrey, H. P., Bugrysheva, J. V. & Newman, S. A. Sleeper cells: the stringent response and persistence in the Borreliella (Borrelia) burgdorferi enzootic cycle. Environ. Microbiol. 19, 3846–3862 (2017).
Hauryliuk, V. et al. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 13, 298–309 (2015).
Mechold, U. et al. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 41, 6175–6189 (2013).
Durfee, T. et al. Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol. 190, 1084–1096 (2008).
Riesenberg, D., Erdei, S., Kondorosi, E. & Kari, C. Positive involvement of ppGpp in derepression of the nif operon in Klebsiella pneumoniae. Mol. Gen. Genet. 185, 198–204 (1982).
Zhou, Y. N. et al. Regulation of cell growth during serum starvation and bacterial survival in macrophages by the bifunctional enzyme SpoT in Helicobacter pylori. J. Bacteriol. 190, 8025–8032 (2008).
Park, S. A., Ko, A. & Lee, N. G. Stimulation of growth of the human gastric pathogen Helicobacter pylori by atmospheric level of oxygen under high carbon dioxide tension. BMC Microbiol. 11, 96 (2011).
Primm, T. P. et al. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182, 4889–4898 (2000).
Betts, J. C. et al. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731 (2002).
Stallings, C. L. et al. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138, 146–159 (2009).
Bryson, D., Hettle, A. G., Boraston, A. B. & Hobbs, J. K. Clinical Mutations That Partially Activate the Stringent Response Confer Multidrug Tolerance in Staphylococcus aureus. Antimicrob Agents Chemother. 64, e02103-19 (2020).
Wood, T. K. & Song, S. Forming and waking dormant cells: the ppGpp ribosome dimerization persister model. Biofilm 2, 100018 (2020).
Svenningsen, M. S. et al. Birth and resuscitation of (p)ppGpp induced antibiotic tolerant persister cells. Sci. Rep. 9, 6056 (2019).
Pu, Y., Ke, Y. & Bai, F. Active efflux in dormant bacterial cells - new insights into antibiotic persistence. Drug Resist Updat 30, 7–14 (2017).
Gerdes, K. & Semsey, S. Microbiology: pumping persisters. Nature 534, 41–42 (2016).
Du Toit, A. Bacterial physiology: persisters are under the pump. Nat. Rev. Microbiol. 14, 332–333 (2016).
AlMatar, M., Albarri, O., Makky, E. A. & Koksal, F. Efflux pump inhibitors: new updates. Pharm. Rep. 73, 1–16 (2021).
Du, D. et al. Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 16, 523–539 (2018).
Ding, Y. H. et al. Role of efflux pumps, their inhibitors, and regulators in colistin resistance. Front. Microbiol. 14, 1207441 (2023).
Pu, Y. et al. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294 (2016).
Zou, J., Peng, B., Qu, J. & Zheng, J. Are bacterial persisters dormant cells only? Front. Microbiol. 12, 708580 (2021).
Kumar, S. et al. Dynamics of efflux pumps in antimicrobial resistance, persistence, and community living of Vibrionaceae. Arch. Microbiol. 206, 7 (2023).
Piddock, L. J. Multidrug-resistance efflux pumps - not just for resistance. Nat. Rev. Microbiol. 4, 629–636 (2006).
Martini, C. L. et al. Cellular growth arrest and efflux pumps are associated with antibiotic persisters in streptococcus pyogenes induced in biofilm-like environments. Front. Microbiol. 12, 716628 (2021).
Ma, X. et al. Local regulator AcrR regulates persister formation by repression of AcrAB efflux pump during exponential growth in aeromonas veronii. Antimicrob. Agents Chemother. 67, e0096922 (2023).
Sulaiman, J. E., Hao, C. & Lam, H. Specific enrichment and proteomics analysis of Escherichia coli persisters from rifampin pretreatment. J. Proteome Res. 17, 3984–3996 (2018).
Adams, K. N. et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53 (2011).
Nolivos, S. et al. Role of AcrAB-TolC multidrug efflux pump in drug-resistance acquisition by plasmid transfer. Science 364, 778–782 (2019).
Rahman, T., Yarnall, B. & Doyle, D. A. Efflux drug transporters at the forefront of antimicrobial resistance. Eur. Biophys. J. 46, 647–653 (2017).
Lamut, A., Peterlin Masic, L., Kikelj, D. & Tomasic, T. Efflux pump inhibitors of clinically relevant multidrug resistant bacteria. Med. Res. Rev. 39, 2460–2504 (2019).
Pumbwe, L., Chang, A., Smith, R. L. & Wexler, H. M. BmeRABC5 is a multidrug efflux system that can confer metronidazole resistance in Bacteroides fragilis. Micro. Drug Resist. 13, 96–101 (2007).
Xu, Y., Liu, S., Zhang, Y. & Zhang, W. DNA adenine methylation is involved in persister formation in E. coli. Microbiol. Res. 246, 126709 (2021).
Riber, L. & Hansen, L. H. Epigenetic memories: the hidden drivers of bacterial persistence? Trends Microbiol. 29, 190–194 (2021).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26–39 (2020).
Wood, W. N., Mohler, K., Rinehart, J. & Ibba, M. Deacylated tRNA accumulation is a trigger for bacterial antibiotic persistence independent of the stringent response. mBio 12, e0113221 (2021).
Salcedo-Sora, J. E. & Kell, D. B. A quantitative survey of bacterial persistence in the presence of antibiotics: towards antipersister antimicrobial discovery. Antibiotics. 9, 508 (2020).
Defraine, V., Fauvart, M. & Michiels, J. Fighting bacterial persistence: current and emerging anti-persister strategies and therapeutics. Drug Resist. Updat 38, 12–26 (2018).
Allison, K. R., Brynildsen, M. P. & Collins, J. J. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 14, 593–598, (2011).
Scorpio, A. & Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 2, 662–667 (1996).
Gopal, P. et al. Pyrazinoic acid inhibits mycobacterial coenzyme A biosynthesis by binding to aspartate decarboxylase PanD. ACS Infect. Dis. 3, 807–819 (2017).
Zhang, Y., Shi, W., Zhang, W. & Mitchison, D. Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2, 1–12 (2013).
Andries, K. et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 (2005).
Yamada, W. et al. Combination therapy to kill mycobacterium tuberculosis in its nonreplicating persister phenotype. Antimicrob. Agents Chemother. 66, e0069522 (2022).
Lanni, A. et al. Activity of drug combinations against mycobacterium abscessus grown in aerobic and hypoxic conditions. Microorganisms 10, 1421 (2022).
Brotz-Oesterhelt, H. et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11, 1082–1087 (2005).
Kirstein, J. et al. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 1, 37–49 (2009).
Gavrish, E. et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21, 509–518 (2014).
Quigley, J. et al. Novel antimicrobials from uncultured bacteria acting against Mycobacterium tuberculosis. mBio. 11, e01516-20 (2020).
Weinhaupl, K. et al. Structure of the drug target ClpC1 unfoldase in action provides insights on antibiotic mechanism of action. J. Biol. Chem. 298, 102553 (2022).
Hosoda, K. et al. Mavintramycin A is a promising antibiotic for treating complex infectious disease. Antimicrob Agents Chemother. 68, e0091723 (2024).
Hurdle, J. G., O’Neill, A. J., Chopra, I. & Lee, R. E. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9, 62–75 (2011).
Hu, Y., Shamaei-Tousi, A., Liu, Y. & Coates, A. A new approach for the discovery of antibiotics by targeting non-multiplying bacteria: a novel topical antibiotic for staphylococcal infections. PLoS One 5, e11818 (2010).
Frapwell, C. J. et al. Antimicrobial activity of the quinoline derivative HT61 against staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 64, e02073-19 (2020).
Amison, R. T. et al. The small quinolone derived compound HT61 enhances the effect of tobramycin against Pseudomonas aeruginosa in vitro and in vivo. Pulm. Pharm. Ther. 61, 101884 (2020).
Hu, Y. & Coates, A. R. Enhancement by novel anti-methicillin-resistant Staphylococcus aureus compound HT61 of the activity of neomycin, gentamicin, mupirocin and chlorhexidine: in vitro and in vivo studies. J. Antimicrob. Chemother. 68, 374–384 (2013).
Hubbard, A. T. et al. Mechanism of action of a membrane-active quinoline-based antimicrobial on natural and model bacterial membranes. Biochemistry 56, 1163–1174 (2017).
Hubbard, A. T., Coates, A. R. & Harvey, R. D. Comparing the action of HT61 and chlorhexidine on natural and model Staphylococcus aureus membranes. J. Antibiot. 70, 1020–1025 (2017).
Ooi, N. et al. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J. Antimicrob. Chemother. 64, 735–740 (2009).
Ooi, N. et al. XF-70 and XF-73, novel antibacterial agents active against slow-growing and non-dividing cultures of Staphylococcus aureus including biofilms. J. Antimicrob. Chemother. 65, 72–78 (2010).
Farrell, D. J., Robbins, M., Rhys-Williams, W. & Love, W. G. In vitro activity of XF-73, a novel antibacterial agent, against antibiotic-sensitive and -resistant Gram-positive and Gram-negative bacterial species. Int. J. Antimicrob. Agents 35, 531–536 (2010).
Zhang, C. et al. Efficacy of a novel antibacterial agent exeporfinium chloride, (XF-73), against antibiotic-resistant bacteria in mouse superficial skin infection models. Infect. Drug Resist. 16, 4867–4879 (2023).
Yendewa, G. A. et al. A two-part phase 1 study to establish and compare the safety and local tolerability of two nasal formulations of XF-73 for decolonisation of Staphylococcus aureus: a previously investigated 0.5 mg/g viscosified gel formulation versus a modified formulation. J. Glob. Antimicrob. Resist. 21, 171–180 (2020).
Mangino, J. E. et al. Exeporfinium chloride (XF-73) nasal gel dosed over 24 h prior to surgery significantly reduced Staphylococcus aureus nasal carriage in cardiac surgery patients: safety and efficacy results from a randomized placebo-controlled phase 2 study. Infect. Control Hosp. Epidemiol. 44, 1–3 (2023).
Board-Davies, E. L. et al. Antimicrobial effects of XF drugs against Candida albicans and its biofilms. Front. Fungal Biol. 4, 1225647 (2023).
Gonzales, F. P., Felgentrager, A., Baumler, W. & Maisch, T. Fungicidal photodynamic effect of a twofold positively charged porphyrin against Candida albicans planktonic cells and biofilms. Future Microbiol. 8, 785–797 (2013).
Vaudaux, P. et al. Extracellular and intracellular bactericidal activities of XF-70 against small-colony variant hemB mutants of meticillin-susceptible and meticillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 37, 576–579 (2011).
Ghosh, C. et al. Aryl-alkyl-lysines: membrane-active small molecules active against murine model of burn infection. ACS Infect. Dis. 2, 111–122 (2016).
Ghosh, C. et al. Aryl-alkyl-lysines: agents that kill planktonic cells, persister cells, biofilms of MRSA and protect mice from skin-infection. PLoS One 10, e0144094 (2015).
Ghosh, C. et al. Aryl-alkyl-lysines: membrane-active fungicides that act against biofilms of. Acs Infect. Dis. 3, 293–301 (2017).
Rajamuthiah, R. et al. Whole animal automated platform for drug discovery against multi-drug resistant Staphylococcus aureus. PLoS One 9, e89189 (2014).
Kim, W. et al. A new class of synthetic retinoid antibiotics effective against bacterial persisters. Nature 556, 103–107 (2018).
Kim, W. et al. Identification of an antimicrobial agent effective against methicillin-resistant staphylococcus aureus persisters using a fluorescence-based screening strategy. PLoS One 10, e0127640 (2015).
Kim, W. et al. NH125 kills methicillin-resistant Staphylococcus aureus persisters by lipid bilayer disruption. Future Med. Chem. 8, 257–269 (2016).
Basak, A. et al. Antimicrobial peptide-inspired NH125 analogues: bacterial and fungal biofilm-eradicating agents and rapid killers of MRSA persisters. Org. Biomol. Chem. 15, 5503–5512 (2017).
Abouelhassan, Y., Basak, A., Yousaf, H. & Huigens, R. W. 3rd Identification of N-Arylated NH125 analogues as rapid eradicating agents against MRSA persister cells and potent biofilm killers of gram-positive pathogens. Chem. Biochem. 18, 352–357 (2017).
Kim, W. et al. The neutrally charged diarylurea compound PQ401 kills antibiotic-resistant and antibiotic-tolerant staphylococcus aureus. mBio. 11, e01140-20 (2020).
Kim, W. et al. Discovery and optimization of nTZDpa as an antibiotic effective against bacterial persisters. ACS Infect. Dis. 4, 1540–1545 (2018).
Luca, V. et al. Esculentin(1-21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell Mol. Life Sci. 70, 2773–2786 (2013).
Scotti, R. et al. Derivatives of Esculentin-1 peptides as promising candidates for fighting infections from Escherichia coli O157:H7. Antibiotics 11, 656 (2022).
Scotti, R. et al. Fighting microbial infections from Escherichia coli O157:H7: the combined use of three essential oils of the cymbopogon genus and a derivative of esculentin-1a peptide. Antibiotics 13, 86 (2024).
Bahar, A. A. et al. Synthetic dendrimeric peptide active against biofilm and persister cells of Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 99, 8125–8135 (2015).
Libardo, M. D. J. et al. Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. FEBS J. 284, 3662–3683 (2017).
Chen, X. et al. Control of bacterial persister cells by Trp/Arg-containing antimicrobial peptides. Appl. Environ. Microbiol. 77, 4878–4885 (2011).
Defraine, V. et al. Efficacy of artilysin art-175 against resistant and persistent acinetobacter baumannii. Antimicrob. Agents Chemother. 60, 3480–3488 (2016).
Briers, Y. et al. Art-175 is a highly efficient antibacterial against multidrug-resistant strains and persisters of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 58, 3774–3784 (2014).
Jennings, M. C. et al. Biofilm-eradicating properties of quaternary ammonium amphiphiles: simple mimics of antimicrobial peptides. Chembiochem 15, 2211–2215 (2014).
Mohamed, M. F. et al. Targeting biofilms and persisters of ESKAPE pathogens with P14KanS, a kanamycin peptide conjugate. Biochim. Biophys. Acta Gen. Subj. 1861, 848–859 (2017).
Schmidt, N. W. et al. Engineering persister-specific antibiotics with synergistic antimicrobial functions. ACS Nano 8, 8786–8793 (2014).
Defraine, V. et al. Antibacterial Activity of 1-[(2,4-Dichlorophenethyl)amino]-3-Phenoxypropan-2-ol against antibiotic-resistant strains of diverse bacterial pathogens, biofilms and in pre-clinical infection models. Front. Microbiol. 8, 2585 (2017).
Liebens, V. et al. Identification of 1-((2,4-Dichlorophenethyl)Amino)-3-Phenoxypropan-2-ol, a novel antibacterial compound active against persisters of pseudomonas aeruginosa. Antimicrob Agents Chemother. 61, e00836-17 (2017).
Narayanaswamy, V. P. et al. Polycationic glycopolymer demonstrates activity against persisters and biofilms of non-tuberculosis mycobacteria cystic fibrosis clinical isolates in vitro. Front. Microbiol. 13, 821820 (2022).
Mukherjee, D. et al. Membrane-targeting AM-0016 kills mycobacterial persisters and shows low propensity for resistance development. Future Microbiol. 11, 643–650 (2016).
Moreira, W., Aziz, D. B. & Dick, T. Boromycin kills mycobacterial persisters without detectable resistance. Front. Microbiol. 7, 199 (2016).
Feng, J. et al. Selective essential oils from spice or culinary herbs have high activity against stationary phase and biofilm Borrelia burgdorferi. Front. Med. 4, 169 (2017).
Ghosh, C. et al. Designing simple lipidated lysines: bifurcation imparts selective antibacterial activity. ChemMedChem 11, 2367–2371 (2016).
Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007).
Singh, S. B. Confronting the challenges of discovery of novel antibacterial agents. Bioorg. Med. Chem. Lett. 24, 3683–3689 (2014).
Strathdee, S. A., Hatfull, G. F., Mutalik, V. K. & Schooley, R. T. Phage therapy: from biological mechanisms to future directions. Cell 186, 17–31 (2023).
Dan, J. M. et al. Development of host immune response to bacteriophage in a lung transplant recipient on adjunctive phage therapy for a multidrug-resistant pneumonia. J. Infect. Dis. 227, 311–316 (2023).
Onsea, J. et al. Bacteriophage therapy for difficult-to-treat infections: the implementation of a multidisciplinary phage task force (The PHAGEFORCE Study Protocol). Viruses 13, 1543 (2021).
Verbeken, G. & Pirnay, J. P. European regulatory aspects of phage therapy: magistral phage preparations. Curr. Opin. Virol. 52, 24–29 (2022).
Dedrick, R. M. et al. Phage therapy of mycobacterium infections: compassionate use of phages in 20 patients with drug-resistant mycobacterial disease. Clin. Infect. Dis. 76, 103–112 (2023).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019).
Eskenazi, A. et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 13, 302 (2022).
Nick, J. A. et al. Host and pathogen response to bacteriophage engineered against Mycobacterium abscessus lung infection. Cell 185, 1860–1874.e1812 (2022).
De Soir, S. et al. Exploiting phage-antibiotic synergies to disrupt Pseudomonas aeruginosa PAO1 biofilms in the context of orthopedic infections. Microbiol. Spectr. 12, e0321923 (2024).
Gordillo Altamirano, F. et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 6, 157–161 (2021).
Fowler, V. G., Jr. et al. Exebacase in addition to standard-of-care antibiotics for staphylococcus aureus bloodstream infections and right-sided infective endocarditis: a phase 3, Superiority-Design, Placebo-Controlled, Randomized Clinical Trial (DISRUPT). Clin. Infect. Dis. ciae043 (2024). Online ahead of print
Gilmer, D. B., Schmitz, J. E., Euler, C. W. & Fischetti, V. A. Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 57, 2743–2750 (2013).
Schuch, R. et al. Combination therapy with lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant Staphylococcus aureus-induced murine bacteremia. J. Infect. Dis. 209, 1469–1478 (2014).
Schuch, R. et al. Bacteriophage Lysin CF-301, a Potent Antistaphylococcal Biofilm Agent. Antimicrob. Agents Chemother. 61, e02666-16 (2017).
Gutierrez, D. et al. Effective removal of staphylococcal biofilms by the endolysin LysH5. PLoS One 9, e107307 (2014).
Lu, T. K. & Collins, J. J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl Acad. Sci. USA 106, 4629–4634 (2009).
Kwan, B. W., Chowdhury, N. & Wood, T. K. Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 17, 4406–4414 (2015).
Cruz-Muniz, M. Y. et al. Repurposing the anticancer drug mitomycin C for the treatment of persistent Acinetobacter baumannii infections. Int .J. Antimicrob. Agents 49, 88–92 (2017).
Sharma, B. et al. Borrelia burgdorferi, the causative agent of lyme disease, forms drug-tolerant persister cells. Antimicrob. Agents Chemother. 59, 4616–4624 (2015).
Chowdhury, N. et al. DNA-crosslinker cisplatin eradicates bacterial persister cells. Biotechnol. Bioeng. 113, 1984–1992 (2016).
Feng, J., Shi, W., Zhang, S. & Zhang, Y. Identification of new compounds with high activity against stationary phase Borrelia burgdorferi from the NCI compound collection. Emerg. Microbes. Infect. 4, e31 (2015).
Pascoe, B. et al. Dormant cells of Staphylococcus aureus are resuscitated by spent culture supernatant. PLoS One 9, e85998 (2014).
Marques, C. N., Morozov, A., Planzos, P. & Zelaya, H. M. The fatty acid signaling molecule cis-2-decenoic acid increases metabolic activity and reverts persister cells to an antimicrobial-susceptible state. Appl. Environ. Microbiol. 80, 6976–6991 (2014).
Pan, J., Bahar, A. A., Syed, H. & Ren, D. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PLoS One 7, e45778 (2012).
Pan, J. et al. (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one sensitizes Escherichia coli persister cells to antibiotics. Appl Microbiol. Biotechnol. 97, 9145–9154 (2013).
Kim, J. S. et al. Selective killing of bacterial persisters by a single chemical compound without affecting normal antibiotic-sensitive cells. Antimicrob. Agents Chemother. 55, 5380–5383 (2011).
Barraud, N., Buson, A., Jarolimek, W. & Rice, S. A. Mannitol enhances antibiotic sensitivity of persister bacteria in Pseudomonas aeruginosa biofilms. PLoS One 8, e84220 (2013).
Meylan, S. et al. Carbon sources tune antibiotic susceptibility in pseudomonas aeruginosa via tricarboxylic acid cycle control. Cell Chem. Biol. 24, 195–206 (2017).
Radlinski, L. C. et al. Chemical Induction of aminoglycoside uptake overcomes antibiotic tolerance and resistance in Staphylococcus aureus. Cell Chem. Biol. 26, 1355–1364.e1354 (2019).
Lv, B. et al. Mechanosensitive channels mediate hypoionic shock-induced aminoglycoside potentiation against bacterial persisters by enhancing antibiotic uptake. Antimicrob. Agents Chemother. 66, e0112521 (2022).
Zhao, Y. et al. Rapid freezing enables aminoglycosides to eradicate bacterial persisters via enhancing mechanosensitive channel MscL-mediated antibiotic uptake. mBio. 11, e03239-19 (2020).
Duan, X. et al. l-Serine potentiates fluoroquinolone activity against Escherichia coli by enhancing endogenous reactive oxygen species production. J. Antimicrob. Chemother. 71, 2192–2199 (2016).
Lebeaux, D. et al. pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms. J. Infect. Dis. 210, 1357–1366 (2014).
Morones-Ramirez, J. R., Winkler, J. A., Spina, C. S. & Collins, J. J. Silver enhances antibiotic activity against gram-negative bacteria. Sci. Transl. Med. 5, 190ra181 (2013).
Starkey, M. et al. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 10, e1004321 (2014).
Maura, D. & Rahme, L. G. Pharmacological inhibition of the pseudomonas aeruginosa MvfR quorum-sensing system interferes with biofilm formation and potentiates antibiotic-mediated biofilm disruption. Antimicrob. Agents Chemother. 61, e01362-17(2017).
Wexselblatt, E. et al. Relacin, a novel antibacterial agent targeting the Stringent Response. Plos Pathog. 8, e1002925 (2012).
Yanling, C. et al. Efficacy of relacin combined with sodium hypochlorite against Enterococcus faecalis biofilms. J. Appl. Oral. Sci. 26, e20160608 (2018).
Danchik, C., Wang, S. & Karakousis, P. C. Targeting the mycobacterium tuberculosis stringent response as a strategy for shortening tuberculosis treatment. Front. Microbiol. 12, 744167 (2021).
Vilcheze, C. et al. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 114, 4495–4500 (2017).
Leszczynska, D. et al. The formation of persister cells in stationary-phase cultures of Escherichia coli is associated with the aggregation of endogenous proteins. PLoS One 8, e54737 (2013).
Dahl, J. U. et al. The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat. Microbiol. 2, 16267 (2017).
Finzi, D. et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5, 512–517 (1999).
Gao, Y., Kraft, J. C., Yu, D. & Ho, R. J. Y. Recent developments of nanotherapeutics for targeted and long-acting, combination HIV chemotherapy. Eur. J. Pharm. Biopharm. 138, 75–91 (2019).
Zheng, E. J., Stokes, J. M. & Collins, J. J. Eradicating bacterial persisters with combinations of strongly and weakly metabolism-dependent antibiotics. Cell Chem. Biol. 27, 1544–1552.e1543 (2020).
Beloin, C., Renard, S., Ghigo, J. M. & Lebeaux, D. Novel approaches to combat bacterial biofilms. Curr. Opin. Pharm. 18, 61–68, (2014).
Tyers, M. & Wright, G. D. Drug combinations: a strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 17, 141–155 (2019).
Feng, J. et al. Stationary phase persister/biofilm microcolony of Borrelia burgdorferi causes more severe disease in a mouse model of Lyme arthritis: implications for understanding persistence, Post-treatment Lyme Disease Syndrome (PTLDS), and treatment failure. Discov. Med. 27, 125–138 (2019).
Yuan, Y. et al. Identification of Persister Drug Combination Clinafloxacin + Cefuroxime + Gentamicin That Eradicates Persistent Pseudomonas aeruginosa Infection in a Murine Cystic Fibrosis Model. Infect. Microbes Dis. 5, 8 (2023).
Lázár, V., Snitser, O., Barkan, D. & Kishony, R. Antibiotic combinations reduce Staphylococcus aureus clearance. Nature 610, 540–546 (2022).
Liu, J. et al. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 367, 200–204 (2020).
Dewachter, L., Fauvart, M. & Michiels, J. Bacterial heterogeneity and antibiotic survival: understanding and combatting persistence and heteroresistance. Mol. Cell 76, 255–267 (2019).
Gefen, O. & Balaban, N. Q. The importance of being persistent: heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol. Rev. 33, 704–717 (2009).
Mascio, C. T., Alder, J. D. & Silverman, J. A. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob. Agents Chemother. 51, 4255–4260 (2007).
Wannigama, D. L. et al. A rapid and simple method for routine determination of antibiotic sensitivity to biofilm populations of Pseudomonas aeruginosa. Ann. Clin. Microbiol. Antimicrob. 19, 8 (2020).
Zhang, L. et al. Combination and nanotechnology based pharmaceutical strategies for combating respiratory bacterial biofilm infections. Int. J. Pharm. 616, 121507 (2022).
Somoskovi, A., Wade, M. M., Sun, Z. & Zhang, Y. Iron enhances the antituberculous activity of pyrazinamide. J. Antimicrob. Chemother. 53, 192–196 (2004).
Bartell, J. A. et al. Bacterial persisters in long-term infection: emergence and fitness in a complex host environment. Plos Pathog. 16, e1009112 (2020).
Claudi, B. et al. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158, 722–733 (2014).
Helaine, S. et al. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014).
Beam, J. E. et al. Macrophage-produced peroxynitrite induces antibiotic tolerance and supersedes intrinsic mechanisms of persister formation. Infect. Immun. 89, e0028621 (2021).
Peyrusson, F. et al. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat. Commun. 11, 2200 (2020).
Anderson, G. G. et al. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301, 105–107 (2003).
Justice, S. S. et al. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl Acad. Sci. USA 101, 1333–1338 (2004).
Lowrie, D. B. et al. Therapy of tuberculosis in mice by DNA vaccination. Nature 400, 269–271 (1999).
Choudhary, G. S. et al. Human granulocyte macrophage colony-stimulating factor enhances antibiotic susceptibility of persister cells. Sci Rep. 5, 17315 (2015).
Hu, Y. et al. Bedaquiline kills persistent Mycobacterium tuberculosis with no disease relapse: an in vivo model of a potential cure. J. Antimicrob. Chemother. 74, 1627–1633 (2019).
Karau, M. et al. Locally delivered antistaphylococcal lysin exebacase or CF-296 is active in methicillin-resistant Staphylococcus aureus implant-associated osteomyelitis. J. Bone Jt Infect. 7, 169–175 (2022).
Wang, Y. et al. High antipersister activity of a promising new quinolone drug candidate in eradicating uropathogenic Escherichia coli persisters and persistent infection in mice. J. Appl. Microbiol. 134, lxad193 (2023).
Acknowledgements
We acknowledge the support by Zhejiang Provincial Natural Science Foundation of China (LY24H290004); National Natural Science Foundation of China Youth Foundation (81701969), Natural Science Youth Exploration Program (2022JKZKTS03), and Open fund for the State Key Laboratory of Pathogenic Biology of Animal Diseases (SKLVEB2021KFKT002) for HXN; and National Center for Infectious Disease startup fund (B2022011-1), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2021R01012), and Jinan Microecological Biomedicine Shandong Laboratory project (JNL-2022050B) for YZ.
Author information
Authors and Affiliations
Contributions
Hongxia Niu and Ying Zhang were responsible for searching the literature, making graphics and writing the manuscript. Jiaying Gu assisted with the literature search. All authors have read and approved the article for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niu, H., Gu, J. & Zhang, Y. Bacterial persisters: molecular mechanisms and therapeutic development. Sig Transduct Target Ther 9, 174 (2024). https://doi.org/10.1038/s41392-024-01866-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01866-5
- Springer Nature Limited