
Abstract
Rheumatoid arthritis (RA) is an incurable systemic autoimmune disease. Disease progression leads to joint deformity and associated loss of function, which significantly impacts the quality of life for sufferers and adds to losses in the labor force. In the past few decades, RA has attracted increased attention from researchers, the abnormal signaling pathways in RA are a very important research field in the diagnosis and treatment of RA, which provides important evidence for understanding this complex disease and developing novel RA-linked intervention targets. The current review intends to provide a comprehensive overview of RA, including a general introduction to the disease, historical events, epidemiology, risk factors, and pathological process, highlight the primary research progress of the disease and various signaling pathways and molecular mechanisms, including genetic factors, epigenetic factors, summarize the most recent developments in identifying novel signaling pathways in RA and new inhibitors for treating RA. therapeutic interventions including approved drugs, clinical drugs, pre-clinical drugs, and cutting-edge therapeutic technologies. These developments will hopefully drive progress in new strategically targeted therapies and hope to provide novel ideas for RA treatment options in the future.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a well-known systemic autoimmune disease. The general features of RA are demonstrated in Fig. 1. The original terminology for ‘rheumatoid arthritis’ is derived from the Greek word for inflamed and watery joints.1,2 The first person to describe and classify this debilitating disease was the French doctor Augustin Jacob Landré-Beauvais in 1880. Landré-Beauvais recorded the important manifestations of the disease with “asthenic gout,” indicating that the condition occurred well in women.3 Later, the British rheumatologist Dr. Alfred Baring Garrod coined the term “rheumatoid arthritis” in 1859.4 Critically, it is now known that global, the incidence of RA is ~1%,5,6,7,8,9,10 with prevalence increasing with age; the disease commonly comes up between the ages of 40 and 50 in individuals with the condition three to five times more in women than in men.6,11,12,13 Repeated and symmetrical multiple micro arthritis is the primary clinical manifestation of the disease, occurring in the hand, wrist, foot, knee, and other joints. In the early stages of the disease, redness, swelling, heat, pain, and joint dysfunction are common.14 The European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) developed new classification criteria for RA: according to joint symptoms, serology indicators (RF or ACPA), duration of symptoms, acute phase reactants, each of these categories has scoring criteria.15 Methotrexate therapy was initiated by identifying disease characteristics, a consensus decision was made, and a scoring system was created to predict which patients would develop erosive and/or persistent disease.15,16,17 In the late stages of the condition, different degrees of rigidity and deformity of joints are seen and, finally, drive several degrees of bone corrosion and skeletal muscle atrophy, synovitis invasion of articular cartilage, sub-cartilage bone erosion, and damage to ligaments and tendons.18 The disease seriously affects the quality of daily life and suffers have high disability rates, and this can impact the loss of labor in the general population. RA also occurs in other tissues and organs, viz. extra-articular tissues and organs, including the eyes, nerves, skin, kidney, lungs, liver, heart, and bones.19,20,21,22

General features of rheumatoid arthritis. Rheumatoid arthritis is an incurable autoimmune disease that occurs most frequently in women, usually in the small joints, with systemic complications that ultimately lead to disability
The cause of RA remains unknown, but it is generally considered related to environmental and genetic factors. The mechanism(s) of action include the joints attacking by body’s immune system by mistake, which causes joint capsule inflammation and thickening, and promotes damage to bones and cartilage at these sites. In the clinical, RA diagnosis is based on the patient’s physical manifestations and symptoms.23,24,25,26,27 X-rays and laboratory tests can assist in the diagnosis or exclusion of some similar disorders, viz. lupus erythematosus, psoriatic arthritis, and fibromyalgia.28,29,30,31,32,33 Since RA is incurable, it burdens individuals and society.34,35,36,37,38 The personal burden arises from musculoskeletal defects, accompanied by a decline in physical function and quality of life.39,40 In addition to direct medical costs, the socioeconomic burden results from RA patients having dysfunction and decreased working ability, and reduced social participation.41 A recent survey in China showed that the average annual direct cost per RA patient was $1917.21 ± $2559.06.42 The Burden of RA across Europe: a Socioeconomic Survey (BRASS) by using Work Productivity and Activity Impairment questionnaire (WPAI) scores indicated that RA people with severe (60%) or moderate pain (48%) experienced additional work obstacles compared with those with mild (34%) or no pain (19%), a statistically remarkable correlation was found between severity, pain, disability, and early retirement.43 Similar survey results in Latin America, China, and other Countries have also been reported.44,45,46
The pathogenesis of RA is tightly related to many characterized signaling pathways. Therefore, in recent years, research attention has focused on developing molecules that function as inhibitors of RA-linked signaling systems. This review will generally introduce the risk factor and pathogenesis of RA, mainly describing the signal transduction pathways that impact RA, drugs in clinical use, potential drugs in clinical/pre-clinical studies, and technologies for targeted therapy.
Risk factors and symptoms
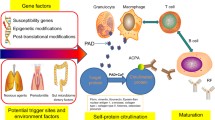
Over the past few decades, RA etiology has been numerously explored, and the available evidence indicating environmental and genetic factors are important in inducing RA. Indeed, the susceptibility genes HLA-DRB1, TNFRSF14, and PTPN22 are closely related to the occurrence of RA.47,48,49,50 HLA-DRB1 is the most widely explored gene and forms part of the HLA complex, the major histocompatibility complex (MHC) human version.51,52,53 The susceptibility and outcome of RA may be related to specific HLA-DR alleles; however, these alleles vary by ethnicity and geographic region.54,55,56 The HLA-DRB1 allele constitutes the strongest genetic association linked to RA, and the allele associated with the disease is a “shared epitope” with a conserved sequence of five amino acids.47 The shared epitope hypothesis indicates that some alleles with this conserved sequence are in connection with the pathogenesis of RA because they allow antigen-presenting cells to incorrectly present their antigens to T cells, which results in T-cell-mediated autoimmune responses that directly contribute to the RA pathogenesis.57,58 Environmental factors are also key points in causing RA, such as smoking, personal dietary pattern, and hygiene, which directly affects the post-transcriptional modification of certain genes or indirectly affects susceptibility genes via epigenetic mechanisms.59,60,61,62 The interaction of environmental factors, epigenetics, and susceptibility genes will drive changes in the relative levels and expression of coded proteins, which could promote autoimmune tolerance disorders.
While RA mainly affects the joints, it may also influence other organ systems,63,64 including the eyes, skin, lungs, liver, heart, and bones (Fig. 2). RA usually presents signs of inflammation, swelling, fever, pain, and stiffness in the affected joints. In general, these processes occur in the small joints of the feet and hands but may also occur in larger joints such as the shoulder and knees.65,66,67,68,69,70 These symptoms are more pronounced after long periods of inactivity, and a conspicuous feature of the disease is increased stiffness in the morning.71,72,73,74,75 Pain related to RA is caused at the inflammation site and is classified as nociceptive rather than neuropathic.76,77 As the pathological condition progresses, continued inflammation results in tendon binding and erosion and destruction of the articular surface; this can impair the range of motion and lead to deformity,65,78,79,80 and local osteoporosis often occurs around the inflamed joints of RA patients.81,82 Sustained production of inflammatory mediators creates a pro-inflammatory cycle, a situation common to many chronic diseases and this likely explains why RA patients are at greater risk of cardiovascular diseases.83,84,85,86 In addition, untreated chronic inflammation may lead to renal amyloidosis,87,88 rheumatoid nodules in the skin,89,90,91 and interstitial lung disease (ILD).92,93,94,95 Moreover, in the eye, episcleritis is common,96,97,98 liver problems like autoimmune hepatitis can also trigger problems,99,100,101 and peripheral neuropathy caused by wrist swelling and median nerve compression is a common problem in carpal tunnel syndrome. Rheumatic diseases of the spine can also contribute to myelopathy, atlantoaxial subluxation may occur due to erosion of the transverse ligament, and can progress to paralysis and even death.102

Risk factors and systemic complications of RA. Genetic and environmental factors are important in inducing RA. While RA mainly affects the joints, it can also influence other organ systems, including the eyes, nerves, skin, kidneys, lungs, liver, heart, and bones
The pathogenesis of RA
RA is initially a state of continuous cellular activation that results in autoimmunity in joints or other organs.103,104 The clinical manifestations of the disease occur predominantly following synovial inflammation and joint injury. Fibroblast-like synoviocytes (FLS) play a crucial role in these pathological courses.105,106,107 Three stages of RA progression are reported and include a non-specific inflammatory stage, amplified by T-cell activation in the synovium, the chronic inflammatory stage, and a tissue damage stage mediated by cytokines like IL-1, IL-6, and TNF-α, respectively.108,109,110,111
Autoimmune response and inflammation
The production of autoantibodies has been linked to severe symptoms like joint injury and increased mortality.112,113,114,115,116 This is likely because of the generation of immune complexes by autoantibodies against citrullinated peptides (ACPAs) with citrulline-containing antigens. These complexes subsequently bind to rheumatoid factors (RF), leading to complement activation.117,118,119,120,121,122 In recent times, the capacity to detect autoimmune responses to citrullinated self-proteins has been a major advance.123 In RA patients, the degree of association between ACPA-positive and ACPA-negative and shared epitopes are different. The non-HLA genes correlated to RA susceptibility between the two genomes are only partially the same. Therefore, some researchers believe ACPA-positive and ACPA-negative RA may be two genetically distinct disease types of RA.124 Some studies have shown that when certain factors in the environment change, arginine is converted into citrulline under the catalysis of peptidylarginine deiminases (PADs), and citrullinated proteins can, through antigens presenting cells (APCs) present to T cells by certain MHC, produce ACPAs and simultaneously elicit autoimmune responses to citrullinated self-antigens in RA patients.125,126 Peptidyl arginine deiminase type4 (PADI4) is also identified as the non-MHC genetic risk factor of RA. Meanwhile, the PADI4 risk allele was associated with bone damage regardless of ACPA positivity in Asian RA patients.127 ACPA binds citrullinated residues on many of the body’s own proteins, including histones, vimentin, fibronectin, fibrinogen, type II collagen, and alpha-enolase, the activated immune responses tissue is uncertain.128 Circulated ACPAs could be detected up to 10 years before diagnosis known as pre-RA.129,130,131,132,133 As time progresses, the epitope diversity and concentration of ACPAs increase, and so do the concentrations of serum cytokine. With effective treatment, ACPA and RF concentrations decrease; however, patients rarely turn into ACPA negative. In contrast, RF drops are more profound and more frequent, and the patients may seroconvert to RF negativity.134 Anti-carbamylated protein (CarP) and acetylated protein autoantibodies also have been identified in RA patients135,136; moreover, other-directed against additional post-translational protein modifications autoantibodies may emerge.
Joint swelling in RA is the result of synovial inflammation caused by immune activation. The swelling is characterized by the entry of leukocytes into the synovial compartment. The cellular composition of RA synovitis is manifested by the accumulation of innate immune cells (e.g., dendritic cells, monocytes, mast cells, and innate lymphoid cells) and adaptive immune cells (e.g., T-helper-1 and T-helper-17 cells, B cells, plasmablasts, and plasma cells). Innate immunity can be initiated by provoking dendritic cells (DCS) in certain environmental or genetic factors. DCs then recruit and activate T cells, which stimulate B cells, macrophages, synoviocytes, chondrocytes, and osteoclasts, and secrete pro-inflammatory and bone-destroying cytokines i.e., IL-1β, IL-6, TNF -α, and matrix metalloproteinases (MMPs).137,138,139 Therefore, in the adjacent bone marrow and synovium, the integration of innate and adaptive immune pathways promotes tissue injury and remodeling.140 This cascade drives chronic inflammation in RA and promotes circulating leukocytes to migrate into the inflamed joint; this process needs angiogenesis to supply nutrients and oxygen to the hypertrophic joint. Proangiogenic factors trigger angiogenesis.141,142,143 Fibroblast-like synoviocytes (FLS) in the synovium intima form a unique invasive phenotype that promotes extracellular matrix invasion and further exacerbates joint injury.144,145,146 (Fig. 3).

Normal and rheumatoid arthritis joints. Joint swelling in RA reflects synovial inflammation due to immune activation. The cellular composition of RA synovitis is characterized by the accumulation of innate and adaptive immune cells (e.g., T cells, dendritic cells, B cells, macrophages, and osteoclasts). Pro-inflammatory and bone-destructive factors of the immune response led to the loss of bone or cartilage with synovial thickening, angiogenesis, and muscle wasting
FLS and immune cells in RA
Currently, studies on RA have analyzed the character of immune cells in the occurrence and course of the disease. More recently, attention has also focused on the local interstitial cells and the role these cell types play in the pathogenesis of RA. Stromal cells constitute the structural framework of organs and tissues.147 Stromal cells are thought to have immune functions, can recognize pathogens, and trigger immune responses. Fibroblasts of the intestine, skin, gums, and synovium are typical stromal cells. They have been proven to express innate immune receptors, especially Toll-like receptors (TLR).148,149,150,151,152 These stromal cells present and express antigens through histocompatibility complex (MHC) II receptors and secrete cytokines and chemokines. Therefore, these stromal cells are components of the innate immune system.
In non-pathological synovial tissue, the normal physiological function of FLS is to build the lining layer of the synovium, secrete synovial fluid, lubricate proteins in the joint, and provide plasma protein for the adjacent cartilage and joint cavity.153 In addition, FLS participates in the continuous remodeling of the synovium by producing matrix components, like collagen, thereby maintaining synovium homeostasis. Under the conditions of RA inflammation, FLS undergoes a profound change from harmless mesenchymal cells to destructive and aggressive tumor-like cells. These transformed RA FLS play a leading role in the production and progression of RA and show a special phenotype characterized by reduced sensitivity to apoptosis, overexpression of adhesion molecules, and abnormal production of cytokines, chemokines, and matrix metalloproteinases (MMPs).149,154,155
A complex network of cytokine and chemokine regulates the synovial compartment inflammatory environment; several clinical interventions (Fig. 4) suggest that among these components, granulocyte-monocyte colony-stimulating factor, interleukin-6 (IL-6), and tumor necrosis factor (TNF) are essential to the process.156,157 Cytokines and chemokines promote inflammation by activating endothelial cells, attracting immune cells accumulation in the synovial compartment, activating fibroblasts, and accumulating activated T cells and B cells. Activated B cells with the assistance of antigen-presenting cells and Th cells, then differentiate into plasma cells to synthesize and secrete various immunoglobulins, helper T cells (Th) differentiate into, Th1, Th2, Th17, and Treg cells on the basis of the cytokine microenvironment. Th1 cytokine secretion was observed to contribute to the increase of Th17 infiltration and IL-17 production in synovial tissues during RA.158 Follicular helper T (Tfh) cells, a subset of CD4+ T cells, promote germinal center (GC) responses by providing the signals needed for high-affinity antibody generation and production of long-life antigen-secreting plasma cells.159 In various systemic autoimmune diseases, uncontrolled expansion of Tfh cells has been observed, and in particular, the frequency of circulating Tfh-like cells, their subtypes, and synovially infiltrating T helper cells correlates with the disease process in patients with RA160; Osteoclast generation is triggered by monocytes and macrophages via receptor activator of nuclear factor κB ligand (RANKL), and fibroblasts following direct interaction with the RANK receptor on dendritic cells, macrophages, and pre-osteoclasts.161,162,163 Bony erosions occur at the so-called bare area of the junction between cartilage, periosteal synovial membrane, and bone.164 Cytokines bind to homologous receptors to trigger plenty of intracellular signal transduction events, causing the activation of genes coding for systems that can aggravate inflammation and cellular and tissue damage.165

Cytokine signaling and anti-rheumatic drugs in RA. In the presence of certain environmental or genetic factors, a stepwise progression from activation of innate immunity can be achieved by stimulating DCs, then recruiting and activating T cells, which in turn stimulate B cells, macrophages, synoviocytes, chondrocytes, and osteoclasts to secrete pro-inflammatory and bone-destroying cytokines (i.e., IL-1β, IL-6, TNF-α, and matrix metalloproteinases (MMPs), resulting in bone and cartilage damage accompanied by synovial membranes thickening and angiogenesis, in the synovium and adjacent bone marrow, and the integration of adaptive and innate immune pathways to promote tissue remodeling and damage drives the chronic phase of RA. Clinically, drugs that target inflammatory cytokine signaling are commonly applied
Signaling pathways in the pathogenesis of RA
Multiple signal transduction pathways are involved in the disease progression of rheumatoid arthritis, the major signaling pathways are shown in Fig. 5, and the abnormal signals are often targets for drug discovery.

Main signaling pathways and their inhibitors related to RA. The JAK signaling pathway, Notch signaling pathway, MAPK signaling pathway, Wnt signaling pathway, PI3K signaling pathway, and SYK signaling pathway are the main signaling pathways involved in the process of RA. Related signalings are often the potential targets for drug discovery
The JAK-STAT signaling pathway
The JAK (Janus-activated Kinase)- STAT (Signal Transduction and Activator of Transcription) is one of the most crucial signaling pathways for cytokine signaling, with well-known regarding how TNF-α rapidly induces the target genes expression, for example, the interferon family, gp130 family, common-γ chain family, receptor tyrosine kinases, and some G protein-coupled receptors can induce transduction through the JAK-STAT pathway.166 This signaling pathway is believed to play a crucial character in cell differentiation, proliferation, apoptosis, and immune function and is especially important in regulating inflammation and immune function. Numerous recent studies found that the JAK-STAT signaling pathway is abnormally activated during RA.167,168,169 JAK family has four members: JAK1, JAK2, JAK3, and TyK2 (tyrosine Kinase 2). The four members have different molecular weights and are highly conserved in the evolutionary process. Although JAK3 is only expressed in blood, vascular smooth muscle, and endothelial cells, JAK1, JAK2, and TyK2 are all widely expressed in multi-tissue and multi-system.170,171 Studies have shown that JAK plays an important role in RA.172,173,174 STAT is a family of cytoplasmic proteins with both transcriptional activation and signal transduction functions. The STAT family including STAT1-4, STAT5A, STAT5B, and STAT6. STAT contains six highly conserved functional domains, the N-terminal conserved domain, the helix domain, the DNA-binding domain, the ligation domain, the SH2 domain, and the C-terminal transcriptional activation domain.175,176 Among these regions, the most conserved and functionally important domain is the SH2 domain. The SH2 domain allows for the specific recognition and docking of phosphorylated tyrosines on the cytokine receptors, JAK, and other STAT molecules.177 The N-terminal is important in controlling STAT interaction with other transcription factors while the DNA-binding region determines where STAT interacts with DNA.178
In recent years, the occurrence and progression of RA have been considered highly correlated to the abnormal activation of the JAK-STAT pathway. This pathway is involved in many pathological conditions and seems important in abnormal hyperplasia of RA FLS, synovial inflammation, and bone destruction.179,180,181 Among these components, synovitis is the pathological basis of RA. Persistent synovitis leads to abnormal hyperplasia of synovitis, which leads to the destruction of bone and cartilage. Many inflammatory reactions have been observed in RA synoviums, such as the activation of adhesion molecule genes and cytokines, which are closely correlated to transcription factors in specific signaling pathways.182,183
JAK1 is involved in signal transduction associated with various cytokines, like IFN-γ and IL-6, that bind and form receptor complexes that activate JAK1 kinase and take part in the pathogenesis of RA, vitiligo, and psoriasis.184,185 JAK3 is participated in the signaling pathways linked to IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, and plays an important role in the differentiation, proliferation, and growth of T cells.168 Tyk2 could be activated by IFNs, IL-6, IL-10, IL-12, IL-23, and selective inhibition of Tyk2 plays a role in the RA treatment.186 JAK2 induces downstream activation of STAT3 and STAT5. It is responsible for signaling through multiple receptors, including receptors that play a function in inflammatory and autoimmune responses, such as IL-6R, IL-12Rβ, and IFN-γ R2. JAK2 is known to be related to a variety of diseases, including blood diseases, diabetes, cancer, and autoimmune diseases. Compared with normal healthy people, the expression of JAK2 in synovial tissue of RA patients is significantly increased.187 Similar expression patterns are also reported in collagen-induced arthritis rats, adjuvant arthritis rats, and other animal models. Indeed, Kristine S et al. used CEP-33779 (highly selective JAK2 inhibitor) to intervene in collagen antibody-induced and collagen-induced mouse arthritis models. The levels of cytokines (IFN-γ, IL-12, and TNF-α) and serum IL-2, IL-12, and p-Stat 3 in the synovial fluid of the model mice were significantly decreased following treatment. Indeed, CEP-33779 significantly reduced several histological parameters that demonstrated improvement in arthritis, including matrix erosion, subchondral osteolysis, osteogenesis, synovial hyperplasia, vasculitis, and synovial inflammation.188 These results indicated that JAK2 took part in the pathogenesis of RA, and inhibition of JAK2 could treat RA by inhibiting the generation of cytokines and the T and B-lymphocytes activation. Additionally, receptors phosphorylated by JAKs can also recruit PI3K, thereby activating the PI3K-AKT pathway.182,189
STAT1 is mainly activated by cytokines such as IL-6, IL-10, and IFN-γ and participates in body activities through IFN-γ-mediated signaling pathways. Currently, STAT1 has been proven to play both protective and pathogenic roles in RA synovitis. Still, its expression generally rises in inflammatory arthritis, indicating that the anti-inflammatory and pro-apoptotic effects of STAT1 are insufficient to counteract its pro-inflammatory effects. IFN-λ and IFN-α/β only activate STAT2. It has been shown that STAT2 is involved in RA-associated inflammation through the combination of STAT1 and interferon regulatory factor 9 (IRF-9) to form a heterodimer transcription complex of interferon-stimulated gene factor (ISGF3).190 STAT4 regulates the balance of IL-12 and IL-23 and participates in RA inflammation through the differentiation of CD4+ T cells into Th17 and Th1 cells. Multiple meta-analyses results191,192 have shown that a single nucleotide polymorphism at rs7574865 of the STAT4 gene potentially correlated with RA susceptibility however, more research is needed to confirm this observation. Other components like STAT5, a Treg cells transcription factor, which, together with Foxp3, is responsible for the differentiation of Treg cells. Various studies have found that the effect of STAT5 may be opposite to that of STAT3 since the inhibition of STAT3 can increase the activity of STAT5 and Foxp3. Thus, it can promote the differentiation of Treg cells and control RA arthritis.193
Similarly, STAT6 is activated by IL-4 while IL-13 is activated by IFN-α in B-lymphocytes. In a proteoglycan-induced mouse arthritis model, IL-4 and STAT6 deficiency can significantly increase the severity of arthritis.194 STAT3 is the primary downstream regulator of the gp130 receptor and can be activated by IL-6, IL-10, IFN-α/β, and other cytokines. It can promote chronic arthritis, regulate the abnormal growth and survival characteristics of RA synovial cells, and further aggravate the clinical symptoms of RA. Wang et al.195 and colleagues were the first to observe that STAT3 showed DNA-binding activity in synovial mononuclear cells from patients with inflammatory arthritis. Moreover, Lee et al.196 found that STAT3 can inhibit FLS apoptosis, increase the activity of T cells and promote the production of antibodies, indicating that STAT3 is involved in multiple links of RA pathogenesis. Oike et al.197 found that in collagen-induced arthritis model mice, p-STAT3 was highly expressed in synovium and cartilage. In addition, the inflammatory cytokines IL-17 and IL-6 in serum were significantly reduced after STAT3 inhibitor treatment. Ji Hyeonet al.193 found that STAT3 was strongly expressed in both RA CD4+ T and synovial cells. The activation of STAT3 made synovial cells have tumor-like characteristics and made synovial cells proliferate fast, survive long, and infiltrate surrounding joint tissues. Notably, STAT3 plays an important role in determining RA helper cell differentiation. Indeed, transfection of STAT3 siRNA inhibited CD4+ T-cell differentiation into Th17 cells and increased the proportion of Treg cells.198 All these results show that STAT3 is closely related to articular inflammation and lymphocyte differentiation, and STAT3 might be a new target for the treatment of RA.197
The MAPK signaling pathway and RA
The MAPK (Mitogen-Activated Protein Kinase) signaling pathway contributes to the regulation of various cellular activities, including gene expression, metabolism, migration, survival, cell cycle progression, apoptosis, and differentiation, which plays a key role in the pathological process of RA.199 Its overactivation is closely correlated to the articular cartilage destruction and inflammatory hyperplasia of the synovial tissues. MAPK regulates the expression of multiple genes and has been considered a potential target for treating RA or other immune-mediated chronic inflammatory diseases.200 It can transduce extracellular signals such as growth factors, neurotransmitters, hormones, stress conditions, viruses, and inflammatory factors into the cells201,202 and play a key role in the transduction of extracellular stimulation to drive intracellular responses.203 P38 MAPK, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) are the three main subfamilies of the MAPK pathway.204
The ERK1/2 activates stimuli in response to ischemia, oxidative stress, and neurotransmitters. ERK1 and ERK2 are key to the regulation of cell differentiation, proliferation, and survival.205 The main effect of JNK MAPKs in RA is cartilage destruction mediated by matrix metalloproteinase (MMP).206 Similarly, P38 is the most important member of the MAPK family linked to the inflammatory response in rheumatoid arthritis. After inflammatory stimulation, p38 is activated and induced in endogenous immune cells such as neutrophils and monocytes. P38 then undergoes nuclear translocation, where it phosphorylates and activates many protein kinases and transcription factors that play key roles in the regulation of humoral and cellular autoimmune responses. In the synovial tissue of RA, p38 is activated and highly expressed by MKK3 and MKK6,207 and commonly used p38 MAPK inhibitors reduce the generation of pro-inflammatory cytokines in neutrophils, macrophages/monocytes, and T lymphocytes.203 P38 MAPK activates and moves into the nucleus, phosphorylating transcription factors such as ATF2, MEF2C,208,209 and these initiate cascades that induce a large increase in inflammatory chemokines like IL-8 and monocyte chemoattractant protein-1 (MCP-1), resulting in synovial thickening. p38 MAPK can also inhibit cell apoptosis. Yu et al.210 found that Integrin activation-induced phosphorylation of p38 MAPK inhibited Fas protein-mediated-cell apoptosis, resulting in a large number of T cells infiltration in synovial tissue, aggravating RA patients’ disease process. Many T cells infiltrate the synovial tissue, most of which are helper T (Th) cells. Studies have shown that the imbalance between Th1/Th2 cells is a crucial pathogenic factor in RA, and the imbalance of Th1/Th2 cells leads to IL-2 and IL-4, and other cytokines being abnormally secreted. Pujari et al. found that Th1/Th2 cytokine secretion is achieved through the P38 MAPK pathway, and inhibition of p38 activity can change the balance of natural CD4+ T cells preventing the differentiation of Th1/Th2 cell types viz. to inhibit their differentiation to Th1 cell variants. Th17 is a newly discovered helper T-cell subset and is featured by the secretion and production of the inflammatory factor IL-17. In the occurrence and development of RA, the role of Th17/IL-17 is also controversial.211 Hot et al.212 found that IL-17 isoform IL-17A can induce three signaling pathways of MAPK family ERK, p38, and JNK and downregulate transcription factors p65 NF-κB and AP-1. These indicate that MAPK and T-cell-mediated RA have a very complex relationship.213 Therefore, P38 is considered a candidate target for treating rheumatoid arthritis.214
The PI3K-AKT signaling pathway in RA
The PI3K (phosphatidylinositol 3 kinase)-AKT (also known as PKB) pathway is an intracellular pathway that regulates proliferation, metabolism, angiogenesis, and cell survival in response to extracellular signals. The key involved genes are PI3K and Protein kinase B (PKB).215,216,217 PI3K can phosphorylate PIP2 to PIP3 by adding a phosphate group, and phosphatases, such as PTEN, can dephosphorylate PIP3 back to PIP2.218 This cycle thereby terminates PI3K signaling. The downstream effects of PI3K are mainly reflected in the regulation of PIP3. In the PI3K-AKT pathway, the phosphate group at position 3 of PIP3 can simultaneously recruit PDK1 and AKT proteins to the plasma membrane, causing PDK1 to phosphorylate threonine at position 308 (T308) of AKT protein. This contributes to the activation of AKT, which further activates the downstream regulatory pathways.215,219
It has been proven that the PI3K/AKT signaling pathway is correlated with the occurrence and development of RA. It can participate in the unusual proliferation of FLS cells and synovial inflammation by stimulating the expression of inflammatory molecules like IL-1β, IL-6, IL-17, IL-21, IL-22, and TNF-α, which constitute the most important pathogenesis of RA pathological changes.220,221,222,223,224 IL-17, TNF-α, and other cytokines are involved in the generation of osteoclasts, which destroy articular cartilage and bone, resulting in joint stiffness and deformity.225 Abnormal PI3K/AKT pathway activation will also stimulate the expression of VEGF and HIF-1α to promote angiogenesis, which not only isolates bones from getting nutrients through the synovium but also gets involved in the release of diverse inflammatory mediators, aggravating the condition of RA.226,227,228 There is evidence indicating that the PI3K/AKT/mTOR pathway participates in the process of RA, the mammalian target of rapamycin (mTOR) inhibits autophagy in FLS, promotes continuous abnormal proliferation of synovial cells, and is also critical for the survival and differentiation of osteoclasts, aggravating RA and mTOR might be a target for RA or other autoimmune diseases.227,229,230,231
SYK signaling pathway in RA
SYK (spleen tyrosine kinase) is a central molecule of B-cell receptor signaling. The level of phosphorylated SYK in peripheral blood B cells of RA patients is dramatically increased. Among these patients, also show strong positive autoantibodies against citrullinated peptides.232 B cells and autoantibodies are produced in most patients of RA and play a crucial role in the pathogenesis of RA.
BTK (Bruton’s tyrosine kinase) belongs to the Tec family of non-receptor tyrosine kinases, which is expressed in all hematopoietic cells, such as B cells and myeloid cells, except T cells and natural killer cells. BTK is a key molecule linking B-cell receptor (BCR) signaling, chemokine receptor signaling, and Toll-like receptor (TLR) signaling, and is involved in regulating B cells.233
In antigen-dependent BCR signaling, BTK can be activated by SYK or PI3K and participate in regulating B-cell survival and proliferation.234 In antigen-independent TLR signaling, most TLRs recruit MYD88 in response to the TLR ligand.235 In chemokine receptor signaling, CXCL12, which is overexpressed in the germinal centers and bone marrow, directly interacts with CXCR4-linked heterotrimeric G protein subunits through BTK, binds to CXCR4, and induces BTK activation.236 In addition, BTK can directly interact with five distinct molecules to promote antibody secretion, class switch recombination, cell proliferation, and generation of pro-inflammatory cytokines, regulating B-cell migration, adhesion, and tumor microenvironment forces.233 As previously described, RA is a systemic autoimmune disease involving dysregulation of T and B lymphocyte proliferation, and dysregulation of B cells via BCR signaling drives the generation of autoantibodies and inflammatory cytokines, thus promoting the progression of RA.237 Elevated levels of phosphorylated BTK have been found in peripheral B cells of RA patients. Meanwhile, in RA patients with rheumatoid factor (RF) positive, the level of phosphorylated BTK was correlated with RF titer.237,238 BTK mediates bone resorption by RANK and regulates osteoclast proliferation and differentiation, which is the main factor in the pathophysiological level of BTK phosphorylation by peripheral B cells in RA patients.237,239 Therefore, BTK is one of the most attractive targets for treating autoimmune diseases including RA.240,241,242,243
Wnt signaling pathway in RA
The Wnt (Wingless/Integrated) signaling pathway is a complicated protein network that normally functions in cancer and embryonic development.244 The Wnt/β-catenin signaling pathway is activated and takes part in a variety of pathological symptoms such as maintenance, differentiation, proliferation, and self-renewal in RA.245 Wnt also plays a key role in synovial inflammation and in the regulation of bone metabolism in RA.246 Wnt family secreted proteins, Frizzled family transmembrane receptor protein Dishevelled (Dsh), glycogen synthesis kinase 3 (GSK3), β-catenin, APC, Axin, and TCF/LEF family transcriptional regulators constitute the classical wnt pathway247; In the non-classical Wnt pathway such as Wnt-Frizzled/PCP signal conduction, Dsh signals through the Rac1 axis and Daam1-RhoA axis.248
NAV2 belongs to the neuro-guiding protein family, and the proteins encoded by it contain multiple functional domains, such as the CH domain, CC domain, CSID domain, and AAA domain.249 These functional domains involve a series of cellular processes, including signal transduction, gene expression regulation, protein degradation, membrane fusion, microtubule and filament dynamics, and cell migration.250,251,252,253 If the above cellular processes are abnormal, they may affect the normal cell function of biological individuals and lead to diseases. Previous studies have proven that NAV2 plays a key role in the development of the mammalian nervous system, resulting in abnormal nerve fiber density and in causing developmental defects of nerves in early embryos following NAV2 deletion.254 NAV2 is also an indispensable protein molecule in the outward growth of human neuroblastoma cells induced by all-trans retinoic acid.255
Our group demonstrates for the first time that NAV2 promotes the fibrocyte-like synoviocytes inflammatory response by activating Wnt/β-catenin signaling256 and the SSH1L/Cofilin-1 signaling pathway in rheumatoid arthritis. We also hypothesized that NAV2 might affect inflammation during RA disease progression and the cell-cell interaction in sensitizing joint-innervating neurons that contribute to arthritis pain.257 Although our studies first indicate that inhibition of NAV2 expression prevents RA progression and reverses inflammation-related phenotypes, we proposed that NAV2 is a novel promising intervention target for RA treatment.
Notch signaling pathway in RA
Notch genes encode a class of cell-surface receptors which is highly conserved and regulate the development of cells in various organisms, from sea urchins to humans. Notch signaling affects numerous processes of normal cell morphogenesis, including cell proliferation, the differentiation of pluripotent progenitors, apoptosis, and the formation of cell boundaries.258 The Notch signaling pathway comprises Notch receptors, Notch ligands (DSL proteins), CSL (CBF-1, Suppressor of hairless, Lag), DNA-binding proteins, other effectors, and Notch regulatory molecules. Mammals have 4 Notch receptors (Notch-1-4) and 5 Notch ligands (Delta-like 1, 3, 4, Jagged1, and Jagged2). The Notch signal is generated by interacting with the Notch ligand of the adjacent cell and the receptor. The Notch protein undergoes cleavages and released the Notch intracellular domain (NICD) into the cytoplasm, followed enters into the nucleus to combine with the transcription factor CSL to form NICD/ CSL transcriptional activation complex activates the target genes of the basic-helix-loop-helix (bHLH) transcriptional repressor family such as HES, HEY, and HERP, and plays a biological role.259 Notch signaling expression and activation stimulate synoviocytes, macrophages, and fibroblast-like synoviocytes to secrete pro-inflammatory cytokines that exacerbate RA.260,261,262,263 Th17 cell differentiation is impaired when blocked Notch signaling.264,265,266 Notch-1 directly binds to the IL-17 and ROR-γT promoters to regulate Th17 differentiation.267 Notch-3 plays a key role in the antigen-specific T-cell differentiation, and Notch-3 blockade inhibits Th17 and Th1 cell activation in CIA mice.268 Notch-3 was also found to be remarkably upregulated in synovial fibroblasts, and in mice model, blocking Notch-3 signaling reduces inflammation and prevents joint injury.269 Targeting Notch was found to minimize associated tissue damage while reducing inflammation.262,270
NF-κB and other transcription factors in RA
A variety of transcription factors, like NF-κB, Nrf2, HIF, and AP-1, are closely related to the pathogenesis of RA.271 The expression of NF-κB in the synovium of RA patients was significantly increased. Activated NF-κB induces the generation of several pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, thus accelerating the development of RA. Upregulation of pro-inflammatory cytokines could also modulate NF-κb activation through positive feedback, thereby, a vicious loop is formed, which intensifies RA development.272,273 At the same time, excessive NF-κB activation also induces apoptosis of abnormal FLS cells in RA.274 In the RA synovium inflammatory microenvironment, aberrant apoptosis of FLS is the major factor associated with RA synovium hyperplasia. In FLS, abnormal cell apoptosis further accumulates in joint tissues and debris adheres to cartilage and bone, exacerbating the articular cartilage and bone destruction.275 The expression of NF-κB-dependent genes further activates NF-κB, translocations NF-κB to the nucleus, and induces the target genes expression. HIF is critical for activating inflammatory cells and angiogenesis in RA.276 Ap-1 regulates MMP, cytokine production, and synovial hyperplasia, which is also an essential process in RA.277,278 The transcription factor Fra-1 enhances macrophage-mediated arthritic inflammation by targeting arginase 1.279 Nrf2 is related to chondrogenesis, prostaglandin secretion, osteoblast formation, and ROS production in RA.280 Thus, targeting transcription factor signaling represents a useful treatment strategy for RA. It has been reported that inhibition of NF-κB can inhibit inflammation, angiogenesis, pannus formation, leukocyte maturation, and activation, and osteoclast differentiation, targeting HIF-1α can induce dysregulation of MMP production, inflammatory cell recruitment, and angiogenesis, inhibition of AP-1 can inhibit the production of MMP-1, MMP-3, MMP-9, MMP-13, and IL-1β.271,281 New agents that regulate transcription factor pathways will be potential candidates for treating RA.
The transcription factor GATA4 is an important regulator of the expression of genes specific to cardiac differentiation. Our research group found increased levels of GATA4 in the synovium of patients with RA. This study is the first time to demonstrate that GATA4 plays a key role in regulating VEGF from RA FLS to induce cell migration, promote cell proliferation, and the formation of angiogenic tubes.282 In addition, this study provided evidence that GATA4 has a previously unknown function as a modulator of RA angiogenesis, and data validate GATA4 as the therapeutic target in RA mice. E2F1, the first transcription factor discovered in the E2F family, mainly exists in dimer binding with Dimerization proteins (DP), which could bind to the promoter region of target genes and regulate the transcription of target genes.283 We found that the transcription factor E2F1 can bind to the Neuron Navigator 2 (NAV2) promoter region, activate NAV2 transcription and expression, and regulate RA through the Wnt/β-catenin signaling pathway.256,284 At the same time, the STAT3-NAV2 axis was found to be a novel therapeutic target for rheumatoid arthritis by activating the SSH1L/ cofilin-1 signaling pathway,285 which may provide therapeutic avenues for reducing pain in RA patients.
Epigenetics regulation in RA signaling pathway
Epigenetics are heritable changes of gene expression without altering the DNA sequence; epigenetics determines which genes are turned on or off. The main mechanisms linked to this process include histone modification, DNA methylation, and non-coding RNA mechanisms.286 These modifications define specific gene expression patterns (Fig. 6). Genetic and environmental factors interact to determine gene expression, especially, cigarette smoking,287,288 a lifestyle that is closely related to the pathogenesis of RA.289,290,291 Fortunately, these epigenetic modifications could be reversed, and the corresponding enzymes which control histone modification or DNA methylation have now been proposed as drug targets for RA.292,293,294

Epigenetic modifications and rheumatoid arthritis. DNA methylation, histone modifications, and-coding RNA mechanisms are often involved in the development of RA
Histone modifications and RA
Histones are proteins that help DNA package to form nucleosomes, and these structures further assemble into chromosomes in the nucleus of cells. Histone modifications at the N-terminal tail include ubiquitination, acetylation, methylation, phosphorylation, deamidation, and ADP ribosylation.295 The key modification sites are lysine and arginine. Histone modifications could inhibit or activate gene expression. Currently, many studies have applied histone modification to multiple diseases, especially in the field of cancer research, which offers new ideas for treating these diseases. Several studies have compared the differences in histone lysine methylation patterns between Osteoarthrosis synovial fibroblasts (OASFs) and Rheumatoid arthritis synovial fibroblasts (RASFs). These studies have detected dozens of histone lysine methyltransferases (HKMTs) and histone lysine demethylases (HKDMs). The results indicated that the expression of HKMTs and HKDMs in OASF and RASF were different at the mRNA level, suggesting that histone lysine methylation (HKM) could influence RASF gene expression.296 NAD-dependent deacetylase sirtuin-1 (SIRT1) is the most frequently explored member of the Nuclear-localized type III histone deacetylases (Sirtuin) family. SIRT1 is participated in several stages of rheumatoid arthritis, in which overexpression leads to-inflammatory cytokine generation and apoptosis resistance in the synovium of rheumatoid arthritis.297 Histone deacetylases (HDAC) are another Star family that has been widely studied. Studies have proven that HDAC1 participates in producing pro-inflammatory factors, and the elimination of HDAC1 in T cells has a protective function on mice with collagen arthritis.298 HDAC inhibitors can inhibit the activation of FLS, and the HDAC1 and HDAC2 expression in RA synovial fibroblasts (RA-SF) is higher than that in OA synovial fibroblasts (OASF).299
Research from our group has demonstrated that HDAC6 protein levels in the adjuvant-induced arthritic rats’ synovium tissues are increased.300 Interestingly, in animal RA models, HDAC inhibitors can improve joint swelling and synovial inflammation and reduce RA symptoms. This evidence provides novel ideas for RA treatment.301,302 Our team also found that in PDGF-induced FLS, the expression of the Jumonji C histone demethylase family (JMJD3) is increased through the Akt signaling pathway, meanwhile, the migration and proliferation ability of FLS is weakened after inhibiting or silencing of JMJD3. Cumulatively, this reduced the rates of CIA.303
Additionally, Non-histone modifications were also observed; for example, Yin et al.304 found that expression of Jmjd1c (a member of the JmjC domain histone demethylase) in B cells was found to protect mice from rheumatoid arthritis. In human B cells with RA, the expression levels of Jmjd1c are inversely correlated with plasma cell levels and disease severity, and Jmjd1c demethylates STAT3 but not histones to inhibit plasma cell differentiation. Meanwhile, our group found histone methyltransferase Smyd2-mediated TRAF2 methylation promotes inflammatory diseases (including RA) through the NF-κB signaling pathway.305 which might also provide some insight for treatment strategies.
DNA methylation and RA
DNA methylation is under DNA methyltransferase (DNMT) catalytic action and transfers the methyl group from S-adenosine methionine (SAM) to the DNA sequence. DNA methylation occurs at the Cytosine of CpG (Cytosine-phosphoric acid guanine) islands to produce 5MC, most of which are located in the promoter region.306 Abnormal hypermethylation of the CpG islands will prevent transcription factors from binding to the promoter and lead to gene silencing.307,308 DNA methylation is considered a potential therapeutic target due to its reversibility. Indeed, DNA methylation levels in synovial tissue of RA and OA patients are not significantly different. However, DNA methylation levels are reported to be lower in RA patients with peripheral blood mononuclear cells (PBMC).309 Furthermore, in PBMC, abnormal cytosine methylation occurs in the promoter regions of IL-6 and IL-10, affecting transcription.310,311 Indeed, studies have shown that DNA methylation levels in T cells and monocytes in RA patients are lower than those in healthy subjects.312,313 Hypermethylation of the promoter region act as a marker of heterochromatin, which affects the binding of DNA to transcription factors and inhibits gene transcription. The specific recombination of methyl groups in the synovial fibroblasts of RA occurs during the development of the disease. DNA methylation reduction is often found in highly proliferative tissues and is related to the methyl group donor molecule, SAM. In addition, the hypomethylation of DNA leads to the increased expression of extracellular matrix proteins, growth factors/receptors, matrix-degrading enzymes, and adhesion molecules. Therefore, in proliferating tissues, these are usually used as markers to identify whether cell proliferation is occurring. The methylation level of cells seems to be affected by the inflammatory environment in which they are located. Some studies stimulated FLS with IL-1 and TNF-α, and the results show that the methylation level of RA-FLS is significantly lower than that of OA-FLS. Moreover, 5-methylcytosine levels were increased.314,315 In RA-FLS, the T-box transcription factor 5 promoter region differs from OA-FLS in methylation status.
Furthermore, the promoter region of its downstream gene Chemokine CxCL12 also shows high rates of hypomethylation in RA-FLS.315 Cribbs et al. revealed that the compromised function of Treg in RA patients is associated with the hypermethylation of a specific region in the promoter of the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4;−658 CpG) as compared with healthy controls.316 DNA hypermethylation prevents the binding of NFATc2 to CTLA-4 and decreases CTLA-4 expression. Consequently, Treg cells lose their function to promote the activation and expression of tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO). In turn, these cells fail to activate the immunomodulatory pathway. These results indicate that small changes in methylation can impact different cell types in RA.
MicroRNAs and RA
MicroRNAs occupy an important position in modifying non-coding RNAs; MicroRNA (miRNA) is a kind of non-coding RNA molecule.317,318 Initially, the miRNA gene is transcribed to form a primary miRNA in the nucleus, cleaved by Drosha to form a precursor -miRNA. By Exportin-5, miRNA is exported to the cytoplasm and cleaved by Dicer to form mature miRNA duplexes, which are then unfolded, and a miRNA strand is added to the RNA-induced silencing complex.319,320 Combining miRNAs and their target mRNAs result in transcriptional repression or mRNA decay.321 MicroRNAs have been implicated in the occurrence and progression of many diseases, especially cancer,322 but now research points focus on their roles in immunological diseases.323 The expression of various miRNAs changed during the development of RA, including miR-146a, miR-155, miR-222, miR-223, miR-203,324,325,326,327,328,329 and miR-132, miR-155, and miR-146a might be potential biomarkers of response to methotrexate treatment in patients with RA.330 Some miRNAs can influence RA by regulating the function of FLS. For instance, miR-203 is upregulated in RA-FLS and induces RA by promoting the generation of MMP-1 and IL-6.331 The expression of miR-19 was upregulated in TNF-α-stimulated FLS, and the inflammatory response was mediated by regulating TLR2, IL-6, and MMP-3.332,333 Many miRNAs are also down-regulated in RA, for example, miR-10a by RAFLS, and can regulate the production of inflammatory factors through the NF-κB signaling pathway.334 The expression of miR-19 decreased in lipopolysaccharide-stimulated RAFLS, and its anti-inflammatory effect is induced by the regulation of IL-1β, IL-6, and other inflammatory factors.335 In animal models, miR-124a can inhibit RA symptoms in rat AIA models by reducing synovial cell proliferation and alleviating cartilage or bone destruction.336 MicroRNAs levels before and after anti-TNF-α combination therapy are potential new biomarkers for monitoring and predicting intervention outcomes. For example, miRNA-23-3p, miRNA-16-5p, miRNA125b-5p, miRNA-146A-5P, miRNA-126-3p, and miRNA-223-3p were found to be significantly upregulated after anti-TNF-α treatment. Interestingly, only responders showed an increase in these miRNAs after treatment, consistent with a decrease in C-reactive protein (CRP), rheumatoid factor (RF), TNF-α, interleukin (IL)-6, and IL-17.337
Targeted therapy for RA
Treatment for rheumatoid arthritis could help relieve pain, reduce joint inflammation, prevent or slow joint injury, reduce disability and keep patients as active as possible. Although rheumatoid arthritis has no cure, an early drug intervention could reduce the risk and pain of joint damage and slows the progression of the disease. In general, non-steroidal anti-inflammatory drugs (NSAID), glucocorticoids (GCs), and disease-modifying anti-rheumatic drugs (DMARDs) are applied in clinical RA treatment, and some cutting-edge technology therapies are emerging for targeted therapy.
DMARDs are subdivided into conventional synthetic DMARDs (csDMARDs), biologic DMARDs (bDMARDs), and targeted synthetic DMARDs (tsDMARDs).338 The principal approved drugs, the drugs currently being assessed in clinical trials, and various pre-clinical drugs for RA treatment are highlighted in Table 1.
Approved drugs
In clinical, NSAIDs like Naprelan (naproxen sodium), Mobic (meloxicam), and Duexis (ibuprofen and famotidine) inhibit cyclooxygenase (COX) activity, thereby inhibiting prostaglandins (PGs) synthesis and producing antipyretic and analgesic effects used for relief of the symptoms and pain of rheumatoid arthritis.339,340,341,342,343,344,345 In the case of excessive use or overtime use of NSAIDs, there will be leukopenia, thrombocytopenia, etc., and digestive tract lesions, such as stomach pain, gastric ulcer, and even ulcer bleeding (so famotidine in Duexis is used for inhibition of gastric secretion), liver damage, kidney damage, etc.346 GCs and Rayos (prednisone) delayed-release tablets also help relieve symptoms and benefit RA patients.347,348,349,350 However, the treatment of hormone drugs can easily lead to side effects like endocrine disorders, osteoporosis, obesity, and decreased immunity.351
Methotrexate (MTX) is the most commonly used csDMARDs and has been regarded as a first-line drug for years. The chemical structure of MTX is similar to folic acid, an antifolate drug. This molecule was originally used for tumor chemotherapy, and low dosages are used for rheumatoid therapy. MTX appears to involve multiple mechanisms, including inhibition of interleukin-1-β binding to the cell-surface receptors,352,353 the inhibition of purine metabolism enzymes, leading to adenosine accumulation and inhibition of T-cell activation and the expression of adhesion molecule. Other studies indicate increased sensitivity of activated T cells to CD95, the selective downregulation of B cells, and the inhibition of methyltransferase activity contributing to the inactivation of enzymes related to immune system function.354,355 Leflunomide is a prodrug, which can be rapidly converted into active metabolites in the body after taking it to inhibit the dihydroorotate dehydrogenase activity and affect the synthesis of pyrimidine in activated lymphocytes, thereby exerting an anti-inflammatory effect.356,357 Azulfidine (sulfasalazine) has the dual effects of anti-inflammatory and antibacterial and could also inhibit the synthesis of immune complexes and rheumatoid factors, thereby alleviating the immunopathological damage of rheumatoid arthritis.358,359,360,361
The bDMARDs, Actemra (tocilizumab) and Kevzara (sarilumab) are interleukin-6 (IL-6) receptor antagonists,362,363,364,365,366,367,368 and the IL-1 receptor antagonist Kineret (anakinra)369,370,371,372 were approved by the FDA for moderate to severely active rheumatoid arthritis adult patients. Enbrel (etanercept) relieves inflammation in RA patients by binding tumor necrosis factor (TNF).373,374,375 Humira (adalimumab) is also working as a tumor necrosis factor (TNF) blocker,376,377,378 and Remicade (infliximab) blocks the activity of TNF-α.379,380,381 In addition, Simponi (golimumab) binds to both the transmembrane and soluble bioactive forms of human TNF-α, thus preventing TNF-α binding to its receptors382,383,384 in the case of treating RA. Orencia (Abatacept) is a selective costimulatory regulator that inhibits T-cell (T lymphocyte) activation by binding to CD86 and CD80, thus blocking the interaction with CD28.385,386,387 This interaction provides the necessary costimulatory signals to fully activate T lymphocytes, which are participated in the pathogenesis of RA.388 Rituxan (Rituximab) is a monoclonal antibody targeting the surface CD20 antigen of pre-B and mature B-lymphocytes. Upon binding to CD20, rituximab leads to antibody-dependent cell-mediated cytotoxic (ADCC) and complement-dependent cytotoxic (CDC) lysis of B cells.389,390,391,392 B cells are proven to play a role in the pathogenesis of RA and related chronic synovitis, including the production of RF or other autoantibodies, antigen-presenting, T-cell activation, and related pro-inflammatory cytokine generation.393
The tsDMARDs are the latest drugs for RA treatments; The US Food and Drug Administration (FDA) approved some JAK inhibitors in clinical use. These small molecules help prevent an individual’s immune system from producing certain enzymes that stimulate inflammation.394 Tofacitinib, a JAK pathway inhibitor developed in 2012, is FDA-approved and marketed under the brand name Xeljanz. Tofacitinib citrate is approved for medical use “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to or are intolerant of methotrexate”.395,396,397,398,399 Baricitinib, sold under Oluminant, acts as a JAK1 and JAK2 inhibitor. In May 2018, the FDA-approved baritinib for treating moderate-to-severe active rheumatoid arthritis patients who have not responded adequately to treatment with one or more TNF antagonists.400,401,402,403 Upadacitinib, marketed under the brand name Rinvoq, is a JAK inhibitor drug FDA-directed for the treatment of adults with psoriatic arthritis and moderate to severely active rheumatoid arthritis who do not or may not respond to methotrexate.404,405,406 JAK1 inhibitor Filgotinib has been approved for the treatment of RA in the European Union and Japan.407,408,409,410 Peficitinib is a JAK3 inhibitor for treating RA recently approved in Japan.411,412 Peficitinib attenuates RA symptoms and inhibits joint destruction in Japanese RA patients who do not respond adequately to MTX.413,414 Iguratimod, which inhibits the activation of NF-κB, is a novel DMARDs approved for the treatment of RA in Japan and China.415,416,417,418,419
NSAIDs are generally only used in the early stage to reduce symptoms of the disease, or until the diagnosis of RA is established, methotrexate is usually combined with GCS for a period of time to control inflammation and gradually reduces the use of GCs, under the initial treatment regimen, about 30–50% of patients with rheumatoid arthritis are in remission, and other csDMARDs are usually added if treatment purpose is not achieved within 3–6 months with methotrexate monotherapy.26,420 If treatment has not achieved the desired results, it is usually combined with methotrexate and biological or targeted synthetic DMARDs.421 This combination therapy could control an additional 30%- 40% of patients with rheumatoid arthritis.5
Common side effects of currently available DMARDs, in addition to cytopenia, liver damage, and elevated cholesterol, both targeted synthesis and biological DMARDs lead to increased frequency of infection, which may be caused by inhibition of their respective inflammatory mediators.338
Of note, targeted synthetic or biologics DMARDs are not supposed to be regarded as first-line therapy because most patients who respond to these drugs also respond to methotrexate treatment alone. Meanwhile, methotrexate is associated with lower cost, lower side effects, and infection frequency compared with targeted synthetic or biological DMARDs.422 Individual patient disease status and treatment outcomes are continually reassessed throughout treatment; it is essential to make adjustments in time.
Drugs in clinical trials
Cohen et al. are developing an experimental drug called Fenebrutinib that blocks the action of Bruton’s tyrosine kinase (BTK). When recently assessed in a phase 2 trial, Fenebrutinib effectively treated patients with RA who had no response to other therapies. Compared with the popular RA drug Humira (Adalimumab), Finebrutinib showed similar efficacy.423 Although more research is needed, scientists are excited about the potential of BTK inhibitors to help RA patients. Similarly, JAK inhibitors are still popular targets for developing RA drugs, and several other JAK inhibitors have proceeded into clinical trials for RA treatment. Ruxolitinib is a selective JAK1/JAK2 inhibitor424 and has been demonstrated for treating psoriasis and myeloproliferative diseases.425 Rusolitinib is generally safe in patients of RA and normal volunteers and has now completed phase 2 clinical trials (NCT00550043). A recent study has assessed the safety and efficacy of a selective inhibitor of JAK1 SHR0302426 in rheumatoid arthritis patients, and this molecule has now entered a Phase 3 clinical trial (NCT04333771). In addition, a phase 2/3 study is underway to assess the safety and long-term efficacy of the JAK3 inhibitor VX-509427,428,429 in rheumatoid arthritis patients (NCT01830985). These drugs are expected to be available soon.
In recent years, many P38 kinase inhibitors have entered clinical trials, unfortunately, no effective inhibitors have been identified. The P38 MAPK inhibitor, VX-702, has shown mediocrity clinical efficacy and transiently inhibits inflammatory factors. However, but appears not to promote sustained inhibition of the chronic inflammation in RA.430 Other compounds like SCIO-469 show no differences in efficacy when compared to the placebo treatment in RA patients.431 Moreover, the therapeutics, Ph-797804,432 SB-681323,433 and BMS-582949,434 inhibitors of P38, are now in RA treatment clinical trials, but results from these interventions have yet to be reported.
Drugs in pre-clinical studies
PI3K takes part in inflammatory processes and is a potential therapeutic target for RA. GS9901 is a selective oral PI3Kδ inhibitor that proved efficacious in a RA animal model.435 The inhibition of PI3KC2γ expression in macrophages and synovial fibroblasts by PBT-6 suggests that it can be used as a novel inhibitor of PI3KC2γ in inflammatory diseases, including rheumatoid arthritis.436 Inhibition of PI3K by ZSTK474 may inhibit bone destruction and synovial inflammation in RA patients, and the Inhibition efficiency of ZSTK474 is much better than that of LY294002, a commonly used PI3K inhibitor.437 The mTOR inhibitor rapamycin may apply in the treatment of RA, aiming to reduce FLS-mediated joint injury and erosive changes. The combination of mTOR inhibitor and vitamin D3 prevents bone destruction in RA.438,439 Notch signaling inhibitor LY411575 inhibits −1 and Notch-3 for treating collagen-induced arthritis (CIA) in rats.440 Ahmad et al441 have reported that STAT3 inhibitor STA-21 reduced the expression of TNF-α and IL-6 in the peripheral blood of collagen-induced arthritis rats and increased the expression of IL-27 produced by CD14+ cells.
Many researchers have indicated that DNA methylation inhibitors like Azacitidine (5’-AzaC) have the potential to inhibit RA progression. In Table 1, we list several DNMT inhibitors that have been investigated in RA drugs (trials or studies). Azacitidine blocked the release of inflammatory cytokines (TNF-α and IL-6) in RAFLS.442 Decitabine Exhibited a decreasing production of Th1 and Th17 pro-inflammatory cytokines and can reduce anti-type II collagen autoantibodies. In the CIA mouse model,443 Zebularine produced a sustained reduction in the severity of arthritis and promoted the generation of Treg.444 Epigallocatechin-3 gallate (EGCG) inhibits MMP-2, IL-6, and IL-8 production and selectively inhibits COX2 expression in human RAFLS.445 Importantly, these drugs have been shown to inhibit the activation of DNMTs in RA-related studies. 5’-AzaC, zebularine, and decitabine are the same class of drugs that belong to nucleoside-derived inhibitors, which were initially investigated and approved for the treatment of cancer.446,447 With the development of nucleoside-derived inhibitors, they were also studied for use in treating RA patients. It was confirmed that treatment with 5’-AzaC decreased the expression of inflammatory cytokines (i.e., TNF-α and IL-6) in RAFLS.442 Furthermore, 5’-AzaC elevated the anti-inflammatory cytokine IL-10 expression in PBMCs isolated from RA patients. This finding was related to the hypomethylation of the IL-10 promoter.311 In a murine CIA model, decitabine showed inhibitory effects towards the anti-type II collagen autoantibodies production and Th1 or Th17 pro-inflammatory cytokines.443
Different HDAC inhibitors target a few members of the HDAC enzyme family and were claimed to have beneficial effects in the treatment of RA. The selective HDAC3 inhibitor MI192 was proven to inhibit the expression of TNF-α and IL-1β induced by LPS in PBMCs, which were derived from healthy donors and RA patients.448 TSA, a Class I and Class II HDAC inhibitor, induced a remarkable decrease in nuclear retention of NF-κB in RAFLS in the presence of IL-1β stimulation, resulting in the temporal reduction of IL-6 mRNA accumulation.449 In another report, nicotinamide, a Class III HDAC inhibitor reduced LPS-stimulated IL6 and TNF-α expression and TNF-α-induced expression of IL-6 in macrophages isolated from RA patients.450 Furthermore, MS-275 and SAHA, then on-specific HDAC inhibitors suppressed the NF-κB p65 nuclear accumulation, induced by LPS in human RA synovial fibroblastic E11 cells THP-1 monocytes, leading to a reduction in pro-inflammatory cytokines.451 Largazole, a Class I HDAC inhibitor, Enhanced TNF-α-induced expression of VCAM-1 and ICAM-1 in RASF; inhibited TNF-α-induced MMP-2 activity; modulated Class II HDAC expression levels.452 MPT0G009, a Class I HDAC and HDAC6 inhibitor, Reduced PGE2 and IL-6 secretion in RAFLS; reduced paw swelling; reduced osteoclast formation and arthritis scores in AIA rats.453 NK-HDAC1, HDAC1inhibitor, reduced proliferation rates of RAFLS and suppressed TNF-α-induced MMP-3 and IL-6 secretion; increased apoptosis of synoviocytes and delayed the progression of disease in CIA mice.454 Recently, HDAC6-specific inhibitors CKD-L and tubA inhibited the expression of IL-1β and TNF-α and increased the IL-10 expression in PBMCs from RA patients.455 Meanwhile, these compounds inhibited TNF-α secretion in THP-1 cells and reduced the arthritis score in CIA mice. CKD-506, an HDAC6 inhibitor, Reduced the production of IL-6 and TNF-α by activated PBMCs from RA patients; inhibited the production of IL-8, IL-6, MMP-1, and MMP-3 by activated FLS; inhibited the severity of arthritis in a murine model of AIA.456 Interestingly, these studies could not show an association between the use of these HDAC inhibitors and the deacetylation of H3 and H4. Since p53 has been reported as a non-histone protein that can be acetylated by HAT,457 a drive in research to study other non-histone targets of HDACs and HATs, such as c-MYC, NF-κB, STAT3, α-tubulin has occurred.458 The non-histone proteins acetylation and deacetylation play roles in all sorts of human diseases, including RA, cancer, and Parkinson’s disease (PD).459,460,461 Therefore, targeting non-histone proteins might be a promising therapeutic strategy for the treatment of RA.
Additionally, Our research has revealed that the hydrogen sulfide (H2S) donor S-propargyl-cysteine (SPRC, named also as ZYZ-802) could alleviate inflammatory response and inhibit HDAC6 expression in vivo via the HDAC6/MyD88/NF-κB signaling pathway,300 or Nrf2-ARE signaling pathway.462 At the same time, we also found that CSE/H2S can reduce the expression of JMJD3 by inhibiting transcription factor SP-1 and alleviating arthritis.463 SPRC might serve as a potential drug for RA treatment. We have developed two sustained-release donors of hydrogen sulfide,464,465 which has solved the problem of hydrogen sulfide release too fast in conventional formulations. ZYZ-802 is filing for clinical trials by CFDA and FDA now.
Cutting-edge technology therapy
Some cutting-edge technologies are emerging for RA treatment, for example: Targeting protein degradation as a new therapeutic approach by using Proteolysis-targeting chimeras (PROTAC) technology to address diseases caused by abnormal expression of pathogenic proteins. PROTAC molecule can bind both the E3-ubiquitin ligase and the target protein, thereby causing the target protein ubiquitination and degradation.466,467 However, PROTAC delivery and bioavailability remain the biggest obstacles to clinic.468 Addressing these issues will be the focus of many laboratories in the coming years. PROTAC -mediated degradation of JAK has been proposed as a novel and promising therapeutic strategy for rheumatoid arthritis.469
Nanoparticles are new and promising drug delivery systems (DDSs) that are designed to deliver a specific dose of the desired medicine to a particular part of the body. They make it safe to increase the bioavailability of drug compounds by allowing drug-controlled release over time. Targeted drug delivery nanomaterials for RA therapy focus on efficacy at the lesion site through local delivery of active ingredients while sparing normal cells and tissues from off-target toxicity. Polylactic-co-glycolic acid (PLGA) is the most widely used nanoparticle because of its biocompatibility, and the FDA has approved PLGA as a drug carrier,470 some pre-clinical studies indicated a combination treatment of MTX-loaded PLGA nanoparticles and near-infrared irradiated showed a durable and superior therapeutic effect in suppressing arthritis compared with MTX single administration.471 Yang et al. announce the first example of RA treatment using bioactive nanoparticles without any drug loading and highlight the role of folic acid-modified silver nanoparticles (FA-AgNPs) through M1 macrophage apoptosis and M1-to-M2 Macrophage repolarization for targeted RA therapy.472
A research team from St. Louis, USA, used CRISPR-Cas9 genome editing technology to transform induced pluripotent stem cells (iPSCs) and constructed cartilage stem cells called “SMART” (Stem cells Modified for Autonomous Regenerative Therapy) in which cells are implanted with a synthetic gene circuit that is regulated by IL-1 to produce an IL-1 receptor antagonist (IL-1Ra). IL-1 promotes inflammation in arthritis by activating inflammatory cells in the joints. When inflammation occurs, intracellular gene circuits that sense changes in endogenous IL-1 cytokine levels are activated to secrete therapeutic levels of IL-1Ra.473 If one therapeutic drug works better than another in a particular patient, it may be possible to develop individualized therapies by reprogramming chondrocytes.
Although RA remains incurable, the development of DMARDs and refined treatments make RA a generally manageable disease. By using different combinations of DMARDs, many patients experience remission of symptoms. However, there are still a large number of patients still do not respond to available therapies to date, indicating the necessity to develop new drugs and treatment strategies. It is hoped that in the near future, some pre-clinical drugs and strategies will successfully move toward clinical studies, providing more options for RA patients.
Conclusions and prospects
Rheumatoid arthritis is a systemic chronic autoimmune disease,474 characterized by symmetrical articular synovitis. The repeated attacks of articular synovitis and the formation of synovial pannus cause the erosion and destruction of the cartilage and subchondral bone in the affected joints, eventually leading to various deformities of the affected joints and the dysfunction of joint function.475,476,477,478 Generally, NSAIDs, GCs, and DMARDs are used for clinical RA treatment. However, they can only delay the disease progression or improve inflammatory symptoms. Furthermore, since the treatment of RA is a long-term process, the side effects of these drugs are inevitable, including immunosuppression, gastrointestinal ulcers, osteoporosis, nausea, fatigue, cytopenia, rashes, liver damage, infections, and psoriasis.479,480,481,482 Therefore, it is an urgent need to develop novel therapeutic strategies that enhance efficacy and reduce toxicity.
During the disease progression of RA, some pro-inflammatory cytokines trigger signal transductions associated with RA, which lead to the recruitment of innate and adaptive immune cells and the activation of synovial cells. These systems release inflammatory mediators, including IL-1, IL-6, and TNF-α, leading to synovial inflammation and exacerbating disease progression.483 A deeper understanding of the involvement of abnormal signal transduction in RA will provide us with novel strategies to prevent and treat this disease class. With the approval of JAK inhibitors for the treatment of RA,484 kinase inhibitors have become a hot spot in drug research. In this review, we summarized the signaling pathways involved in the RA pathogenesis, new potential targets, and associated inhibitors, such as MAPK, WNT, PI3K/AKT, SYK, and JAK/STAT pathways, respectively. We also indicate new targets such as NAV2. Furthermore, it is raised that P38 inhibitors applied in the treatment of RA are not ideal.430,431 These highlights future challenges in treating RA and the need to identify new specific targets to drive developments in the synthesis of newer selective inhibitors.
Several cutting-edge technologies are appearing for RA therapy, for instance: Targeting protein degradation as a new therapeutic approach by using Proteolysis-targeting chimeras (PROTAC) technology; Nanoparticles were used for drug targeting and sustained-release delivery; CRISPR-Cas9 genome editing technology et.al. Advances in RA treatment have taught us that “one size does not fit all” and that personalized therapy is now the consensus goal.473,485,486
In closing, the current review highlights specific signal transduction pathways and molecular targets that may hold promise in the treatment of RA, also raised the developments in new drugs for use and prospect some cutting-edge technologies in treating RA, hope to provide new ideas for RA’s therapy in the future.
Methods
We reviewed the literature on rheumatoid arthritis up to 2022. PubMed was searched using the terms “rheumatoid arthritis” plus “signaling pathways”, “molecular mechanisms”, “genetic factors”, “epigenetics”, and “therapeutic interventions”. A literature review of the retrieved papers was presented herein.
References
Paget, S. A., Lockshin, M. D. & Loebl, S. The hospital for special surgery rheumatoid arthritis handbook: everything you need to know. (Wiley, 2002).
Senthelal, S., Li, J., Ardeshirzadeh, S. & Thomas, M. A. in StatPearls (StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC., 2022).
Landré-Beauvais, A. J. The first description of rheumatoid arthritis. Unabridged text of the doctoral dissertation presented in 1800. Jt. Bone Spine 68, 130–143 (2001).
Garrod, A.B. The nature and treatment of gout and rheumatic gout. Br. Foreign Med. Chir. Rev. 25, 419–435 (1860).
Smolen, J. S., Aletaha, D. & McInnes, I. B. Rheumatoid arthritis. Lancet 388, 2023–2038 (2016).
Radu, A. F. & Bungau, S. G. Management of rheumatoid arthritis: an overview. Cells 10, 2857 (2021).
Otón, T. & Carmona, L. The epidemiology of established rheumatoid arthritis. Best. Pract. Res. Clin. Rheumatol. 33, 101477 (2019).
Wasserman, A. Rheumatoid arthritis: common questions about diagnosis and management. Am. Fam. Physician 97, 455–462 (2018).
Almutairi, K. et al. The global prevalence of rheumatoid arthritis: a meta-analysis based on a systematic review. Rheumatol. Int. 41, 863–877 (2021).
Safiri, S. et al. Global, regional and national burden of rheumatoid arthritis 1990-2017: a systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 78, 1463–1471 (2019).
Azer, S. A., AlSwaidan, N. M., Alshwairikh, L. A. & AlShammari, J. M. Accuracy and readability of cardiovascular entries on Wikipedia: are they reliable learning resources for medical students? BMJ Open. 5, e008187 (2015).
Alamanos, Y., Voulgari, P. V. & Drosos, A. A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic review. Semin Arthritis Rheum. 36, 182–188 (2006).
Goodson, N. & Symmons, D. Rheumatoid arthritis in women: still associated with an increased mortality. Ann. Rheum. Dis. 61, 955–956 (2002).
Kolarz, K., Targońska-Stępniak, B. & Majdan, M. [Early reumatoid arthritis]. Wiad. Lek. 71, 1061–1065 (2018).
Kay, J. & Upchurch, K. S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 51, vi5–vi9 (2012).
Bykerk, V. P. & Massarotti, E. M. The new ACR/EULAR classification criteria for RA: how are the new criteria performing in the clinic? Rheumatology. 51, vi10–vi15 (2012).
Mjaavatten, M. D. & Bykerk, V. P. Early rheumatoid arthritis: the performance of the 2010 ACR/EULAR criteria for diagnosing RA. Best. Pract. Res. Clin. Rheumatol. 27, 451–466 (2013).
Coutant, F. & Miossec, P. Evolving concepts of the pathogenesis of rheumatoid arthritis with focus on the early and late stages. Curr. Opin. Rheumatol. 32, 57–63 (2020).
Kurko, J. et al. Genetics of rheumatoid arthritis - a comprehensive review. Clin. Rev. Allergy Immunol. 45, 170–179 (2013).
Sparks, J. A. Rheumatoid arthritis. Ann. Intern Med. 170, Itc1–itc16 (2019).
Figus, F. A. et al. Rheumatoid arthritis: extra-articular manifestations and comorbidities. Autoimmun. Rev. 20, 102776 (2021).
Pope, J. E. Management of fatigue in rheumatoid arthritis. RMD Open. 6, e001084 (2020).
Majithia, V. & Geraci, S. A. Rheumatoid arthritis: diagnosis and management. Am. J. Med. 120, 936–939 (2007).
Cush, J. J. Rheumatoid arthritis: early diagnosis and treatment. Med. Clin. North Am. 105, 355–365 (2021).
Zhao, J. & Li, Z. G. The challenges of early diagnosis and therapeutic prediction in rheumatoid arthritis. Int J. Rheum. Dis. 21, 2059–2062 (2018).
Littlejohn, E. A. & Monrad, S. U. Early diagnosis and treatment of rheumatoid arthritis. Prim. Care 45, 237–255 (2018).
Li, X. & Su, Y. [Diagnosis and treatment of early rheumatoid arthritis]. Zhonghua Nei Ke Za Zhi 59, 724–727 (2020).
Cush, J. J. Rheumatoid arthritis: early diagnosis and treatment. Rheum. Dis. Clin. North Am. 48, 537–547 (2022).
da Mota, L. M. et al. Imaging diagnosis of early rheumatoid arthritis. Rev. Bras. Reumatol. 52, 757–766 (2012).
Galvez-Sánchez, C. M. et al. Attentional function in fibromyalgia and rheumatoid arthritis. PLoS One 16, e0246128 (2021).
Thomas, D. C. et al. Orofacial manifestations of rheumatoid arthritis and systemic lupus erythematosus: a narrative review. Quintessence Int. 52, 454–466 (2021).
Rose, J. Autoimmune connective tissue diseases: systemic lupus erythematosus and rheumatoid arthritis. Emerg. Med. Clin. North Am. 40, 179–191 (2022).
Coates, L. C., FitzGerald, O., Helliwell, P. S. & Paul, C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: is all inflammation the same? Semin. Arthritis Rheum. 46, 291–304 (2016).
Cross, M. et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 73, 1316–1322 (2014).
Roodenrijs, N. M. T. et al. Difficult-to-treat rheumatoid arthritis: contributing factors and burden of disease. Rheumatology 60, 3778–3788 (2021).
Kvien, T. K. Epidemiology and burden of illness of rheumatoid arthritis. Pharmacoeconomics 22, 1–12 (2004).
Furneri, G. et al. Systematic literature review on economic implications and pharmacoeconomic issues of rheumatoid arthritis. Clin. Exp. Rheumatol. 30, S72–S84 (2012).
Kobelt, G. & Jönsson, B. The burden of rheumatoid arthritis and access to treatment: outcome and cost-utility of treatments. Eur. J. Health Econ. 8, 95–106 (2008).
Kitas, G. D. & Gabriel, S. E. Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives. Ann. Rheum. Dis. 70, 8–14 (2011).
Woolf, A. D. & Pfleger, B. Burden of major musculoskeletal conditions. Bull. World Health Organ. 81, 646–656 (2003).
Sokka, T. et al. Work disability remains a major problem in rheumatoid arthritis in the 2000s: data from 32 countries in the QUEST-RA study. Arthritis Res. Ther. 12, R42 (2010).
Hu, H., Luan, L., Yang, K. & Li, S. C. Burden of rheumatoid arthritis from a societal perspective: a prevalence-based study on cost of this illness for patients in China. Int J. Rheum. Dis. 21, 1572–1580 (2018).
Galloway, J. et al. The impact of disease severity and duration on cost, early retirement and ability to work in rheumatoid arthritis in Europe: an economic modelling study. Rheumatol. Adv. Pract. 4, rkaa041 (2020).
Xavier, R. M. et al. Burden of rheumatoid arthritis on patients’ work productivity and quality of life. Adv. Rheumatol. 59, 47 (2019).
Langley, P. C. et al. The impact of rheumatoid arthritis on the burden of disease in urban China. J. Med. Econ. 14, 709–719 (2011).
Zhang, W. & Anis, A. H. The economic burden of rheumatoid arthritis: beyond health care costs. Clin. Rheumatol. 30, S25–S32 (2011).
Gregersen, P. K., Silver, J. & Winchester, R. J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 30, 1205–1213 (1987).
Wysocki, T., Olesińska, M. & Paradowska-Gorycka, A. Current understanding of an emerging role of HLA-DRB1 gene in rheumatoid arthritis-from research to clinical practice. Cells 9, 1127 (2020).
Abbasifard, M., Imani, D. & Bagheri-Hosseinabadi, Z. PTPN22 gene polymorphism and susceptibility to rheumatoid arthritis (RA): Updated systematic review and meta-analysis. J. Gene Med. 22, e3204 (2020).
Perdigones, N. et al. Evidence of epistasis between TNFRSF14 and TNFRSF6B polymorphisms in patients with rheumatoid arthritis. Arthritis Rheum. 62, 705–710 (2010).
Raychaudhuri, S. et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44, 291–296 (2012).
Weyand, C. M. & Goronzy, J. J. Association of MHC and rheumatoid arthritis. HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res. 2, 212–216 (2000).
Kerlan-Candon, S. et al. HLA-DRB1 gene transcripts in rheumatoid arthritis. Clin. Exp. Immunol. 124, 142–149 (2001).
Kapitány, A. et al. Association of rheumatoid arthritis with HLA-DR1 and HLA-DR4 in hungary. Ann. N. Y Acad. Sci. 1051, 263–270 (2005).
Becart, S. et al. The role of posttranslational modifications in generating neo-epitopes that bind to rheumatoid arthritis-associated HLA-DR alleles and promote autoimmune T cell responses. PLoS One 16, e0245541 (2021).
Auger, I. & Roudier, J. HLA-DR and the development of rheumatoid arthritis. Autoimmunity 26, 123–128 (1997).
van der Helm-van Mil, A. H. et al. An independent role of protective HLA class II alleles in rheumatoid arthritis severity and susceptibility. Arthritis Rheum. 52, 2637–2644 (2005).
Weyand, C. M. & Goronzy, J. J. HLA polymorphisms and T cells in rheumatoid arthritis. Int Rev. Immunol. 18, 37–59 (1999).
McInnes, I. B. & Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219 (2011).
Klein, K. & Gay, S. Epigenetics in rheumatoid arthritis. Curr. Opin. Rheumatol. 27, 76–82 (2015).
Giannini, D. et al. One year in review 2020: pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 38, 387–397 (2020).
Smolen, J. S. et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 4, 18001 (2018).
Turesson, C. et al. Extra-articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46 years. Ann. Rheum. Dis. 62, 722–727 (2003).
Conforti, A. et al. Beyond the joints, the extra-articular manifestations in rheumatoid arthritis. Autoimmun. Rev. 20, 102735 (2021).
Grassi, W., De Angelis, R., Lamanna, G. & Cervini, C. The clinical features of rheumatoid arthritis. Eur. J. Radiol. 27, S18–S24 (1998).
Sharif, K. et al. Rheumatoid arthritis in review: Clinical, anatomical, cellular and molecular points of view. Clin. Anat. 31, 216–223 (2018).
Jevtic, V. & Lingg, G. Differential diagnosis of rheumatoid and psoriatic arthritis at an early stage in the small hand and foot joints using magnetic resonance imaging. Handchir. Mikrochir. Plast. Chir. 44, 163–170 (2012).
Meng, X. H. et al. Rheumatoid arthritis of knee joints: MRI-pathological correlation. Orthop. Surg. 10, 247–254 (2018).
Imagama, T., Tokushige, A., Seki, K. & Taguchi, T. Weight bearing joints destruction in rheumatoid arthritis. Curr. Rheumatol. Rev. 13, 37–42 (2017).
Chen, A. L., Joseph, T. N. & Zuckerman, J. D. Rheumatoid arthritis of the shoulder. J. Am. Acad. Orthop. Surg. 11, 12–24 (2003).
Ngian, G. S. Rheumatoid arthritis. Aust. Fam. Physician 39, 626–628 (2010).
Mok, C. C. Morning stiffness in elderly patients with rheumatoid arthritis: what is known about the effect of biological and targeted agents? Drugs Aging 35, 477–483 (2018).
Boeth, H. et al. Quantification of morning stiffness to assess disease activity and treatment effects in rheumatoid arthritis. Rheumatology 60, 5282–5291 (2021).
Orange, D. E. et al. Rheumatoid arthritis morning stiffness is associated with synovial fibrin and neutrophils. Arthritis Rheumatol. 72, 557–564 (2020).
Jain, S., Mishra, D. & Dhir, V. Determinants of morning stiffness in rheumatoid arthritis: comment on the Article by Orange et al. Arthritis Rheumatol. 73, 174–175 (2021).
Suresh, E. Diagnosis of early rheumatoid arthritis: what the non-specialist needs to know. J. R. Soc. Med. 97, 421–424 (2004).
Schaible, H. G. Nociceptive neurons detect cytokines in arthritis. Arthritis Res. Ther. 16, 470 (2014).
Dave, M. H., Mason, L. W. & Hariharan, K. Forefoot deformity in rheumatoid arthritis: a comparison of shod and unshod populations. Foot Ankle Spec. 8, 378–383 (2015).
Smith, G. C. & Amirfeyz, R. The flexible swan neck deformity in rheumatoid arthritis. J. Hand Surg. Am. 38, 1405–1407 (2013).
Louwerens, J. W. & Schrier, J. C. Rheumatoid forefoot deformity: pathophysiology, evaluation and operative treatment options. Int. Orthop. 37, 1719–1729 (2013).
Adami, G. et al. Osteoporosis in rheumatic diseases. Int J. Mol. Sci. 20, 5867 (2019).
Wysham, K. D., Baker, J. F. & Shoback, D. M. Osteoporosis and fractures in rheumatoid arthritis. Curr. Opin. Rheumatol. 33, 270–276 (2021).
Adawi, M., Firas, S. & Blum, A. Rheumatoid arthritis and atherosclerosis. Isr. Med. Assoc. J. 21, 460–463 (2019).
Liao, K. P. Cardiovascular disease in patients with rheumatoid arthritis. Trends Cardiovasc. Med. 27, 136–140 (2017).
England, B. R., Thiele, G. M., Anderson, D. R. & Mikuls, T. R. Increased cardiovascular risk in rheumatoid arthritis: mechanisms and implications. BMJ 361, k1036 (2018).
Meyer, P. W., Anderson, R., Ker, J. A. & Ally, M. T. Rheumatoid arthritis and risk of cardiovascular disease. Cardiovasc. J. Afr. 29, 317–321 (2018).
Kapoor, T. & Bathon, J. Renal manifestations of rheumatoid arthritis. Rheum. Dis. Clin. North Am. 44, 571–584 (2018).
Pruzanski, W. Renal amyloidosis in rheumatoid arthritis. J. Rheumatol. 34, 889 (2007). author reply 889.
Tilstra, J. S. & Lienesch, D. W. Rheumatoid nodules. Dermatol. Clin. 33, 361–371 (2015).
Lora, V., Cerroni, L. & Cota, C. Skin manifestations of rheumatoid arthritis. G. Ital. Dermatol. Venereol. 153, 243–255 (2018).
Böcher, W., Galle, P. R. & Märker-Hermann, E. [Skin nodules and ulcers of the limbs in a patient with rheumatoid arthritis]. Dtsch. Med. Wochenschr. 127, 735–738 (2002).
Kadura, S. & Raghu, G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 30, 210011 (2021).
Dai, Y., Wang, W., Yu, Y. & Hu, S. Rheumatoid arthritis-associated interstitial lung disease: an overview of epidemiology, pathogenesis and management. Clin. Rheumatol. 40, 1211–1220 (2021).
England, B. R. & Hershberger, D. Management issues in rheumatoid arthritis-associated interstitial lung disease. Curr. Opin. Rheumatol. 32, 255–263 (2020).
Spagnolo, P. et al. The lung in rheumatoid arthritis: focus on interstitial lung disease. Arthritis Rheumatol. 70, 1544–1554 (2018).
Zagora, S. L. et al. Inflammatory eye and rheumatic disease. Int J. Rheum. Dis. 22, 2091–2095 (2019).
Promelle, V., Goeb, V. & Gueudry, J. Rheumatoid arthritis associated episcleritis and scleritis: an update on treatment perspectives. J. Clin. Med. 10, 2118 (2021).
Ouederni, M., Sassi, H. & Cheour, M. Multinodular scleritis in a patient with rheumatoid arthritis. Clin. Rheumatol. 40, 4359–4360 (2021).
Selmi, C., De Santis, M. & Gershwin, M. E. Liver involvement in subjects with rheumatic disease. Arthritis Res. Ther. 13, 226 (2011).
Weinblatt, M. E., Tesser, J. R. & Gilliam, J. H. 3rd The liver in rheumatic diseases. Semin. Arthritis Rheum. 11, 399–405 (1982).
Radovanović-Dinić, B., Tešić-Rajković, S., Zivkovic, V. & Grgov, S. Clinical connection between rheumatoid arthritis and liver damage. Rheumatol. Int. 38, 715–724 (2018).
Wasserman, B. R., Moskovich, R. & Razi, A. E. Rheumatoid arthritis of the cervical spine–clinical considerations. Bull. NYU Hosp. Jt. Dis. 69, 136–148 (2011).
Porchas-Quijada, M. et al. IgG anti-ghrelin immune complexes are increased in rheumatoid arthritis patients under biologic therapy and are related to clinical and metabolic markers. Front. Endocrinol. (Lausanne). 10, 252 (2019).
Weyand, C. M. & Goronzy, J. J. The immunology of rheumatoid arthritis. Nat. Immunol. 22, 10–18 (2021).
Nygaard, G. & Firestein, G. S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 16, 316–333 (2020).
Bustamante, M. F., Garcia-Carbonell, R., Whisenant, K. D. & Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 19, 110 (2017).
Wu, Z. et al. Fibroblast-like synoviocytes in rheumatoid arthritis: surface markers and phenotypes. Int. Immunopharmacol. 93, 107392 (2021).
Marcucci, E. et al. Extra-articular rheumatoid arthritis. Reumatismo 70, 212–224 (2018).
Ridgley, L. A., Anderson, A. E. & Pratt, A. G. What are the dominant cytokines in early rheumatoid arthritis? Curr. Opin. Rheumatol. 30, 207–214 (2018).
McInnes, I. B. & Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 7, 429–442 (2007).
Mateen, S. et al. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 455, 161–171 (2016).
Honda, K. & Littman, D. R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 30, 759–795 (2012).
Aletaha, D., Alasti, F. & Smolen, J. S. Rheumatoid factor, not antibodies against citrullinated proteins, is associated with baseline disease activity in rheumatoid arthritis clinical trials. Arthritis Res. Ther. 17, 229 (2015).
van Delft, M. A. M. & Huizinga, T. W. J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 110, 102392 (2020).
Derksen, V., Huizinga, T. W. J. & van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol. 39, 437–446 (2017).
Scherer, H. U., Häupl, T. & Burmester, G. R. The etiology of rheumatoid arthritis. J. Autoimmun. 110, 102400 (2020).
Zhao, X. et al. Circulating immune complexes contain citrullinated fibrinogen in rheumatoid arthritis. Arthritis Res. Ther. 10, R94 (2008).
Anquetil, F. et al. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J. Immunol. 194, 3664–3674 (2015).
Umeda, N., Matsumoto, I. & Sumida, T. [The pathogenic role of ACPA in rheumatoid arthritis]. Nihon Rinsho Meneki Gakkai Kaishi. 40, 391–395 (2017).
Degboé, Y. Pre-rheumatoid arthritis and ACPA: Contribution of ACPAs in the pathogeny of pre-disease stage. Jt. Bone Spine 88, 105098 (2021).
Malmström, V., Catrina, A. I. & Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat. Rev. Immunol. 17, 60–75 (2017).
Pertsinidou, E. et al. Rheumatoid arthritis autoantibodies and their association with age and sex. Clin. Exp. Rheumatol. 39, 879–882 (2021).
Schellekens, G. A. et al. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. 1998. J. Immunol. 195, 8–16 (2015).
Padyukov, L. et al. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 70, 259–265 (2011).
Muller, S. & Radic, M. Citrullinated autoantigens: from diagnostic markers to pathogenetic mechanisms. Clin. Rev. Allergy Immunol. 49, 232–239 (2015).
Pratesi, F. et al. Fingerprinting of anti-citrullinated protein antibodies (ACPA): specificity, isotypes and subclasses. Lupus 24, 433–441 (2015).
Seri, Y. et al. [Peptidylarginine deiminase type4 (PADI4) role in immune system]. Nihon Rinsho Meneki Gakkai Kaishi. 37, 154–159 (2014).
Reynisdottir, G. et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann. Rheum. Dis. 75, 1722–1727 (2016).
Nielen, M. M. et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 50, 380–386 (2004).
Petrovská, N. et al. The pre-clinical phase of rheumatoid arthritis: from risk factors to prevention of arthritis. Autoimmun. Rev. 20, 102797 (2021).
Molendijk, M., Hazes, J. M. & Lubberts, E. From patients with arthralgia, pre-RA and recently diagnosed RA: what is the current status of understanding RA pathogenesis? RMD Open. 4, e000256 (2018).
Paul, B. J., Kandy, H. I. & Krishnan, V. Pre-rheumatoid arthritis and its prevention. Eur. J. Rheumatol. 4, 161–165 (2017).
Greenblatt, H. K., Kim, H. A., Bettner, L. F. & Deane, K. D. Preclinical rheumatoid arthritis and rheumatoid arthritis prevention. Curr. Opin. Rheumatol. 32, 289–296 (2020).
Böhler, C., Radner, H., Smolen, J. S. & Aletaha, D. Serological changes in the course of traditional and biological disease modifying therapy of rheumatoid arthritis. Ann. Rheum. Dis. 72, 241–244 (2013).
Shi, J. et al. Carbamylation and antibodies against carbamylated proteins in autoimmunity and other pathologies. Autoimmun. Rev. 13, 225–230 (2014).
Wu, C. Y., Yang, H. Y., Luo, S. F. & Lai, J. H. From rheumatoid factor to anti-citrullinated protein antibodies and anti-carbamylated protein antibodies for diagnosis and prognosis prediction in patients with rheumatoid arthritis. Int J. Mol. Sci. 22, 686 (2021).
Wehr, P., Purvis, H., Law, S. C. & Thomas, R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin. Exp. Immunol. 196, 12–27 (2019).
Noack, M. & Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 39, 365–383 (2017).
Burrage, P. S., Mix, K. S. & Brinckerhoff, C. E. Matrix metalloproteinases: role in arthritis. Front. Biosci. 11, 529–543 (2006).
Shim, J. H., Stavre, Z. & Gravallese, E. M. Bone loss in rheumatoid arthritis: basic mechanisms and clinical implications. Calcif. Tissue Int. 102, 533–546 (2018).
Elshabrawy, H. A. et al. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 18, 433–448 (2015).
Wang, Y., Wu, H. & Deng, R. Angiogenesis as a potential treatment strategy for rheumatoid arthritis. Eur. J. Pharmacol. 910, 174500 (2021).
Szekanecz, Z., Besenyei, T., Paragh, G. & Koch, A. E. Angiogenesis in rheumatoid arthritis. Autoimmunity 42, 563–573 (2009).
Bartok, B. & Firestein, G. S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 233, 233–255 (2010).
Neumann, E. et al. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol. Med. 16, 458–468 (2010).
Ospelt, C., Neidhart, M., Gay, R. E. & Gay, S. Synovial activation in rheumatoid arthritis. Front. Biosci. 9, 2323–2334 (2004).
Weyand, C. M. & Goronzy, J. J. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 13, 291–301 (2017).
Owens, B. M. et al. CD90(+) stromal cells are non-professional innate immune effectors of the human colonic mucosa. Front. Immunol. 4, 307 (2013).
Ospelt, C. et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 58, 3684–3692 (2008).
Umetsu, D. T., Katzen, D., Jabara, H. H. & Geha, R. S. Antigen presentation by human dermal fibroblasts: activation of resting T lymphocytes. J. Immunol. 136, 440–445 (1986).
Uehara, A. & Takada, H. Functional TLRs and NODs in human gingival fibroblasts. J. Dent. Res. 86, 249–254 (2007).
Samarpita, S., Kim, J. Y., Rasool, M. K. & Kim, K. S. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res. Ther. 22, 16 (2020).
Mor, A., Abramson, S. B. & Pillinger, M. H. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin. Immunol. 115, 118–128 (2005).
Jones, D. S. et al. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat. Chem. Biol. 13, 38–45 (2017).
Clanchy, F. I. L. & Williams, R. O. Ibudilast inhibits chemokine expression in rheumatoid arthritis synovial fibroblasts and exhibits immunomodulatory activity in experimental arthritis. Arthritis Rheumatol. 71, 703–711 (2019).
Feldmann, M. & Maini, S. R. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol. Rev. 223, 7–19 (2008).
Pandolfi, F. et al. Interleukin-6 in rheumatoid arthritis. Int J. Mol. Sci. 21, 5238 (2020).
Kosmaczewska, A., Swierkot, J., Ciszak, L. & Wiland, P. [The role of Th1, Th17, and Treg cells in the pathogenesis of rheumatoid arthritis including anti-inflammatory action of Th1 cytokines]. Postepy Hig. Med. Dosw. 65, 397–403 (2011).
Lu, J. et al. Follicular helper T cells: potential therapeutic targets in rheumatoid arthritis. Cell Mol. Life Sci. 78, 5095–5106 (2021).
Wang, X. et al. Imbalance of circulating Tfr/Tfh ratio in patients with rheumatoid arthritis. Clin. Exp. Med. 19, 55–64 (2019).
Pettit, A. R. et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159, 1689–1699 (2001).
Redlich, K. et al. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Invest. 110, 1419–1427 (2002).
Geusens, P. The role of RANK ligand/osteoprotegerin in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 4, 225–233 (2012).
Martel-Pelletier, J., Welsch, D. J. & Pelletier, J. P. Metalloproteases and inhibitors in arthritic diseases. Best. Pract. Res. Clin. Rheumatol. 15, 805–829 (2001).
Bergström, B. et al. The rheumatoid arthritis risk gene AIRE is induced by cytokines in fibroblast-like synoviocytes and augments the pro-inflammatory response. Front. Immunol. 10, 1384 (2019).
Bolli, R., Dawn, B. & Xuan, Y. T. Role of the JAK-STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 13, 72–79 (2003).
Simon, L. S. et al. The Jak/STAT pathway: a focus on pain in rheumatoid arthritis. Semin. Arthritis Rheum. 51, 278–284 (2021).
Malemud, C. J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 10, 117–127 (2018).
Angelini, J. et al. JAK-inhibitors for the treatment of rheumatoid arthritis: a focus on the present and an outlook on the future. Biomolecules 10, 1002 (2020).
Lai, S. Y. & Johnson, F. M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist. Updat. 13, 67–78 (2010).
Rawlings, J. S., Rosler, K. M. & Harrison, D. A. The JAK/STAT signaling pathway. J. Cell Sci. 117, 1281–1283 (2004).
Stabile, H. et al. JAK/STAT signaling in regulation of innate lymphoid cells: the gods before the guardians. Immunol. Rev. 286, 148–159 (2018).
Xin, P. et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80, 106210 (2020).
Banerjee, S. et al. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77, 521–546 (2017).
Kisseleva, T., Bhattacharya, S., Braunstein, J. & Schindler, C. W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285, 1–24 (2002).
de Araujo, E. D. et al. Structural Implications of STAT3 and STAT5 SH2 domain mutations. Cancers (Basel). 11, 1757 (2019).
Darnowski, J. W. et al. Stat3 cleavage by caspases: impact on full-length Stat3 expression, fragment formation, and transcriptional activity. J. Biol. Chem. 281, 17707–17717 (2006).
Müller, C. W. DNA recognition by NF kappa B and STAT transcription factors. Ernst Schering Res. Found. Workshop 34, 143–166 (2001).
Hammaker, D. et al. Joint location-specific JAK-STAT signaling in rheumatoid arthritis fibroblast-like synoviocytes. ACR Open Rheumatol. 1, 640–648 (2019).
Qin, Y. et al. Age-associated B cells contribute to the pathogenesis of rheumatoid arthritis by inducing activation of fibroblast-like synoviocytes via TNF-α-mediated ERK1/2 and JAK-STAT1 pathways. Ann. Rheum. Dis. 81, 1504–1514 (2022).
Emori, T. et al. Role of JAK-STAT signaling in the pathogenic behavior of fibroblast-like synoviocytes in rheumatoid arthritis: effect of the novel JAK inhibitor peficitinib. Eur. J. Pharmacol. 882, 173238 (2020).
Bousoik, E. & Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A closer look at JAK/STAT signaling pathway. Front. Oncol. 8, 287 (2018).
Malemud, C. J. Intracellular signaling pathways in rheumatoid arthritis. J. Clin. Cell Immunol. 4, 160 (2013).
Leonard, W. J. & Lin, J. X. Cytokine receptor signaling pathways. J. Allergy Clin. Immunol. 105, 877–888 (2000).
Abdou, A. G., Maraee, A., Yassien, H. & Sarhan, M. Immunohistochemistry of janus kinase 1 (JAK1) expression in vitiligo. J. Pathol. Transl. Med. 52, 363–368 (2018).
He, X. et al. Selective Tyk2 inhibitors as potential therapeutic agents: a patent review (2015-2018). Expert Opin. Ther. Pat. 29, 137–149 (2019).
Monari, C. et al. A microbial polysaccharide reduces the severity of rheumatoid arthritis by influencing Th17 differentiation and proinflammatory cytokines production. J. Immunol. 183, 191–200 (2009).
Stump, K. L. et al. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res. Ther. 13, R68 (2011).
Gotthardt, D., Trifinopoulos, J., Sexl, V. & Putz, E. M. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front. Immunol. 10, 2590 (2019).
Blaszczyk, K. et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 466, 511–524 (2015).
Gao, W. et al. Association between rs7574865 polymorphism in STAT4 gene and rheumatoid arthritis: an updated meta-analysis. Eur. J. Intern. Med. 71, 101–103 (2020).
El-Lebedy, D. et al. Association of STAT4 rs7574865 and PTPN22 rs2476601 polymorphisms with rheumatoid arthritis and non-systemically reacting antibodies in Egyptian patients. Clin. Rheumatol. 36, 1981–1987 (2017).
Ju, J. H. et al. Modulation of STAT-3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of Treg cells. Arthritis Rheum. 64, 3543–3552 (2012).
Finnegan, A. et al. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat-dependent mechanisms. J. Immunol. 169, 3345–3352 (2002).
Wang, F., Sengupta, T. K., Zhong, Z. & Ivashkiv, L. B. Regulation of the balance of cytokine production and the signal transducer and activator of transcription (STAT) transcription factor activity by cytokines and inflammatory synovial fluids. J. Exp. Med. 182, 1825–1831 (1995).
Lee, S. Y. et al. IL-17-mediated Bcl-2 expression regulates survival of fibroblast-like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Res. Ther. 15, R31 (2013).
Oike, T. et al. Stat3 as a potential therapeutic target for rheumatoid arthritis. Sci. Rep. 7, 10965 (2017).
Tripathi, S. K. et al. Genome-wide analysis of STAT3-mediated transcription during early human Th17 cell differentiation. Cell Rep. 19, 1888–1901 (2017).
Ralph, J. A. & Morand, E. F. MAPK phosphatases as novel targets for rheumatoid arthritis. Expert Opin. Ther. Targets 12, 795–808 (2008).
Sujitha, S. & Rasool, M. MicroRNAs and bioactive compounds on TLR/MAPK signaling in rheumatoid arthritis. Clin. Chim. Acta 473, 106–115 (2017).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Kyriakis, J. M. & Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737 (2012).
Gaestel, M. MAPK-activated protein kinases (MKs): novel insights and challenges. Front. Cell Dev. Biol. 3, 88 (2015).
Whitaker, R. H. & Cook, J. G. Stress relief techniques: p38 MAPK determines the balance of cell cycle and apoptosis pathways. Biomolecules 11, 1444 (2021).
Ohori, M. ERK inhibitors as a potential new therapy for rheumatoid arthritis. Drug N. Perspect. 21, 245–250 (2008).
Kanai, T. et al. The JNK pathway represents a novel target in the treatment of rheumatoid arthritis through the suppression of MMP-3. J. Orthop. Surg. Res. 15, 87 (2020).
Zhang, L. L. et al. BAFF, involved in B cell activation through the NF-κB pathway, is related to disease activity and bone destruction in rheumatoid arthritis. Acta Pharm. Sin. 42, 1665–1675 (2021).
Terazawa, S. et al. The attenuated secretion of hyaluronan by UVA-exposed human fibroblasts is associated with up- and downregulation of HYBID and HAS2 expression via activated and inactivated signaling of the p38/ATF2 and JAK2/STAT3 cascades. Int J. Mol. Sci. 22, 2057 (2021).
Zhang, J. et al. Inhibition against p38/MEF2C pathway by Pamapimod protects osteoarthritis chondrocytes hypertrophy. Panminerva Med. (2020).
Lin, Y. P. et al. Aberrant integrin activation induces p38 MAPK phosphorylation resulting in suppressed Fas-mediated apoptosis in T cells: implications for rheumatoid arthritis. Mol. Immunol. 46, 3328–3335 (2009).
Samarpita, S. & Rasool, M. Cyanidin attenuates IL-17A cytokine signaling mediated monocyte migration and differentiation into mature osteoclasts in rheumatoid arthritis. Cytokine 142, 155502 (2021).
Hot, A. et al. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann. Rheum. Dis. 70, 341–348 (2011).
Alam, M. S. et al. Unique properties of TCR-activated p38 are necessary for NFAT-dependent T-cell activation. PLoS Biol. 16, e2004111 (2018).
Pargellis, C. & Regan, J. Inhibitors of p38 mitogen-activated protein kinase for the treatment of rheumatoid arthritis. Curr. Opin. Investig. Drugs 4, 566–571 (2003).
Markman, B., Dienstmann, R. & Tabernero, J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget 1, 530–543 (2010).
Sun, K. et al. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: a narrative review. Osteoarthr. Cartil. 28, 400–409 (2020).
Ersahin, T., Tuncbag, N. & Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 11, 1946–1954 (2015).
Chen, C. Y., Chen, J., He, L. & Stiles, B. L. PTEN: tumor suppressor and metabolic regulator. Front. Endocrinol. (Lausanne). 9, 338 (2018).
Sathe, A. & Nawroth, R. Targeting the PI3K/AKT/mTOR pathway in bladder cancer. Methods Mol. Biol. 1655, 335–350 (2018).
Tsai, C. H. et al. Osteopontin inhibition of miR-129-3p enhances IL-17 expression and monocyte migration in rheumatoid arthritis. Biochim. Biophys. Acta Gen. Subj. 1861, 15–22 (2017).
Shoda, H. et al. Increased serum concentrations of IL-1 beta, IL-21 and Th17 cells in overweight patients with rheumatoid arthritis. Arthritis. Res. Ther. 19, 111 (2017).
Kwok, S. K. et al. Interleukin-21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen-induced arthritis. Arthritis Rheum. 64, 740–751 (2012).
Mitra, A., Raychaudhuri, S. K. & Raychaudhuri, S. P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 60, 38–42 (2012).
Dinesh, P. & Rasool, M. Berberine inhibits IL-21/IL-21R mediated inflammatory proliferation of fibroblast-like synoviocytes through the attenuation of PI3K/Akt signaling pathway and ameliorates IL-21 mediated osteoclastogenesis. Cytokine 106, 54–66 (2018).
Xuan, W. et al. Osteoclast differentiation gene expression profiling reveals chemokine CCL4 mediates RANKL-induced osteoclast migration and invasion via PI3K pathway. Cell Biochem. Funct. 35, 171–177 (2017).
Zou, L. et al. Relationship between PI3K pathway and angiogenesis in CIA rat synovium. Am. J. Transl. Res. 8, 3141–3147 (2016).
Ba, X. et al. WTD attenuating rheumatoid arthritis via suppressing angiogenesis and modulating the PI3K/AKT/mTOR/HIF-1α pathway. Front. Pharmacol. 12, 696802 (2021).
Li, G. Q. et al. PI3 kinase/Akt/HIF-1α pathway is associated with hypoxia-induced epithelial-mesenchymal transition in fibroblast-like synoviocytes of rheumatoid arthritis. Mol. Cell Biochem. 372, 221–231 (2013).
Feng, F. B. & Qiu, H. Y. Effects of Artesunate on chondrocyte proliferation, apoptosis and autophagy through the PI3K/AKT/mTOR signaling pathway in rat models with rheumatoid arthritis. Biomed. Pharmacother. 102, 1209–1220 (2018).
Du, H. et al. A novel phytochemical, DIM, inhibits proliferation, migration, invasion and TNF-α induced inflammatory cytokine production of synovial fibroblasts from rheumatoid arthritis patients by targeting MAPK and AKT/mTOR signal pathway. Front. Immunol. 10, 1620 (2019).
Suto, T. & Karonitsch, T. The immunobiology of mTOR in autoimmunity. J. Autoimmun. 110, 102373 (2020).
Iwata, S. et al. Activation of Syk in peripheral blood B cells in patients with rheumatoid arthritis: a potential target for abatacept therapy. Arthritis Rheumatol. 67, 63–73 (2015).
Corneth, O. B. J., Klein Wolterink, R. G. J. & Hendriks, R. W. BTK signaling in B cell differentiation and autoimmunity. Curr. Top. Microbiol. Immunol. 393, 67–105 (2016).
Smith, C. I. E. & Burger, J. A. Resistance mutations to BTK inhibitors originate from the NF-κB but Not From the PI3K-RAS-MAPK arm of the B cell receptor signaling pathway. Front. Immunol. 12, 689472 (2021).
Rip, J. et al. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front. Immunol. 10, 95 (2019).
Pavlasova, G. & Mraz, M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica 105, 1494–1506 (2020).
Arneson, L. C., Carroll, K. J. & Ruderman, E. M. Bruton’s tyrosine kinase inhibition for the treatment of rheumatoid arthritis. Immunotargets Ther. 10, 333–342 (2021).
Wang, S. P. et al. Amplification of IL-21 signalling pathway through Bruton’s tyrosine kinase in human B cell activation. Rheumatology 54, 1488–1497 (2015).
Shinohara, M. et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132, 794–806 (2008).
Gabizon, R. & London, N. A fast and clean BTK inhibitor. J. Med. Chem. 63, 5100–5101 (2020).
Gaballa, S. & Pinilla-Ibarz, J. BTK inhibitors in chronic lymphocytic leukemia. Curr. Hematol. Malig. Rep. 16, 422–432 (2021).
Liang, C. et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur. J. Med. Chem. 151, 315–326 (2018).
Zhang, D., Gong, H. & Meng, F. Recent advances in BTK inhibitors for the treatment of inflammatory and autoimmune diseases. Molecules 26, 4907 (2021).
Lie, D. C. et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 437, 1370–1375 (2005).
Rabelo Fde, S. et al. The Wnt signaling pathway and rheumatoid arthritis. Autoimmun. Rev. 9, 207–210 (2010).
Miao, C. G. et al. Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling. Cell Signal. 25, 2069–2078 (2013).
Sun, J. et al. MicroRNA-26b inhibits cell proliferation and cytokine secretion in human RASF cells via the Wnt/GSK-3β/β-catenin pathway. Diagn. Pathol. 10, 72 (2015).
Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell Signal. 22, 717–727 (2010).
Muley, P. D. et al. The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev. Neurobiol. 68, 1441–1453 (2008).
Coy, J. F. et al. Pore membrane and/or filament interacting like protein 1 (POMFIL1) is predominantly expressed in the nervous system and encodes different protein isoforms. Gene 290, 73–94 (2002).
Maes, T., Barceló, A. & Buesa, C. Neuron navigator: a human gene family with homology to unc-53, a cell guidance gene from Caenorhabditis elegans. Genomics 80, 21–30 (2002).
Nakaya, Y., Sukowati, E. W., Wu, Y. & Sheng, G. RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nat. Cell Biol. 10, 765–775 (2008).
Wang, K. S. et al. Family-based association analysis of NAV2 gene with the risk and age at onset of Alzheimer’s disease. J. Neuroimmunol. 310, 60–65 (2017).
McNeill, E. M., Roos, K. P., Moechars, D. & Clagett-Dame, M. Nav2 is necessary for cranial nerve development and blood pressure regulation. Neural Dev. 5, 6 (2010).
Marzinke, M. A., Mavencamp, T., Duratinsky, J. & Clagett-Dame, M. 14-3-3ε and NAV2 interact to regulate neurite outgrowth and axon elongation. Arch. Biochem Biophys. 540, 94–100 (2013).
Wang, R. et al. NAV2 positively modulates inflammatory response of fibroblast-like synoviocytes through activating Wnt/β-catenin signaling pathway in rheumatoid arthritis. Clin. Transl. Med. 11, e376 (2021).
Chakrabarti, S. et al. Sensitization of knee-innervating sensory neurons by tumor necrosis factor-α-activated fibroblast-like synoviocytes: an in vitro, coculture model of inflammatory pain. Pain 161, 2129–2141 (2020).
Keewan, E. & Naser, S. A. The role of notch signaling in macrophages during inflammation and infection: implication in rheumatoid arthritis? Cells 9, 111 (2020).
Šućur, A. et al. Notch receptors and ligands in inflammatory arthritis - a systematic review. Immunol. Lett. 223, 106–114 (2020).
Nakazawa, M. et al. Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 44, 1545–1554 (2001).
Ando, K. et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene 22, 7796–7803 (2003).
Park, J. S. et al. Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann. Rheum. Dis. 74, 267–274 (2015).
Jiao, Z. et al. Notch signaling mediates TNF-α-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin. Dev. Immunol. 2012, 350209 (2012).
Keerthivasan, S. et al. Notch signaling regulates mouse and human Th17 differentiation. J. Immunol. 187, 692–701 (2011).
Meyer Zu Horste, G. et al. RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep. 16, 392–404 (2016).
Zhang, W. et al. γ-secretase inhibitor alleviates acute airway inflammation of allergic asthma in mice by downregulating Th17 cell differentiation. Mediators Inflamm. 2015, 258168 (2015).
Ma, L. et al. Notch1 signaling regulates the Th17/Treg immune imbalance in patients with psoriasis vulgaris. Mediators Inflamm. 2018, 3069521 (2018).
Jiao, Z. et al. Engagement of activated Notch signalling in collagen II-specific T helper type 1 (Th1)- and Th17-type expansion involving Notch3 and Delta-like1. Clin. Exp. Immunol. 164, 66–71 (2011).
Wei, K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264 (2020).
Bai, X. et al. MicroRNA-146a protects against LPS-induced organ damage by inhibiting Notch1 in macrophage. Int. Immunopharmacol. 63, 220–226 (2018).
Le Rossignol, S., Ketheesan, N. & Haleagrahara, N. Redox-sensitive transcription factors play a significant role in the development of rheumatoid arthritis. Int. Rev. Immunol. 37, 129–143 (2018).
Noort, A. R., Tak, P. P. & Tas, S. W. Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. Ther. 17, 15 (2015).
Maracle, C. X. et al. Targeting non-canonical nuclear factor-κB signalling attenuates neovascularization in a novel 3D model of rheumatoid arthritis synovial angiogenesis. Rheumatology 56, 294–302 (2017).
Jimi, E., Fei, H. & Nakatomi, C. NF-κB signaling regulates physiological and pathological chondrogenesis. Int J. Mol. Sci. 20, 6275 (2019).
Baum, R. & Gravallese, E. M. Bone as a target organ in rheumatic disease: impact on osteoclasts and osteoblasts. Clin. Rev. Allergy Immunol. 51, 1–15 (2016).
Gong, Y. et al. Effect of moxibustion on HIF-1α and VEGF levels in patients with rheumatoid arthritis. Pain. Res. Manag. 2019, 4705247 (2019).
Shiozawa, S. & Tsumiyama, K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle 8, 1539–1543 (2009).
Han, Z., Boyle, D. L., Manning, A. M. & Firestein, G. S. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 28, 197–208 (1998).
Hannemann, N. et al. Transcription factor Fra-1 targets arginase-1 to enhance macrophage-mediated inflammation in arthritis. J. Clin. Invest. 129, 2669–2684 (2019).
Chadha, S. et al. Role of Nrf2 in rheumatoid arthritis. Curr. Res. Transl. Med. 68, 171–181 (2020).
Westra, J., Molema, G. & Kallenberg, C. G. Hypoxia-inducible factor-1 as regulator of angiogenesis in rheumatoid arthritis - therapeutic implications. Curr. Med. Chem. 17, 254–263 (2010).
Jia, W. et al. GATA4 regulates angiogenesis and persistence of inflammation in rheumatoid arthritis. Cell Death Dis. 9, 503 (2018).
Gao, S. et al. E2F1 mediates the downregulation of POLD1 in replicative senescence. Cell Mol. Life Sci. 76, 2833–2850 (2019).
Wang, R. et al. Neuron navigator 2 is a novel mediator of rheumatoid arthritis. Cell Mol. Immunol. 18, 2288–2289 (2021).
Wang, R. et al. STAT3-NAV2 axis as a new therapeutic target for rheumatoid arthritis via activating SSH1L/Cofilin-1 signaling pathway. Signal Transduct. Target Ther. 7, 209 (2022).
Oblak, L. et al. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 69, 101348 (2021).
Chang, K. et al. Smoking and rheumatoid arthritis. Int. J. Mol. Sci. 15, 22279–22295 (2014).
Hussain, M. S. & Tripathi, V. Smoking under hypoxic conditions: a potent environmental risk factor for inflammatory and autoimmune diseases. Mil. Med. Res. 5, 11 (2018).
Elzorkany, B. et al. Does smoking affect level of seropositivity in RA? A post-HOC global and inter-country analysis of COMORA cohort. Rheumatol. Int. 41, 699–705 (2021).
Hedenstierna, L. et al. Effects of alcohol consumption and smoking on risk for RA: results from a Swedish prospective cohort study. RMD Open. 7, e001379 (2021).
Gudelj Gračanin, A. et al. The effect of smoking on disease activity in rheumatoid arthritis - our experience. Acta Clin. Croat. 59, 312–317 (2020).
Barik, R. R. & Bhatt, L. K. Emerging epigenetic targets in rheumatoid arthritis. Rheumatol. Int. 41, 2047–2067 (2021).
Mueller, A. L. et al. Recent advances in understanding the pathogenesis of rheumatoid arthritis: new treatment strategies. Cells 10, 3017 (2021).
Nair, N., Barton, A. & Wilson, A. G. Cell-specific epigenetic drivers of pathogenesis in rheumatoid arthritis. Epigenomics 13, 549–560 (2021).
Zhang, Y. et al. Overview of histone modification. Adv. Exp. Med. Biol. 1283, 1–16 (2021).
Araki, Y. et al. Altered gene expression profiles of histone lysine methyltransferases and demethylases in rheumatoid arthritis synovial fibroblasts. Clin. Exp. Rheumatol. 36, 314–316 (2018).
Niederer, F. et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann. Rheum. Dis. 70, 1866–1873 (2011).
Göschl, L. et al. Histone deacetylase 1 (HDAC1): A key player of T cell-mediated arthritis. J. Autoimmun. 108, 102379 (2020).
Horiuchi, M. et al. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J. Rheumatol. 36, 1580–1589 (2009).
Li, M. et al. Sp1 S-sulfhydration induced by hydrogen sulfide inhibits inflammation via HDAC6/MyD88/NF-κB signaling pathway in adjuvant-induced arthritis. Antioxidants 11, 732 (2022).
Chung, Y. L., Lee, M. Y., Wang, A. J. & Yao, L. F. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol. Ther. 8, 707–717 (2003).
Nishida, K. et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 50, 3365–3376 (2004).
Jia, W. et al. Histone demethylase JMJD3 regulates fibroblast-like synoviocyte-mediated proliferation and joint destruction in rheumatoid arthritis. FASEB J. 32, 4031–4042 (2018).
Yin, Y. et al. Jmjd1c demethylates STAT3 to restrain plasma cell differentiation and rheumatoid arthritis. Nat. Immunol. 23, 1342–1354 (2022).
Wu, W. et al. SMYD2-mediated TRAF2 methylation promotes the NF-κB signaling pathways in inflammatory diseases. Clin. Transl. Med. 11, e591 (2021).
Tammen, S. A., Friso, S. & Choi, S. W. Epigenetics: the link between nature and nurture. Mol. Asp. Med. 34, 753–764 (2013).
Andersen, G. B. & Tost, J. A summary of the biological processes, disease-associated changes, and clinical applications of DNA methylation. Methods Mol. Biol. 1708, 3–30 (2018).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Corvetta, A., Della Bitta, R., Luchetti, M. M. & Pomponio, G. 5-Methylcytosine content of DNA in blood, synovial mononuclear cells and synovial tissue from patients affected by autoimmune rheumatic diseases. J. Chromatogr. 566, 481–491 (1991).
Nile, C. J. et al. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 58, 2686–2693 (2008).
Fu, L. H. et al. Hypomethylation of proximal CpG motif of interleukin-10 promoter regulates its expression in human rheumatoid arthritis. Acta Pharm. Sin. 32, 1373–1380 (2011).
Nemtsova, M. V. et al. Epigenetic changes in the pathogenesis of rheumatoid arthritis. Front. Genet. 10, 570 (2019).
de Andres, M. C. et al. Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res. Ther. 17, 233 (2015).
Doody, K. M., Bottini, N. & Firestein, G. S. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics 9, 479–492 (2017).
Karouzakis, E. et al. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 60, 3613–3622 (2009).
Cribbs, A. P. et al. Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol. 66, 2344–2354 (2014).
Cai, Y., Yu, X., Hu, S. & Yu, J. A brief review on the mechanisms of miRNA regulation. Genomics Proteom. Bioinformatics 7, 147–154 (2009).
Saliminejad, K., Khorram Khorshid, H. R., Soleymani Fard, S. & Ghaffari, S. H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 234, 5451–5465 (2019).
Michlewski, G. & Cáceres, J. F. Post-transcriptional control of miRNA biogenesis. RNA 25, 1–16 (2019).
Jonas, S. & Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433 (2015).
Bernardo, B. C., Ooi, J. Y., Lin, R. C. & McMullen, J. R. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med. Chem. 7, 1771–1792 (2015).
Lee, Y. S. & Dutta, A. MicroRNAs in cancer. Annu Rev. Pathol. 4, 199–227 (2009).
Simpson, L. J. & Ansel, K. M. MicroRNA regulation of lymphocyte tolerance and autoimmunity. J. Clin. Invest. 125, 2242–2249 (2015).
Grabiec, A. M. & Reedquist, K. A. The ascent of acetylation in the epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 9, 311–318 (2013).
Abo ElAtta, A. S., Ali, Y. B. M., Bassyouni, I. H. & Talaat, R. M. Upregulation of miR-221/222 expression in rheumatoid arthritis (RA) patients: correlation with disease activity. Clin. Exp. Med. 19, 47–53 (2019).
Fulci, V. et al. miR-223 is overexpressed in T-lymphocytes of patients affected by rheumatoid arthritis. Hum. Immunol. 71, 206–211 (2010).
Bae, S. C. & Lee, Y. H. MiR-146a levels in rheumatoid arthritis and their correlation with disease activity: a meta-analysis. Int J. Rheum. Dis. 21, 1335–1342 (2018).
Tavasolian, F., Hosseini, A. Z., Soudi, S. & Naderi, M. miRNA-146a improves immunomodulatory effects of MSC-derived exosomes in rheumatoid arthritis. Curr. Gene Ther. 20, 297–312 (2020).
Wang, Y. et al. miR-155 promotes fibroblast-like synoviocyte proliferation and inflammatory cytokine secretion in rheumatoid arthritis by targeting FOXO3a. Exp. Ther. Med. 19, 1288–1296 (2020).
Singh, A., Patro, P. S. & Aggarwal, A. MicroRNA-132, miR-146a, and miR-155 as potential biomarkers of methotrexate response in patients with rheumatoid arthritis. Clin. Rheumatol. 38, 877–884 (2019).
Stanczyk, J. et al. Altered expression of microRNA-203 in rheumatoid arthritis synovial fibroblasts and its role in fibroblast activation. Arthritis Rheum. 63, 373–381 (2011).
Philippe, L. et al. TLR2 expression is regulated by microRNA miR-19 in rheumatoid fibroblast-like synoviocytes. J. Immunol. 188, 454–461 (2012).
Gantier, M. P. et al. A miR-19 regulon that controls NF-κB signaling. Nucleic Acids Res. 40, 8048–8058 (2012).
Mu, N. et al. A novel NF-κB/YY1/microRNA-10a regulatory circuit in fibroblast-like synoviocytes regulates inflammation in rheumatoid arthritis. Sci. Rep. 6, 20059 (2016).
Philippe, L. et al. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann. Rheum. Dis. 72, 1071–1079 (2013).
Nakamachi, Y. et al. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann. Rheum. Dis. 75, 601–608 (2016).
Castro-Villegas, C. et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis Res. Ther. 17, 49 (2015).
Lin, Y. J., Anzaghe, M. & Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 9, 880 (2020).
Shavlovskaya, O. A., Bokova, I. A. & Shavlovskiy, N. I. [Meloxicam clinical effects]. Zh . Nevrol. Psikhiatr Im. S S Korsakova. 122, 36–42 (2022).
Bombardier, C. et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 343, 1520–1528 (2000). 1522 p following 1528.
Ahmed, M., Khanna, D. & Furst, D. E. Meloxicam in rheumatoid arthritis. Expert Opin. Drug Metab. Toxicol. 1, 739–751 (2005).
Khalil, N. Y. & Aldosari, K. F. Meloxicam. Profiles Drug Subst. Excip. Relat. Methodol. 45, 159–197 (2020).
Ruschitzka, F. et al. Differential blood pressure effects of ibuprofen, naproxen, and celecoxib in patients with arthritis: the PRECISION-ABPM (Prospective Randomized Evaluation of Celecoxib Integrated Safety Versus Ibuprofen or Naproxen Ambulatory Blood Pressure Measurement) Trial. Eur. Heart J. 38, 3282–3292 (2017).
A fixed-dose combination ibuprofen and famotidine (Duexis). Med. Lett. Drugs Ther. 53, 85–86 (2011).
Sutton, L. B. Naproxen sodium. J. Am. Pharm. Assoc. Ns36, 663–667 (1996).
Crofford, L. J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 15, S2 (2013).
Buttgereit, F. & Gibofsky, A. Delayed-release prednisone - a new approach to an old therapy. Expert Opin. Pharmacother. 14, 1097–1106 (2013).
Ursini, F., Naty, S., Bruno, C. & Grembiale, R. D. Old but good: modified-release prednisone in rheumatoid arthritis. Rev. Recent Clin. Trials 12, 124–128 (2017).
Krasselt, M. & Baerwald, C. Efficacy and safety of modified-release prednisone in patients with rheumatoid arthritis. Drug Des. Devel Ther. 10, 1047–1058 (2016).
Henness, S. & Yang, L. P. Modified-release prednisone: in patients with rheumatoid arthritis. Drugs 73, 2067–2076 (2013).
van Everdingen, A. A., Jacobs, J. W., Siewertsz Van Reesema, D. R. & Bijlsma, J. W. Low-dose prednisone therapy for patients with early active rheumatoid arthritis: clinical efficacy, disease-modifying properties, and side effects: a randomized, double-blind, placebo-controlled clinical trial. Ann. Intern Med. 136, 1–12 (2002).
Brody, M., Böhm, I. & Bauer, R. Mechanism of action of methotrexate: experimental evidence that methotrexate blocks the binding of interleukin 1 beta to the interleukin 1 receptor on target cells. Eur. J. Clin. Chem. Clin. Biochem. 31, 667–674 (1993).
Pincus, T., Bergman, M. J. & Yazici, Y. Limitations of clinical trials in chronic diseases: is the efficacy of methotrexate (MTX) underestimated in polyarticular psoriatic arthritis on the basis of limitations of clinical trials more than on limitations of MTX, as was seen in rheumatoid arthritis? Clin. Exp. Rheumatol. 33, S82–S93 (2015).
Wessels, J. A., Huizinga, T. W. & Guchelaar, H. J. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology 47, 249–255 (2008).
Böhm, I. Increased peripheral blood B-cells expressing the CD5 molecules in association to autoantibodies in patients with lupus erythematosus and evidence to selectively down-modulate them. Biomed. Pharmacother. 58, 338–343 (2004).
Keen, H. I., Conaghan, P. G. & Tett, S. E. Safety evaluation of leflunomide in rheumatoid arthritis. Expert Opin. Drug Saf. 12, 581–588 (2013).
Pinto, P. & Dougados, M. Leflunomide in clinical practice. Acta Reumatol. Port. 31, 215–224 (2006).
Plosker, G. L. & Croom, K. F. Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs 65, 1825–1849 (2005).
Suarez-Almazor, M. E. et al. Sulfasalazine for rheumatoid arthritis. Cochrane Database Syst. Rev. 1998, Cd000958 (2000).
Rains, C. P., Noble, S. & Faulds, D. Sulfasalazine. A review of its pharmacological properties and therapeutic efficacy in the treatment of rheumatoid arthritis. Drugs 50, 137–156 (1995).
Dale, J., Alcorn, N., Capell, H. & Madhok, R. Combination therapy for rheumatoid arthritis: methotrexate and sulfasalazine together or with other DMARDs. Nat. Clin. Pr. Rheumatol. 3, 450–458 (2007).
Sheppard, M. et al. Tocilizumab (Actemra). Hum. Vaccin Immunother. 13, 1972–1988 (2017).
in Mother To Baby | Fact Sheet (Organization of Teratology Information Specialists (OTIS) Copyright by OTIS, March 1, 2020., 1994).
Scott, L. J. Tocilizumab: a review in rheumatoid arthritis. Drugs 77, 1865–1879 (2017).
Tocilizumab for rheumatoid arthritis. Drug Ther. Bull. 48, 9–12 (2010).
Sanmartí, R., Ruiz-Esquide, V., Bastida, C. & Soy, D. Tocilizumab in the treatment of adult rheumatoid arthritis. Immunotherapy 10, 447–464 (2018).
Crotti, C., Biggioggero, M., Becciolini, A. & Favalli, E. G. Sarilumab: patient-reported outcomes in rheumatoid arthritis. Patient Relat. Outcome Meas. 9, 275–284 (2018).
Lamb, Y. N. & Deeks, E. D. Sarilumab: a review in moderate to severe rheumatoid arthritis. Drugs 78, 929–940 (2018).
Ramírez, J. & Cañete, J. D. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Expert Opin. Drug Saf. 17, 727–732 (2018).
Mertens, M. & Singh, J. A. Anakinra for rheumatoid arthritis: a systematic review. J. Rheumatol. 36, 1118–1125 (2009).
Bedaiwi, M. K., Almaghlouth, I. & Omair, M. A. Effectiveness and adverse effects of anakinra in treatment of rheumatoid arthritis: a systematic review. Eur. Rev. Med. Pharm. Sci. 25, 7833–7839 (2021).
Waugh, J. & Perry, C. M. Anakinra: a review of its use in the management of rheumatoid arthritis. BioDrugs 19, 189–202 (2005).
Zhao, S., Mysler, E. & Moots, R. J. Etanercept for the treatment of rheumatoid arthritis. Immunotherapy 10, 433–445 (2018).
Cobo-Ibáñez, T. & Martín-Mola, E. Etanercept: long-term clinical experience in rheumatoid arthritis and other arthritis. Expert Opin. Pharmacother. 8, 1373–1397 (2007).
Danila, M. I., Hughes, L. B. & Bridges, S. L. Pharmacogenetics of etanercept in rheumatoid arthritis. Pharmacogenomics 9, 1011–1015 (2008).
Zhao, S., Chadwick, L., Mysler, E. & Moots, R. J. Review of biosimilar trials and data on adalimumab in rheumatoid arthritis. Curr. Rheumatol. Rep. 20, 57 (2018).
Lu, X. et al. Efficacy and safety of adalimumab biosimilars: current critical clinical data in rheumatoid arthritis. Front. Immunol. 12, 638444 (2021).
Fleischmann, R. et al. Long-term safety and efficacy of upadacitinib or adalimumab in patients with rheumatoid arthritis: results through 3 years from the SELECT-COMPARE study. RMD Open. 8, e002012 (2022).
Lichtenstein, L. et al. Infliximab-related infusion reactions: systematic review. J. Crohns Colitis 9, 806–815 (2015).
Alten, R. & van den Bosch, F. Dose optimization of infliximab in patients with rheumatoid arthritis. Int J. Rheum. Dis. 17, 5–18 (2014).
Maini, S. R. Infliximab treatment of rheumatoid arthritis. Rheum. Dis. Clin. North Am. 30, 329–347 (2004).
Radner, H. & Aletaha, D. Anti-TNF in rheumatoid arthritis: an overview. Wien. Med. Wochenschr. 165, 3–9 (2015).
Singh, J. A., Noorbaloochi, S. & Singh, G. Golimumab for rheumatoid arthritis. Cochrane Database Syst. Rev. 1, Cd008341 (2010).
Keystone, E. C. et al. Golimumab, a human antibody to tumour necrosis factor {alpha} given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann. Rheum. Dis. 68, 789–796 (2009).
Rubbert-Roth, A. et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N. Engl. J. Med. 383, 1511–1521 (2020).
Pombo-Suarez, M. & Gomez-Reino, J. J. Abatacept for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 15, 319–326 (2019).
Bonelli, M. & Scheinecker, C. How does abatacept really work in rheumatoid arthritis? Curr. Opin. Rheumatol. 30, 295–300 (2018).
Blair, H. A. & Deeks, E. D. Abatacept: a review in rheumatoid arthritis. Drugs 77, 1221–1233 (2017).
Tavakolpour, S. et al. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin. Rheumatol. 38, 2977–2994 (2019).
Lopez-Olivo, M. A. et al. Rituximab for rheumatoid arthritis. Cochrane Database Syst. Rev. 1, Cd007356 (2015).
Wareing, A. An evaluation of rituximab for rheumatoid arthritis. Am. J. Nurs. 116, 22 (2016).
Frampton, J. E. & Scott, L. J. Rituximab: in rheumatoid arthritis. BioDrugs 21, 333–341 (2007).
Salles, G. et al. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv. Ther. 34, 2232–2273 (2017).
Jamilloux, Y. et al. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 18, 102390 (2019).
Dhillon, S. Tofacitinib: a review in rheumatoid arthritis. Drugs 77, 1987–2001 (2017).
van Vollenhoven, R. F. et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 367, 508–519 (2012).
Hodge, J. A. et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 34, 318–328 (2016).
Caporali, R. & Zavaglia, D. Real-world experience with tofacitinib for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 37, 485–495 (2019).
Yamaoka, K. Tofacitinib for the treatment of rheumatoid arthritis: an update. Expert Rev. Clin. Immunol. 15, 577–588 (2019).
Al-Salama, Z. T. & Scott, L. J. Baricitinib: a review in rheumatoid arthritis. Drugs 78, 761–772 (2018).
Taylor, P. C. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Urits, I. et al. Baricitinib for the treatment of rheumatoid arthritis. Reumatologia 58, 407–415 (2020).
Baricitinib for rheumatoid arthritis. Aust Prescr. 42, 34–35, (2019).
Serhal, L. & Edwards, C. J. Upadacitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 15, 13–25 (2019).
Upadacitinib for rheumatoid arthritis. Aust Prescr. 43, 178–179, (2020).
Tanaka, Y. A review of upadacitinib in rheumatoid arthritis. Mod. Rheumatol. 30, 779–787 (2020).
Dhillon, S. & Keam, S. J. Filgotinib: first approval. Drugs 80, 1987–1997 (2020).
Kim, E. S. & Keam, S. J. Filgotinib in rheumatoid arthritis: a profile of its use. Clin. Drug Investig. 41, 741–749 (2021).
Richez, C. & Truchetet, M. E. Evaluating filgotinib for the treatment of rheumatoid arthritis. Expert Opin. Pharmacother. 22, 2435–2444 (2021).
Becciolini, A. et al. Filgotinib as rheumatoid arthritis therapy. Drugs Today (Barc.). 57, 543–550 (2021).
Kaneko, Y. Efficacy and safety of peficitinib in rheumatoid arthritis. Mod. Rheumatol. 30, 773–778 (2020).
Qiu, Q., Feng, Q., Tan, X. & Guo, M. JAK3-selective inhibitor peficitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Pharmacol. 12, 547–554 (2019).
Takeuchi, T. et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann. Rheum. Dis. 78, 1305–1319 (2019).
Tanaka, Y. & Izutsu, H. Peficitinib for the treatment of rheumatoid arthritis: an overview from clinical trials. Expert Opin. Pharmacother. 21, 1015–1025 (2020).
Xie, S., Li, S., Tian, J. & Li, F. Corrigendum: iguratimod as a new drug for rheumatoid arthritis: current landscape. Front. Pharmacol. 11, 488 (2020).
Xie, S., Li, S., Tian, J. & Li, F. Iguratimod as a new drug for rheumatoid arthritis: current landscape. Front. Pharmacol. 11, 73 (2020).
Nozaki, Y. Iguratimod: novel molecular insights and a new csDMARD for rheumatoid arthritis, from Japan to the world. Life (Basel). 11, 457 (2021).
Tanaka, K., Yamaguchi, T. & Hara, M. Iguratimod for the treatment of rheumatoid arthritis in Japan. Expert Rev. Clin. Immunol. 11, 565–573 (2015).
Li, J. et al. Efficacy and safety of iguratimod for the treatment of rheumatoid arthritis. Clin. Dev. Immunol. 2013, 310628 (2013).
Burmester, G. R. & Pope, J. E. Novel treatment strategies in rheumatoid arthritis. Lancet 389, 2338–2348 (2017).
Wasserman, A. M. Diagnosis and management of rheumatoid arthritis. Am. Fam. Physician 84, 1245–1252 (2011).
Aletaha, D. & Smolen, J. S. Diagnosis and management of rheumatoid arthritis: a review. Jama 320, 1360–1372 (2018).
Cohen, S. et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, phase II Trial (ANDES Study). Arthritis Rheumatol. 72, 1435–1446 (2020).
Davis, R. R. et al. Structural insights into JAK2 inhibition by ruxolitinib, fedratinib, and derivatives thereof. J. Med. Chem. 64, 2228–2241 (2021).
Shi, J. G. et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J. Clin. Pharmacol. 51, 1644–1654 (2011).
Wu, H. et al. JAK1-STAT3 blockade by JAK inhibitor SHR0302 attenuates inflammatory responses of adjuvant-induced arthritis rats and decreases Th17 and total B cells. Jt. Bone Spine 83, 525–532 (2016).
Genovese, M. C. et al. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 68, 46–55 (2016).
Genovese, M. C., Yang, F., Østergaard, M. & Kinnman, N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann. Rheum. Dis. 75, 1979–1983 (2016).
Mahajan, S. et al. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J. Pharm. Exp. Ther. 353, 405–414 (2015).
Damjanov, N., Kauffman, R. S. & Spencer-Green, G. T. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 60, 1232–1241 (2009).
Genovese, M. C. et al. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J. Rheumatol. 38, 846–854 (2011).
Xing, L. et al. Discovery and characterization of atropisomer PH-797804, a p38 MAP kinase inhibitor, as a clinical drug candidate. ChemMedChem 7, 273–280 (2012).
Singh, D. et al. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J. Clin. Pharmacol. 50, 94–100 (2010).
Liu, C. et al. Discovery of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38α MAP kinase inhibitor for the treatment of inflammatory diseases. J. Med. Chem. 53, 6629–6639 (2010).
Patel, L. et al. Discovery of orally efficacious phosphoinositide 3-kinase δ inhibitors with improved metabolic stability. J. Med. Chem. 59, 9228–9242 (2016).
Kim, J. et al. PBT-6, a novel PI3KC2γ inhibitor in rheumatoid arthritis. Biomol. Ther. (Seoul.). 28, 172–183 (2020).
Toyama, S. et al. Inhibitory effects of ZSTK474, a novel phosphoinositide 3-kinase inhibitor, on osteoclasts and collagen-induced arthritis in mice. Arthritis Res. Ther. 12, R92 (2010).
Laragione, T. & Gulko, P. S. mTOR regulates the invasive properties of synovial fibroblasts in rheumatoid arthritis. Mol. Med. 16, 352–358 (2010).
Kim, T. H. et al. Combined therapeutic application of mTOR inhibitor and vitamin D(3) for inflammatory bone destruction of rheumatoid arthritis. Med. Hypotheses 79, 757–760 (2012).
Chen, J. et al. Treatment of collagen-induced arthritis rat model by using Notch signalling inhibitor. J. Orthop. Transl. 28, 100–107 (2021).
Ahmad, S. F. et al. STA-21, a STAT-3 inhibitor, attenuates the development and progression of inflammation in collagen antibody-induced arthritis. Immunobiology 222, 206–217 (2017).
Sun, Z. H. et al. MeCP2 regulates PTCH1 expression through DNA methylation in rheumatoid arthritis. Inflammation 40, 1497–1508 (2017).
Petralia, M. C. et al. Effects of treatment with the hypomethylating agent 5-aza-2’-deoxycytidine in murine type II collagen-induced arthritis. Pharm. (Basel). 12, 174 (2019).
Huang, Y. S. et al. Pharmacological modulation of T cell immunity results in long-term remission of autoimmune arthritis. Proc. Natl Acad. Sci. USA 118, e2100939118 (2021).
Fechtner, S., Singh, A., Chourasia, M. & Ahmed, S. Molecular insights into the differences in anti-inflammatory activities of green tea catechins on IL-1β signaling in rheumatoid arthritis synovial fibroblasts. Toxicol. Appl Pharmacol. 329, 112–120 (2017).
Jabbour, E., Issa, J. P., Garcia-Manero, G. & Kantarjian, H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer 112, 2341–2351 (2008).
Cheng, J. C. et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl Cancer Inst. 95, 399–409 (2003).
Gillespie, J. et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. 64, 418–422 (2012).
Grabiec, A. M., Korchynskyi, O., Tak, P. P. & Reedquist, K. A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 71, 424–431 (2012).
Grabiec, A. M. et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 184, 2718–2728 (2010).
Choo, Q. Y., Ho, P. C., Tanaka, Y. & Lin, H. S. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology 49, 1447–1460 (2010).
Ahmed, S. et al. Largazole, a class I histone deacetylase inhibitor, enhances TNF-α-induced ICAM-1 and VCAM-1 expression in rheumatoid arthritis synovial fibroblasts. Toxicol. Appl Pharmacol. 270, 87–96 (2013).
Hsieh, I. N. et al. Preclinical anti-arthritic study and pharmacokinetic properties of a potent histone deacetylase inhibitor MPT0G009. Cell Death Dis. 5, e1166 (2014).
Li, M. et al. Therapeutic effects of NK-HDAC-1, a novel histone deacetylase inhibitor, on collagen-induced arthritis through the induction of apoptosis of fibroblast-like synoviocytes. Inflammation 36, 888–896 (2013).
Oh, B. R. et al. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res. Ther. 19, 154 (2017).
Park, J. K. et al. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 22, 176 (2020).
Gu, W. & Roeder, R. G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 (1997).
Glozak, M. A., Sengupta, N., Zhang, X. & Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 (2005).
Grabiec, A. M., Tak, P. P. & Reedquist, K. A. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res. Ther. 10, 226 (2008).
Singh, B. N. et al. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 10, 935–954 (2010).
Sharma, S. & Taliyan, R. Targeting histone deacetylases: a novel approach in Parkinson’s disease. Parkinsons Dis. 2015, 303294 (2015).
Wu, W. J. et al. S-propargyl-cysteine attenuates inflammatory response in rheumatoid arthritis by modulating the Nrf2-ARE signaling pathway. Redox Biol. 10, 157–167 (2016).
Wu, W. et al. Cystathionine-γ-lyase ameliorates the histone demethylase JMJD3-mediated autoimmune response in rheumatoid arthritis. Cell Mol. Immunol. 16, 694–705 (2019).
Yu, Y. et al. A novel dendritic mesoporous silica based sustained hydrogen sulfide donor for the alleviation of adjuvant-induced inflammation in rats. Drug Deliv. 28, 1031–1042 (2021).
Yu, Y. et al. The preparation of a novel poly(lactic acid)-based sustained H(2)S releasing microsphere for rheumatoid arthritis alleviation. Pharmaceutics 13, 742 (2021).
Neklesa, T. K., Winkler, J. D. & Crews, C. M. Targeted protein degradation by PROTACs. Pharm. Ther. 174, 138–144 (2017).
Martín-Acosta, P. & Xiao, X. PROTACs to address the challenges facing small molecule inhibitors. Eur. J. Med. Chem. 210, 112993 (2021).
Saraswat, A. L. et al. Drug delivery challenges and formulation aspects of proteolysis targeting chimera (PROTACs). Drug Discov. Today 28, 103387 (2022).
Kargbo, R. B. PROTAC-mediated degradation of janus kinase as a therapeutic strategy for cancer and rheumatoid arthritis. ACS Med. Chem. Lett. 12, 945–946 (2021).
Danhier, F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J. Control Release 161, 505–522 (2012).
Ha, Y. J. et al. Methotrexate-loaded multifunctional nanoparticles with near-infrared irradiation for the treatment of rheumatoid arthritis. Arthritis Res. Ther. 22, 146 (2020).
Yang, Y. et al. Targeted silver nanoparticles for rheumatoid arthritis therapy via macrophage apoptosis and Re-polarization. Biomaterials 264, 120390 (2021).
Choi, Y. R. et al. A genome-engineered bioartificial implant for autoregulated anticytokine drug delivery. Sci. Adv. 7, eabj1414 (2021).
Smolen, J. S. & Aletaha, D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nat. Rev. Rheumatol. 11, 276–289 (2015).
Liu, E. & Perl, A. Pathogenesis and treatment of autoimmune rheumatic diseases. Curr. Opin. Rheumatol. 31, 307–315 (2019).
Moudgil, K. D. Advances in the pathogenesis and treatment of autoimmunity. Cell Immunol. 339, 1–3 (2019).
Alam, J., Jantan, I. & Bukhari, S. N. A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 92, 615–633 (2017).
Veale, D. J., Orr, C. & Fearon, U. Cellular and molecular perspectives in rheumatoid arthritis. Semin. Immunopathol. 39, 343–354 (2017).
Davis, J. M. 3rd The patient experience of drug side effects in rheumatoid arthritis: intriguing data from an exploratory online survey. J. Rheumatol. 49, 967–970 (2022).
Albrecht, K. & Müller-Ladner, U. Side effects and management of side effects of methotrexate in rheumatoid arthritis. Clin. Exp. Rheumatol. 28, S95–S101 (2010).
Xue, Y., Cohen, J. M., Wright, N. A. & Merola, J. F. Skin signs of rheumatoid arthritis and its therapy-induced cutaneous side effects. Am. J. Clin. Dermatol. 17, 147–162 (2016).
Hyndman, I. J. Rheumatoid arthritis: past, present and future approaches to treating the disease. Int J. Rheum. Dis. 20, 417–419 (2017).
Chen, Z., Bozec, A., Ramming, A. & Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 15, 9–17 (2019).
Roskoski, R. Jr Properties of FDA-approved small molecule protein kinase inhibitors: a 2022 update. Pharm. Res. 175, 106037 (2022).
Mankia, K. & Emery, P. Should we aim for personalized prevention in individuals at risk of rheumatoid arthritis? Int J. Rheum. Dis. 24, 621–622 (2021).
Wei, M. & Chu, C. Q. Prediction of treatment response: personalized medicine in the management of rheumatoid arthritis. Best. Pract. Res. Clin. Rheumatol. 36, 101741 (2022).
Lyseng-Williamson, K. A. & Foster, R. H. Infliximab: a pharmacoeconomic review of its use in rheumatoid arthritis. Pharmacoeconomics 22, 107–132 (2004).
Acknowledgements
This work was funded by the Macau Science and Technology Development fund (FDCT 0007/2019/AKP, 0021/2020/AGJ, 0011/2020/A1, 0089/2021/A, 0088/2021/A, 003/2022/ALC), The National Natural Science Foundation of China (Nos. 81973320), Innovative research team of high-level local universities and a key laboratory program of the Educational Commission of Shanghai Municipality (no. ZDSYS14005). Special thanks should be delivered to our best friend, famous designer Qian Hua, for her original hand-sketching rheumatoid arthritis joints illustrations.
Author information
Authors and Affiliations
Contributions
Y.Z.Z. and J.M. supervised this work. Q.D. wrote/revised the manuscript and designed the figures. W.H., R.W., Q.Y., M.Z., M.L., and J.C. helped search the database. P.R. checked and polished the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, Q., Hu, W., Wang, R. et al. Signaling pathways in rheumatoid arthritis: implications for targeted therapy. Sig Transduct Target Ther 8, 68 (2023). https://doi.org/10.1038/s41392-023-01331-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01331-9
- Springer Nature Limited
This article is cited by
-
ECM-mimetic, NSAIDs loaded thermo-responsive, immunomodulatory hydrogel for rheumatoid arthritis treatment
BMC Biotechnology (2024)
-
Mechanistic role of quercetin as inhibitor for adenosine deaminase enzyme in rheumatoid arthritis: systematic review
Cellular & Molecular Biology Letters (2024)
-
Long-term effects of abatacept on atherosclerosis and arthritis in older vs. younger patients with rheumatoid arthritis: 3-year results of a prospective, multicenter, observational study
Arthritis Research & Therapy (2024)
-
PTEN: an emerging target in rheumatoid arthritis?
Cell Communication and Signaling (2024)
-
Exploring the mechanism of Celastrol in the treatment of rheumatoid arthritis based on systems pharmacology and multi-omics
Scientific Reports (2024)