
Abstract
Patients with rheumatic diseases, including rheumatoid arthritis and osteoarthritis, almost universally describe pain and stiffness as important contributors to reduced health-related quality of life. Of the treatment options available, NSAIDs are the most widely used agents for symptomatic treatment. NSAIDs are effective anti-inflammatory and analgesic drugs by virtue of their ability to inhibit biosynthesis of prostaglandins at the level of the cyclooxygenase enzyme. However, many of the adverse effects of NSAIDs are also related to inhibition of prostaglandin production, making their use problematic in some patient populations. For the clinician, understanding the biology of prostaglandin as it relates to gastrointestinal, renal, and cardiovascular physiology and the pharmacologic properties of specific NSAIDs is key to using these drugs safely. Of particular importance is the recognition of co-morbid conditions and concomitant drugs that may increase the risk of NSAIDs in particular patients. In patients with risk factors for NSAID toxicity, using the lowest dose of a drug with a short half-life only when it is needed is likely to be the safest treatment option. For those patients whose symptoms cannot be managed with intermittent treatment, using protective strategies is essential.
Similar content being viewed by others

Introduction
The use of NSAIDs is ubiquitous in rheumatology because of their effectiveness as anti-inflammatory and analgesic agents. In addition to their use in rheumatoid arthritis (RA) and osteoarthritis (OA), NSAIDs are widely used in the symptomatic management of other rheumatic diseases characterized by chronic musculo-skeletal pain and diverse forms of acute pain. NSAIDs differ widely in their chemical class, but share the property of blocking production of prostaglandins (PGs) [1]. This is accomplished by inhibiting the activity of the enzyme PGG/H synthase, also called cyclooxygenase (COX). COX occurs in two isoforms, COX-1 and COX-2, which differ in their tissue distribution and regulation. The isoforms serve different biological functions, in that COX-1 is expressed under basal conditions and is involved in the biosynthesis of PG serving homeostatic functions while COX-2 expression is increased during inflammation and other pathologic situations [2]. The clinical effects of NSAIDs are evaluated in terms of effects on the different COX isoforms. Inhibition of COX-2 by NSAIDs blocks PG production at sites of inflammation or other forms of tissue damage, while inhibition of COX-1 in certain other tissues - most importantly, platelets and the gastroduodenal mucosa - can lead to common adverse effects of NSAIDs such as bleeding and gastrointestinal ulceration [3].
COX isoform specificity, however, is only one factor that impacts the efficacy and adverse effect profile of individual NSAIDs. Most traditional NSAIDs inhibit both isoforms, albeit with some differences in the relative potency for COX-1 and COX-2. Some NSAIDs lack inhibition of platelet function, which is the operational definition of COX-2-selective NSAIDs [4]. The pharmacologic properties, including chemical class, formulation, and drug half-life, of individual drugs may be equally important in determining the properties of NSAIDs. In light of the widespread use of NSAIDs for common diseases, which are likely to increase in prevalence with the aging of the population, it is critically important to appreciate the potential adverse events associated with NSAIDs to use them safely in patients with rheumatic diseases.
NSAID classification and pharmacology
NSAIDs generally are grouped according to their chemical structures, plasma half-life, and COX-1 versus COX-2-selectivity (Table 1). Structurally, most NSAIDs are organic acids with low pK values that lend themselves to their accumulation at sites of inflammation, areas that often exhibit lower pH than uninvolved sites. Most often, there is a direct relationship between low pK and short half-life, but there are exceptions - such as nabumetone, which is nonacidic. Classifying NSAIDs based on plasma half-life can be problematic given the fact that these drugs tend to accumulate in synovial fluid, where the concentration of drug may remain more stable than in the plasma. Short half-life NSAIDs potentially could be given less frequently than indicated by their plasma half-life. NSAIDs exhibiting longer half-lives require more time to reach steady-state plasma levels. Drugs with half-life >12 hours can be given once or twice a day, and plasma levels increase for a few days to several weeks (depending on the specific half-life) but then tend to remain constant between doses. NSAIDs with longer half-lives also enable drug concentrations to equilibrate between the plasma and the synovial fluid, although total bound and unbound drug levels are usually lower in synovial fluid because there is less albumin in synovial fluid than in plasma. However, NSAIDs with longer half-life or extended release formulation may be associated with increased propensity to cause adverse effects [5]. COX-isozyme selectivity is likely to be a critically important factor in determining relative gastrointestinal and cardiovascular risk that should also be considered in addition to other pharmacologic properties for each NSAID [6].
Almost all NSAIDs are >90% bound to plasma proteins. If total drug concentrations are increased beyond the point at which the binding sites on albumin are saturated, biologically active free-drug concentrations increase disproportionately to the increasing total drug concentration. The clearance of NSAIDs is usually by hepatic metabolism, with production of inactive metabolites that are excreted in the bile and urine. Most NSAIDs are metabolized through the microsomal cytochrome P450-containing mixed-function oxidase system. NSAIDs are most often metabolized by CYP3A or CYP2C9, or both. However, some are metabolized by other cytosolic hepatic enzymes. Different patients can respond to the same NSAID in a variety of ways and the basis for this individual variability remains unclear. Several pharmacologic factors related to NSAIDs may influence this variability, such as dose response, plasma half-life, enantiomeric conversion, urinary excretion, and pharmaco-dynamic variation [7]. Such drug factors include protein binding, the metabolic profile of the drug, and the percentage of the drug that is available as the active (S) enantiomer. There is also genetic variability in the cytochrome P450 metabolic enzymes such that some individuals or ethnic groups metabolize drugs more slowly. For example, Asians are frequently slow metabolizers through the CYP2C9 pathway. Finally, the pharmaco-kinetics of some NSAIDs are affected by hepatic disease, renal disease or old age.
NSAID mechanism of action
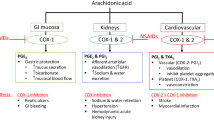
NSAIDs exert their actions by inhibiting enzymatic activity of the COX enzymes. These enzymes are the first committed step in the synthesis of PG from arachidonic acid (Figure 1). Arachidonic acid is an omega-6 poly-unsaturated fatty acid commonly found at the sn-2 position of cell membrane glycerophospholipids and cleaved from cell membranes by one of several different phospholipase A2 enzymes [8]. COX-1 and COX-2 are bifunctional enzymes that mediate a COX reaction whereby arachidonate plus two molecules of oxygen are converted to the cyclic endoperoxide PGG2, followed by a hydroperoxidase reaction in which PGG2 undergoes a two-electron reduction to PGH2 [8]. The unstable inter-mediate PGH2 spontaneously rearranges or is enzymatically converted by specific synthases to biologically active PG, of which there are many isoforms [9]. The overall regulation of the type and amount of PG produced in a given cell or tissue is determined by the expression levels of COX-1, COX-2, and terminal synthase enzymes.

Prostaglandin biosynthetic pathway. Prostaglandins (PGs) are produced from cell membrane phospholipids from the precursor omega-6 polyunsaturated fatty acid, arachidonic acid. The cyclooxygenase enzymes are bifunctional enzymes that generate PGG2 and then the unstable intermediate PGH2. This intermediate is converted by tissue-specific synthases to PG that act on their respective receptors. cPGES, cytosolic prostaglandin E synthase; DP, prostaglandin E receptor; EP, prostaglandin E receptor; FP, prostaglandin F receptor; IP, prostaglandin I receptor; mPGES, microsomal prostaglandin E synthase; PGDS, prostaglandin D synthase; PGIS, prostaglandin I synthase; TP, thromboxane A receptor; TXS, thromboxane synthase; TXA2, thromboxane A2.
All of the NSAIDs are synthetic inhibitors of the COX active site, but subtle mechanistic differences in the manner in which individual NSAIDs interact and bind with the active site are responsible for some of the differences in their pharmacologic characteristics [10]. Acetylsalicylic acid is the only covalent, irreversible modifier of COX-1 and COX-2, whereas all of the other NSAIDs are competitive inhibitors, competing with arachidonic acid for binding in the active site.
Cyclooxygenase-2 selectivity
COX isozyme selectivity is defined most commonly using the concentration of drug required to inhibit PG production by 50% in a particular assay system (inhibitory concentration). Ratios using values obtained for COX-1 50% inhibitory concentrations compared with COX-2 50% inhibitory concentrations can be calculated and used as a standard measure for comparing the degrees of selectivity of a particular NSAID for one or the other COX isoform [6]. PG assay systems can vary widely, however, making it difficult to compare directly results from studies using different assay systems. To circumvent such problems, most clinicians have accepted the use of the in vitro whole-blood assay to compare NSAID selectivities. In this system, COX-1 inhibition is assessed as a function of the reduction of thromboxane made by platelets after clot formation. Inhibition of COX-2 is based on the inhibition of PGE2 production in a heparinized blood sample after lipopolysaccharide stimulation. A COX-2-selective NSAID lacks an inhibitory effect on platelet COX-1 at concentrations at or above those that maximally inhibit COX-2 [11, 12].
Traditional NSAIDs, such as meloxicam, nimesulide, etodolac, and diclofenac show some selectivity for inhibiting COX-2 over COX-1. After the discovery of COX-2, efforts to further enhance COX-2 selectivity led to the development of celecoxib, valdecoxib, rofecoxib, etori-coxib and lumiracoxib. Most COX-2-selective NSAIDs are diaryl compounds containing a sulfonamide (celecoxib, valdecoxib) or a methylsulfone (rofecoxib, etoricoxib) rather than a carboxyl group, while lumiracoxib is an analog of diclofenac and the only acidic COX-2-selective NSAID. Lumiracoxib is available in only a few countries worldwide. Valdecoxib and rofecoxib are no longer available in any country because of concerns for excess cardiovascular adverse effects. Etoricoxib is approved in the European Union but not in the United States, while celecoxib is available worldwide. Celecoxib and etoricoxib are weak time-independent inhibitors of COX-1, but strong time-dependent inhibitors of COX-2 that require entry into and stabilized binding in the catalytic pocket. Because these drugs lack a carboxyl group, arginine 120 is not involved, but multiple sites of hydrogen and hydrophobic binding stabilize drugs at the catalytic site. The sulfur-containing phenyl ring of COX-2-selective NSAIDs plays a pivotal role in binding stability by occupying the hydrophobic side pocket characteristic of the COX-2 catalytic site. If this side pocket is removed by mutagenesis, all isozyme selectivity is lost [13].
NSAID formulation
NSAIDs are produced in a variety of dosage forms, including intravenous, slow-release and sustained-release oral preparations, and topical preparations in various forms including gels and patches, and suppositories. Given the desire to reduce NSAID toxicity while preserving drug delivery to a specific site, efforts continue to alter drug formulation and delivery systems. Nanoparti-cles, liposomes, and microspheres are under investigation to allow dose reduction and specific targeting. Intra-articular delivery is under consideration, but because joints have very efficient lymphatic clearance systems the utility of this form of targeting remains to be proved.
Topical NSAID formulations were developed to reduce systemic exposure while preserving efficacy. Several factors - including the drug, formulation, and site of application - are important for efficacy [14]. Diclofenac, for example, is available as a solution, gel, or patch. The systemic effects are directly proportional to the surface area, and this method of delivery results in a relatively stable systemic diclofenac level compared with oral administration [15]. A recent Chochrane review concludes that topical NSAIDs can provide good levels of pain relief and that gastrointestinal adverse events are reduced compared with oral NSAIDs [16].
NSAIDs have also been combined with agents having gastroprotective effects into polypills that are currently available on the market. This strategy may increase compliance with effective protective agents, thereby reducing adverse effects in clinical practice. Combining diclofenac with the synthetic PGE1 analog misoprostol (arthrotec) is shown to reduce risk of NSAID-related peptic ulcerations and mucosal injury, but utility of the combination is often limited by misoprostol-induced cramping and diarrhea [17]. In population-based studies, arthrotec was more effective than diclofenac and misoprotsol co-prescription in preventing hospitalization for peptic ulcer disease or gastrointestinal hemorrhage [18]. Several poly-pills containing NSAIDs and proton-pump inhibitors are approved for use in rheumatic diseases including ketoprofen with omeprazole (axorid) [19]. The combination of enteric-coated naproxen and the proton pump inhibitor esomeprazole (vimovo) was shown to reduce endoscopically detected gastric ulcers [20]. The combination of ibuprofen and the H2-blocker famotidine (duexis) was also shown to reduce endoscopically detected gastric and duodenal ulcers [21].
A different strategy is nitric oxide-releasing NSAIDs, which are synthesized by the ester linkage of a nitric oxide-releasing moiety to conventional NSAIDs including aspirin, flurbiprofen, diclofenac, sulindac, and others [22]. The nitric oxide moiety is slowly released by enzymatic activity in vivo, probably by esterases, resulting in slow accumulation of the parent NSAID. The lower rate of gastrointestinal ulceration associated with these drugs is probably related to nitric oxide-associated vasodilation and the relatively lower concentration of parent NSAID.
Therapeutic effects of NSAIDS in rheumatic diseases
NSAIDs are frequently used as first-line agents for the symptomatic relief of many different inflammatory conditions. In double-blind, randomized clinical trials of inflammatory arthritis, NSAIDs have been compared with placebo, aspirin, and each other. Clinical trials of NSAID efficacy in RA and OA most often employ a design whereby the current NSAID is discontinued and the patient must have an increase in symptoms or flare to enter the study. Although there is some variation in primary outcome measures, most include parameters that make up the American College of Rheumatology-20. Efficacy superior to that of placebo is easily demonstrated for NSAIDs within 1 to 2 weeks in patients with active RA who are not receiving corticosteroids or other anti-inflammatory medications [23]. Comparisons of adequate doses of traditional NSAIDs or COX-2-selective NSAIDs with one another almost always show comparable efficacy. Despite improvement in pain and stiffness with NSAIDs, these agents do not usually reduce acute-phase reactants, nor do they modify radiographic progression. The anti-inflammatory effects of NSAIDs have also been demonstrated in OA, rheumatic fever, juvenile rheumatoid arthritis, ankylosing spondylitis, gout, and systemic lupus erythematosus. Although not as rigorously proven, their efficacy is also accepted in treatment of reactive arthritis, psoriatic arthritis, acute and chronic bursitis, and tendonitis.
Virtually all NSAIDs relieve pain when used in doses substantially lower than those required to suppress inflammation. The analgesic action of NSAIDs is due to inhibition of PG production in peripheral tissues and in the central nervous system. In the periphery, PGs do not induce pain per se, but sensitize peripheral nociceptors to the effects of mediators such as bradykinin or histamine [24]. PGs released during inflammation or other trauma lower the activation threshold of tetrodotoxin-resistant sodium channels on sensory neurons. In the central nervous system, where NSAIDs and acetaminophen exert analgesic effects, PGs also play an important role in neuronal sensitization. COX-2 is constitutively expressed in the dorsal horn of the spinal cord, and its expression is increased during inflammation [25]. Centrally generated PGE2 activates spinal neurons and also microglia that contribute to neuropathic pain [26]. Both COX-1 and COX-2 play a role in nociception, as demonstrated by reductions of experimental pain in mice deficient in either COX-1 or COX-2 [27].
Adverse effects
NSAIDs share a common spectrum of clinical toxicities, although the frequency of particular side effects varies with the compound (Table 2). The hazards of individual NSAIDs are related to their pharmacologic characteristics, such as bioavailability and half-life, as well as their potency for inhibition of COX-1 and COX-2 [5, 6, 28]. The focus of this review is on renal, hepatic, and cardiovascular adverse effects that are particularly important in patients with rheumatic diseases due to the age of the patients and medication use. Gastrointestinal adverse effects are common and important causes of morbidity and mortality, but are reviewed in detail in other manuscripts in this supplement.
Renal effects
Prostaglandins play a vital role in solute and renovascular homeostasis [29–31]. Sodium retention has been reported to occur in up to 25% of NSAID-treated patients and may be particularly apparent in patients who have an existing avidity for sodium, such as those with mild heart failure or liver disease [32]. Decreased sodium excretion in NSAID-treated patients can lead to weight gain and peripheral edema. This effect may be sufficiently important to cause clinically important exacerbations of congestive heart failure.
NSAIDs may cause altered blood pressure, with average increases in mean arterial pressure of between 5 and 10 mmHg. Using NSAIDs has also been reported to possibly increase the risk of initiating antihypertensive therapy in older patients, with the magnitude of increased risk being proportional to the NSAID dose [33]. Furthermore, in a large (n =351,630) prospective cohort of women aged 44 to 69 without hypertension in 1990, incident hypertension over the following 8 years was significantly more likely in frequent users of aspirin, acetaminophen, and NSAIDs [34]. NSAIDs can attenuate the effects of antihypertensive agents including diuretics, angiotensin-converting enzyme inhibitors, and β-blockers, interfering with blood pressure control.
NSAID-treated patients may develop hyporeninemic hypoaldosteronism that manifests as type IV renal tubular acidosis and hyperkalemia [32]. The degree of hyper-kalemia is generally mild; however, patients with renal insufficiency or those that may otherwise be prone to hyperkalemia (for example, patients with diabetes mellitus and those on angiotensin-converting enzyme inhibitors or potassium-sparing diuretics) may be at greater risk.
Acute renal failure is an uncommon consequence of NSAID treatment. This failure is due to the vasoconstrictive effects of NSAIDs and is reversible. In most cases, renal failure occurs in patients who have a depleted actual or effective intravascular volume (for example, congestive heart failure, cirrhosis, or renal insufficiency) [32]. Marked reduction in medullary blood flow may result in papillary necrosis that may arise from apoptosis of medullary interstitial cells. Inhibition of COX-2 may be a predisposing factor for renal failure [31, 35].
Another adverse renal effect resulting from NSAIDs involves an idiosyncratic reaction accompanied by massive proteinuria and acute interstitial nephritis. Hypersensitivity phenomena, such as fever, rash, and eosinophilia, may occur. This syndrome has been observed with most NSAIDs.
Use of analgesics, particularly acetaminophen and aspirin, has been associated with nephropathy leading to chronic renal failure. In one large case-control study, the regular use of aspirin or acetaminophen was associated with a risk of chronic renal failure 2.53 times as high as that for nonuse, and the risk increased significantly with an increasing cumulative lifetime dose [36]. In subjects regularly using both acetaminophen and aspirin, the risk was also significantly increased compared with users of either agent alone. No association between the use of non-aspirin NSAIDs and chronic renal failure could be detected after adjusting for acetaminophen and aspirin use. Pre-existing renal or systemic disease was a necessary precursor to analgesic-associated renal failure, and those without pre-existing renal disease had only a small risk of end-stage renal disease [36, 37].
Hepatic effects
Small elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs, and notable elevations of ALT or AST (approximately ≥3 times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials of NSAIDs. Patients usually have no symptoms, and discontinuation or dose reduction generally results in normalization of the trans-aminase values - although rare, fatal outcomes have been reported with almost all NSAIDs. Those NSAIDs most likely to be associated with hepatic adverse events are diclofenac and sulindac.
Cardiovascular effects
The risk of adverse cardiovascular effects associated with NSAID use was not widely appreciated until COX-2-selective NSAIDs were introduced into clinical practice. Rofecoxib, a potent highly specific COX-2 inhibitor with a very long half-life, was shown to have a substantially increased risk of myocardial infarction and stroke and was removed from the market because of this adverse effect [28, 38]. The relationship between excess cardiovascular risk for all NSAIDs, not only COX-2-selective NSAIDs, is proposed to be related to the degree of COX-2 inhibition and an absence of complete inhibition of COX-1 [39]. Investigators have shown an increased relative risk of myocardial infarction for drugs that inhibit COX-2 <90% at therapeutic concentrations in the whole blood (relative risk = 1.18, 95% confidence interval = 1.02 to 1.38), whereas drugs that inhibit COX-2 to a greater degree present a relative risk of 1.60 (95% confidence interval3= 1.41 to 1.81) [39].
Relative inhibition of the COX isoforms is not the only mechanism that contributes cardiovascular hazard. Other actions of NSAIDs - including effects on blood pressure, endothelial function, and nitric oxide production, and other renal effects - may play a role in cardiovascular risk [28, 40, 41]. Multiple analyses have demonstrated that the risk for cardiovascular hazard is significantly higher in those patients with pre-existing coronary artery disease. Some NSAIDs, notably ibuprofen, may interfere with the irreversible inhibition of platelet COX-1 by aspirin, thereby increasing cardiovascular hazard in aspirin users [39]. It is prudent to recommend that aspirin be taken 23hours prior to ibuprofen dosing [42, 43].
A number of large-scale randomized controlled trials comparing NSAIDs with placebo or with each other have been performed and analyzed to determine the risk of myocardial infarction, stroke, cardiovascular death, death from any cause, and Antiplatelet Trialists' Collaboration composite outcomes [28]. Because event rates in most of these studies were low, uncertainty regarding absolute and relative risk remains. For example, there were only 554 myocardial infarctions in aggregate across all trials included in the most comprehensive analysis to date. Nevertheless, it appears from analyses of these aggregated clinical trials that all traditional and COX-2-selective NSAIDs except naproxen carry an excess risk >30% compared with placebo [28]. Pairwise comparisons of the most commonly used traditional and COX-2-selective NSAIDs studied in clinical trials also suggest that naproxen may have lower cardiovascular risk [28]. One meta-analysis explored the effects of dose and dosing regimen in a pooled analysis of six randomized placebo controlled trials of celecoxib [44]. Lower doses and once-daily regimens were associated with lower relative risks for the Antiplatelet Trialists' Collaboration outcomes. This finding confirms findings from other studies that suggest avoiding continuous interference with PG biosynthesis is associated with lower cardiovascular risk [39].
Because clinical trials have been underpowered to specifically address relative cardiovascular risk of NSAIDs, investigators have turned to observational data-sets. Using a very large observational database with 8,852 cases of nonfatal myocardial infarction, a recent case-control study also identified a 35% increase in the risk for MI in current use of NSAIDs [39]. This type of study also identifies naproxen as potentially having a lower risk. In this analysis, a long half-life was an independent predictor of MI hazard. The effect of dose and a slow-release formulation demonstrated that risk was a direct consequence of prolonged drug exposure. The risk associated with these pharmacologic factors may be even more important than COX-2 specificity for most NSAIDs [28, 39].
A number of strategies have been suggested to mitigate cardiovascular risks associated with NSAID use (Table 3) [43]. These recommendations take into account a patient's underlying risk, aspirin use, and the interaction between NSAIDs. In addition, the specific choice of NSAID should consider the pharmacologic properties [28, 39].
NSAIDs are associated with reduced sodium excretion, volume expansion, increased preload, and hypertension. As a result of these properties, patients with pre-existing heart failure are at risk of decompensation with a relative risk of 3.8 (95% confidence = 1.1 to 12.7). After adjusting for age, sex, and concomitant medication, the relative risk was 9.9 (95% confidence = 1.7 to 57.0) [45]. Studies disagree about whether NSAIDs are a risk for new heart failure, but older patients may be at particular risk for heart failure exacerbation [45, 46].
Effects of concomitant drugs, diseases, and aging
Because of the widespread use of prescription and non-prescription NSAIDs, there are ample opportunities for interaction with other drugs and for interactions with patient-specific factors [47]. Specific drug interactions are listed on the package inserts of individual agents.
Drug-drug interactions
Since most NSAIDs are extensively bound to plasma proteins, they may displace other drugs from binding sites or may themselves be displaced by other agents. NSAIDs may increase the activity or toxicity of sulfonylurea, hypoglycemic agents, oral anticoagulants, phenytoin, sulfonamides, and methotrexate by displacing these drugs from their protein binding sites and increasing the free fraction of the drug in plasma [47]. However, a recent Cochrane review concluded that concurrent use of NSAIDs with methotrexate appeared safe provided appropriate monitoring was performed [48]. NSAIDs may blunt the antihypertensive effects of β-blockers, angiotensin-converting enzyme inhibitors, and thiazides, leading to de-stabilization of blood pressure control [49]. There is an increased risk of gastrointestinal toxicity when NSAIDs and selective serotonin reuptake inhibitors are taken concomitantly compared with taking either agent alone, and this is greater than the additive risk [50].
Drug-disease interactions
RA and other diseases (for example, hepatic and renal disease) that decrease serum albumin concentrations are associated with increased concentrations of free NSAIDs. Hepatic and renal diseases may also impair drug metabolism or excretion, and thereby increase the toxicity of a given dose of NSAID to an individual patient. Renal insufficiency may be accompanied by accumulated endogenous organic acids that may displace NSAIDs from protein binding sites.
Drug reactions in older people
Aging is accompanied by changes in physiology, resulting in altered pharmacokinetics and pharmacodynamics. Decreased drug clearance may be the consequence of reductions in hepatic mass, enzymatic activity, blood flow, renal plasma flow, glomerular filtration rate, and tubular function associated with aging. Older people are more likely to experience adverse gastrointestinal and renal effects related to NSAIDs. The increased risk of cardiovascular disease in older patients raises concerns of accelerated myocardial infarction or stroke. The use of aspirin for prevention of cardiovascular disease increases the toxicity of NSAIDs and, conversely, the concomitant use of NSAIDs may increase aspirin resistance. Use of proton pump inhibitors for gastroprotection may interfere with the efficacy of antiplatelet agents such as clopidogrel [42]. Older people have more illnesses than younger patients and therefore take more medications, increasing the possibility of drug-drug interactions. Older patients may also be more likely to self-medicate or make errors in drug dosing. For these reasons, frequent monitoring for compliance and toxicity should be a part of the use of NSAIDs in this population.
Choosing anti-inflammatory analgesic therapy
In choosing an NSAID for a particular patient, the clinician must consider efficacy, potential toxicity related to concomitant drugs and patient factors, and cost [1]. A study to assess patient preferences for treatment-related benefits and risks of NSAIDs for OA suggested that reductions in ambulatory pain and difficulty doing daily activities were the most important benefits. The risk of myocardial infarction and stroke were the most important risk outcomes. However, patients were willing to accept a small increased risk of myocardial infarction to reduce ambulatory, but not resting, pain [51]. Furthermore, patient preference for factors such as the dosing regimen may be taken into account. In addition to choices from the perspective of the individual patient and physician, it may be important to take a broader view. Choice of anti-inflammatory analgesic therapy can also be considered from the perspective of healthcare institutions and payers. The symptoms and conditions for which NSAIDs are used are extraordinarily common. Consequently, the cost of NSAIDs as a proportion of total drug costs can be high when drugs are expensive. The increased cost of branded NSAIDs has an important pharmacoeconomic impact. On the other hand, adverse events can have important economic consequences, and improved safety may be cost-effective.
Choosing anti-inflammatory analgesic therapy has become increasingly complex with the increased understanding of their associated toxicities. Prospectively considering the presence of gastrointestinal and cardiovascular risk factors is essential when considering treatment options (Table 4) [1]. Gastrointestinal risks are well known and strategies to prevent ulceration and bleeding are available. There are many questions regarding the risk for cardiovascular events in patients using NSAIDs; in general, the data suggest that physicians should be cautious of using NSAIDs in patients with known cardiovascular disease. In those patients with risks for NSAID toxicity, avoiding potent drugs with a long half-life or extended-release formulations is prudent. Intermittent dosing rather than continuous daily use reduces toxicity.
An absence of anti-inflammatory activity reduces the effectiveness of acetaminophen for diseases accompanied by a significant component of infl ammation (for example, RA, gout). However, acetaminophen is a safe and effective alternative for milder pain conditions, including OA. With respect to patient preference, a survey study demonstrated that only 14% of a large group of rheumatic disease patients (n = 1,799) with RA, OA, or fibromyalgia preferred acetaminophen over NSAIDs, while 60% preferred NSAIDs [52]. In a head-to-head clinical trial of acetaminophen versus diclofenac plus misoprostol, there was significantly greater improvement in pain scores for patients in the diclofenac group. This finding was magnified in those patients with more severe disease at baseline [53].
Acetaminophen should be tried as the initial therapy in patients with mild to moderate pain for reasons of safety and cost. However, if patients have moderate to severe symptoms or if evidence of inflammation is present, moving to treatment with NSAIDs may provide more rapid and effective relief [54].
Key messages
-
NSAIDs are effective treatments for relief of pain, swelling, and stiffness of arthritis and other rheumatic diseases.
-
The chemical class and pharmacology of individual NSAIDs significantlly influence their toxicity.
-
Co-morbid conditions should be considered in prescribing NSAIDs and special care should be taken in prescribing these drugs to older patients.
-
The lowest dose of a short-acting NSAID for the shortest time required is recommended for patients at risk of adverse effects.
Abbreviations
- COX:
-
cyclooxygenase
- NSAID:
-
nonsteroidal anti-inflammatory drug
- OA:
-
osteoarthritis
- PG:
-
prostaglandin
- RA:
-
rheumatoid arthritis.
References
Crofford L: Prostanoid biology and its therapeutic targeting. Kelley's Textbook of Rheumatology. Edited by: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR. 2013, Philadelphia: Saunders, 871-893. 9
Crofford LJ, Lipsky PE, Brooks P, Abramson SB, Simon LS, van de Putte LBA: Basic biology and clinical application of specific COX-2 inhibitors. Arthritis Rheum. 2000, 43: 4-13. 10.1002/1529-0131(200001)43:1<4::AID-ANR2>3.0.CO;2-V.
Lanas A: Nonsteridal antiinflammatory drugs and cyclooxygenase inhibition in the gastrointestinal tract: a trip from peptic ulcer to colon cancer. Am J Med Sci. 2009, 338: 96-106. 10.1097/MAJ.0b013e3181ad8cd3.
Patrono C, Patrignani P, Garcia-Rodrigues LA: Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2002, 108: 7-13.
Massó González EL, Patrignani P, Tacconelli S, García Rodríguez L: Variability among nonsteroidal antiinflammatory drugs in risk of upper gastrointestinal bleeding. Arthritis Rheum. 2010, 62: 1592-1601. 10.1002/art.27412.
Capone ML, Tacconelli S, Rodriguez LG, Patrignani P: NSAIDs and cardiovascular disease: transducing human pharmacology results into clinical read-outs in the general population. Pharmacol Rep. 2010, 62: 530-535. 10.1016/S1734-1140(10)70310-8.
Hinz B, Brune K: A ntipyretic analgesics: nonsteroidal anti-inflammatory drugs, selective COX-2 inhibitors, paracetamol and pyrazolinones. Handb Exp Pharmacol. 2007, 177: 65-93.
Smith WL, DeWitt DL, Garavito RM: Cyclooxygenases: structural, cellular, and molecular biology. Ann Rev Biochem. 2000, 69: 145-182. 10.1146/annurev.biochem.69.1.145.
Simmons DL, Bottin g RM, Hla T: The biology of prostaglandin synthesis and inhibition. Pharmacol Revs. 2004, 56: 387-437. 10.1124/pr.56.3.3.
Llorens O, Perez JJ, Palomar A, Mauleon D: Differential binding mode of diverse cyclooxygenase inhibitors. J Mol Graph Model. 2002, 20: 359-371. 10.1016/S1093-3263(01)00135-8.
FitzGerald GA, Pa trono C: The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001, 345: 433-442. 10.1056/NEJM200108093450607.
Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P: Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007, 82: 85-94. 10.1016/j.prostaglandins.2006.05.019.
Kurumbail RA, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC: Structural basis for selective inhibition of cyclooxygenase-2 by antiinfl ammatory agents. Nature. 1996, 384: 644-648. 10.1038/384644a0.
Barkin RL: Topica l nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. 2012,
Kienzler JL, Gold M, Nollevaux F: Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol. 2010, 50: 50-61. 10.1177/0091270009336234.
Derry S, Moore RA, Rabbie R: Topical NSAIDs for chronic musculoskeletal pain in adults. Cochrane Database Syst Rev. 2012, 9: CD007400-
Lanza PL, Chan FK L, Quigley EMM: Guidelines for prevention of NSAIDrelated ulcer complications. Am J Gastroenterol. 2009, 104: 728-738. 10.1038/ajg.2009.115.
Ashworth NL, Peloso PM, Muhanjarine N, Stang M: Risk of hospitalization with peptic ulcer disease or gastrointestinal hemorrhage associated with nabumetone, arthrotec, diclofenac, and naproxen in a population based cohort study. J Rheumatol. 2005, 32: 2212-2217.
Gigante A, Tagarro I: Non-steroidal anti-inflammatory drugs and gastroprotection with proton pump inhibitors: a focus on ketoprofen/ omeprazole. Clin Drug Investig. 2012, 32: 221-233. 10.2165/11596670-000000000-00000.
Goldstein JL, Hochberg MC, Fort JG, Zhang Y, Hwang C, Sostek M: Clinical trial: the incidence of NSAID-associated endoscopic gastric ulcers in patients treated with PN 400 (naproxen plus esomeprazole magnesium) vs. enteric-coated naproxen alone. Aliment Pharmacol Ther. 2010, 32: 401-413. 10.1111/j.1365-2036.2010.04378.x.
Bello AE: DUEXIS® (ibuprofen 800 mg, famotidine 26.6 mg): a new approach to gastroprotection for patients with chronic pain and inflammation who require treatment with a nonsteroidal anti-inflammatory drug. Ther Adv Musculoskelet Dis. 2012, 4: 327-339. 10.1177/1759720X12444710.
Keeble JE, Moore P K: Pharmacology and potential therapeutic applications of nitric oxide-releasing non-steroidal atni-inflammatory and related nitric oxide-donating drugs. Br J Pharmacol. 2002, 137: 295-310. 10.1038/sj.bjp.0704876.
Hochberg MC: New directions in symptomatic therapy for patients with osteoarthritis and rheumatoid arthritis. Semin Arthritis Rheum. 2002, 32: 4-14. 10.1053/sarh.2002.37215.
Ito S, Okuda-Ashit aka E, Minami T: Central and peripheral roles of prostaglandins in pain and their interactions with novel neuropeptides nociceptin and nocistatin. Neurosci Res. 2001, 41: 299-332. 10.1016/S0168-0102(01)00289-9.
Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD, Isakson PC: The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostglandin E2 is mediated by inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci. 2001, 21: 5847-5853.
Kunori S, Matsumura S, Okuda-Ashtaka E, Katano T, Audoly LP, Urade Y, Ito S: A novel role of prostaglandin E2 in neuropathic pain: blockade of microglial migration in the spinal cord. Glia. 2011, 59: 208-218. 10.1002/glia.21090.
Ballou LR, Botting RM, Goorha S, Zhang J, R VJ: Nociception in cyclooxygenase isozyme-deficient mice. Proc Natl Acad Sci USA. 2000, 97: 10272-10276. 10.1073/pnas.180319297.
Trelle S, Reichenback S, Wandel S, Hildebrand P, Tschannen B, Billiger PM, Egger M, Juni P: Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011, 342: c7086-10.1136/bmj.c7086.
Brater DC: Anti-inflammatory agents and renal function. Semin Arthritis Rheum. 2002, 32: 33-42. 10.1053/sarh.2002.37216.
FitzGerald GA: The choreography of cyclooxygenases in the kidney. J Clin Invest. 2002, 110: 33-34. 10.1172/JCI0216044.
Harris RC, Breyer MD: Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006, 1: 236-245. 10.2215/CJN.00890805.
Brater DC, Harris C, Redfern JS, Gertz BJ: Renal effects of COX-2 selective inhibitors. Am J Nephrol. 2001, 21: 1-15. 10.1159/000046212.
Gurwitz JH, Avorn J, Bonh RL, Glynn RJ, Monane M, Mogun H: Initiation of antihypertensive treatment during nonsteroidal anti-inflammatory drug therapy. JAMA. 1994, 272: 781-786. 10.1001/jama.1994.03520100043031.
Dedier J, Stampfer MJ, Hankinson SE, Willett WC, Speizer FE, Curhan GC: Nonnarcotic analgesic use and the risk of hypertension in US women. Hypertension. 2002, 40: 604-608. 10.1161/01.HYP.0000035856.77718.DA.
Akhund L, Quinet R J, Ishaq S: Celecoxib-related renal papillary necrosis. Arch Intern Med. 2003, 163: 114-115. 10.1001/archinte.163.1.114.
Fored CM, Ejerblad E, Lindblad P, Fryzek JP, Dickman PW, Signorello LB, Lipworth L, Elinder C-G, Blot WJ, McLaughlin JK, Zack MM, Nyrén O: Acetaminophen, aspirin, and chronic renal failure: a nationwide case-control study in Sweden. N Engl J Med. 2001, 345: 1801-1808. 10.1056/NEJMoa010323.
Rexrode KM, Buring JE, Glynn RJ, Stampfer MJ, Youngman LD, Gaziono JM: Analgesic use and renal function in men. JAMA. 2001, 286: 315-321. 10.1001/jama.286.3.315.
Juni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M: Risk of cardiovascular events and rofecoxib: a cumulative metaanalysis. Lancet. 2004, 364: 2021-2029. 10.1016/S0140-6736(04)17514-4.
Garcia Rodriguez LA, Tacconelli S, Patrignani P: Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general populations. J Am Coll Cardiol. 2008, 52: 1628-1636. 10.1016/j.jacc.2008.08.041.
FitzGerald GA: Coxibs and cardiov ascular disease. N Engl J Med. 2004, 351: 1709-1711. 10.1056/NEJMp048288.
Harirforoosh S, Aghazadeh-Habashi A, Jamali F: Extent of renal effect of cyclo-oxygenase-2-selective inhibitors is pharmacokinetic dependent. Clin Exp Pharmacol Physiol. 2006, 33: 917-924. 10.1111/j.1440-1681.2006.04464.x.
Mackenzie IS, Coughtrie MW, MacDo nald TM, Wei L: Antiplatelet drug interactions. J Intern Med. 2010, 268: 516-529. 10.1111/j.1365-2796.2010.02299.x.
Friedewald VE, Bennett JS, Christ o JP, Pool JL, Scheiman Ja, Simon LS, Strand V, White WB, Williams GW, Roberts WC: AJC Editor's Consensus: Selective and nonselective nonsteroidal anti-inflammatory drugs and cardiovascular risk. Am J Cardiol. 2010, 106: 873-884. 10.1016/j.amjcard.2010.04.006.
Solomon SD, Wittes J, Finn PV, Fo wler R, Viner J, Bertagnolli MM, Arber N, Levin B, Meinert CL, Martin B, Pater JL, Goss PE, Lance P, Obara S, Chew EY, Kim J, Arndt G, Hawk E, Cross Trial Safety Assessment Group: Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial sarety analysis. Circulation. 2008, 117: 2104-2113. 10.1161/CIRCULATIONAHA.108.764530.
Feenstra J, Heerdink ER, Grobbee DE, Stricker BR: Association of nonsteroidal anti-inflammatory drugs with first occurrence of heart failure and with replapsing heart failure: the Rotterdam Study. Arch Intern Med. 2002, 162: 265-270. 10.1001/archinte.162.3.265.
Page J, Henry D: Consumption of N SAIDs and the development of congestive heart failure in elderly patients: an under-recognized public health problem. Arch Intern Med. 2000, 160: 777-784. 10.1001/archinte.160.6.777.
Brater DC: Drug-drug and drug-disease interactions with nonsteroidal anti-inflammatory drugs. Am J Med. 1986, 80: 62-
Colebatch AN, Marks JL, van der Heijde DM, Edwards CJ: Safety of nonsteroidal anti-inflammatory drugs and/or paracetamol in people receiving methotrexate for inflammatory arthritis: a Cochrane systematic review. J Rheumatol Suppl. 2012, 90: 62-73. 10.3899/jrheum.120345.
White WB: Defining the problem of treating the patient with hypertension and arthritis pain. Am J Med. 2009, 122 (5 Suppl): S3-S9.
Mort JR, Aparasu RR, Baer RK: Interaction between selective serotonin reuptake inhibitors and nonsteroidal anti-inflammatory drugs: review of the literature. Pharmacotherapy. 2006, 26: 1307-1313. 10.1592/phco.26.9.1307.
Hauber AB, Arden NK, Mohamed AF, Johnson FR, Peloso PM, Watson DJ, Mavros P, Gammaitoni A, Sen SS, Taylor SD: A discrete-choice experiment of United Kingdom patients' willingness to risk adverse events for improved function and pain control in osteoarthritis. Osteoarthritis Cartilage. 2013, 21: 289-297. 10.1016/j.joca.2012.11.007.
Wolfe F, Zhao S, Lane N: Preference for nonsteroidal anti-inflammatory drugs over acetaminophen by rheumatic disease patients: a survey of 1,799 patients with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Arthritis Rheum. 2000, 43: 378-385. 10.1002/1529-0131(200002)43:2<378::AID-ANR18>3.0.CO;2-2.
Pincus T, Koch GG, Sokka T, Lefko with J, Wolfe F, Jordan JM, Luta G, Callahan LF, Wang X, Schwartz T, Abramson SB, Caldwell JR, Harrell RA, Kremer JM, Lautzenheiser RL, Markenson JA, Schnitzer TJ, Weaver A, Cummins P, Wilson A, Morant S, Fort J: A randomized, double-blind, crossover clinical trial of diclofenac plus misoprostol versus acetaminophen in patients with osteoarthritis of the hip or knee. Arthritis Rheum. 2001, 44: 1587-1598. 10.1002/1529-0131(200107)44:7<1587::AID-ART282>3.0.CO;2-X.
Zhang W, Nuki G, Moskowitz RW, Abramson S, Altman RD, Arden NK, Bierma-Zeinstra S, Brandt KD, Croft P, Doherty M, Dougados M, Hochberg M, Hunter DJ, Kwoh K, Lohmander LS, Tugwell P: OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage. 2010, 18: 476-499. 10.1016/j.joca.2010.01.013.
Declaration
This article has been published as part of Arthritis Research & Therapy Volume 15 Suppl 3, 2013: 'Gastroprotective NSAIDS'. The full contents of the supplement are available online at http://arthritis-research.com/supplements/15/S3. The supplement was proposed by the journal and developed by the journal in collaboration with the Guest Editor. The Guest Editor assisted the journal in preparing the outline of the project but did not have oversight of the peer review process. The Guest Editor serves as a clinical and regulatory consultant in drug development and has served as such consultant for companies which manufacture and market NSAIDs including Pizer, Pozen, Horizon Pharma, Logical Therapeutics, Nuvo Research, Iroko, Imprimis, JRX Pharma, Nuvon, Medarx, Asahi. The articles have been through the journal's standard peer review process. Publication of this supplement has been supported by Horizon Pharma Inc. Duexis (ibuprofen and famotidine) is a product marketed by the sponsor.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Rights and permissions
About this article
Cite this article
Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res Ther 15 (Suppl 3), S2 (2013). https://doi.org/10.1186/ar4174
Published:
DOI: https://doi.org/10.1186/ar4174