
Abstract
Lysophosphatidic acid (LPA) is an abundant bioactive phospholipid, with multiple functions both in development and in pathological conditions. Here, we review the literature about the differential signaling of LPA through its specific receptors, which makes this lipid a versatile signaling molecule. This differential signaling is important for understanding how this molecule can have such diverse effects during central nervous system development and angiogenesis; and also, how it can act as a powerful mediator of pathological conditions, such as neuropathic pain, neurodegenerative diseases, and cancer progression. Ultimately, we review the preclinical and clinical uses of Autotaxin, LPA, and its receptors as therapeutic targets, approaching the most recent data of promising molecules modulating both LPA production and signaling. This review aims to summarize the most update knowledge about the mechanisms of LPA production and signaling in order to understand its biological functions in the central nervous system both in health and disease.
Similar content being viewed by others

Introduction
Lysophosphatidic acid biosynthesis and degradation
Lysophosphatidic acid (1- or 2-acyl-sn-glycerol 3-phosphate/radyl-glycerol-phosphate, LPA) is a bioactive phospholipid that is produced during the synthesis of cell membranes and is described as a robust extracellular signaling molecule present in all eukaryotic tissues and blood plasma.1,2 LPA is the smallest bioactive lipid which exerts potent extracellular signaling through its interaction with its six specific G protein-coupled receptors (GPCRs), mediating important responses, such as cell proliferation, migration, and cytoskeletal reorganization.3,4,5 LPA molecules are characterized by a glycerol backbone with the addition of a phosphate group at the sn-3 position and a hydroxyl group and a fatty acid chain in either sn-1 or sn-2 positions, with their variations being due to the different fatty acid chains. LPA can be present both intra- and extracellularly with different production pathways in each one of these contexts.6
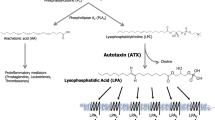
There is one major pathway involved in LPA extracellular synthesis. In this pathway, membrane phospholipids such as phosphatidylcholine, phosphatidylserine, and phosphatidylethanolamine are the precursors’ molecules to produce lysophosphatidylcholine (LPC), lysophosphatidylserine (LPS), and lysophosphatidylethanolamine, respectively through the action of phospholipase A1 (PLA1) or PLA2. These lysophospholipids (LPs) are then converted into LPA by autotoxin (ATX), which is mainly responsible for LPA maintenance at a physiological concentration in plasma after birth and also during vascular and embryonic development.4 It is well established that ATX is the major source of synthetized LPA, which is derived from membrane phospholipids.7 Also, the extracellular production of LPA by ATX is important for their role as bioactive lipids by mediating cellular responses through LPA receptors.8
Regarding the intracellular LPA production, at least four pathways are involved in this process: (1) the monoacyglicerol kinase (MAGK) pathway; (2) the phosphatidic acid-phospholipase A1 (PA-PLA1) or A2 (PA-PLA2); (3) the glycerophosphate acyltransferase (GPAT) synthesis pathway; and (4) the oxidative modification of low-density lipoprotein (LDL) pathway.
The MAGK pathway recycles the monoacylglycerol (MAG) produced by the action of lipid phosphate phosphatases (LPPs), allowing the turnover of already catalyzed LPA for another round of signaling.9,10 The second pathway involves the production of phosphatidic acid (PA) from phospholipids due to the activity of phospholipase D (PLD1 and PLD2) or from diacylglycerol by diacylglycerol kinase. Afterwards, PA is converted into LPA through the action of PLA1 or PLA2 (Fig. 1).4,11 The difference between these enzymes is that PLA1 removes 1-acyl and thus produces 2-acyl-LPA, whereas PLA2 removes 2-acyl and thus produces 1-acyl-LPA. Tissue or cell specific expression of these enzymes can be important for signaling transduction when considering that different LPA receptors can have different binding to 1- or 2-acyl LPA as, for example, LPAR3 and LPAR6 prefer 2-acyl LPA.12,13,14

LPA intra- and extracellular biosynthesis and degradation. Several mechanisms are implicated in LPA biosynthesis. Several mechanisms are implicated in LPA biosynthesis. Extracellularly, LPA can be produced by two different mechanisms from phospholipids (ATX dependent or independent mechanisms), and signals through six different transmembrane G protein-coupled receptors. Intracellularly, at least four pathways are involved in this process: (1) the monoacyglicerol kinase (MAGK) pathway; (2) the phosphatidic acid-phospholipase A1 (PA-PLA1) or A2 (PA-PLA2); (3) the glycerophosphate acyltransferase (GPAT) synthesis pathway; and (4) the oxidative modification of low-density lipoprotein (LDL) pathway. Finally, these pathways of intracellular LPA synthesis are the same which leads to its degration. Scheme created using Biorender.com
Also, LPA is an intermediate in glycerolipid synthesis, being produced by GPAT-mediated conversion of glycerol-3-phosphate (G3P). This production of LPA has probably no signaling importance, as this LPA is rapidly converted into PA by acylglycerophosphate acyltransferase (AGPTA, or lysophosphatidic acid acyltransferase) in microssomes (Fig. 1).15,16,17,18
Finally, all three intracellular mechanisms of LPA production can also be important for its degradation: (1) generation of MAG by LPPs; (2) conversion into PA by AGPTA; and (3) G3P production by lysophosphatases/ lysophospholipases. As we can notice, the intracellular pathways and either produce or degrade LPA depending on metabolites production, expression of the intermediaty enzimes and activation of glycerolipid syntesis.9,10 Also, it is important to note that most current evidence suggests that intracellular LPA synthesis and degradation serves as a source of precursors for glycerolipid synthesis and not extracellular signaling molecules.17,19
The most common LPA form known in research is described as 18:1 oleoyl-LPA (1-acyl-2-hydroxy-sn-glycero-3-phosphate). There are different LPA species depending on the acyl chain length and so their molecular mass can vary between 430 and 480 Da. Nonetheless, there are many different isoforms of LPA known in biological organisms depending on its phospholipid precursor.4,20,21 So, nowadays, there are two different LPA species known, saturated one’s fatty acids (16:0, 18:0) and unsaturated one’s fatty acids (16:1, 18:1, 18:2, 20:4) and, all these LPA species performs distinct biological activities.22,23,24,25,26,27 Moreover, as being so different those different LPA types are recognized by different LPA receptors.27
LPA is present in all eukaryotic tissues and fluids under both physiological and pathophysiological conditions. For instance, it has been found in the cerebrospinal fluid (CSF), neural tissue, seminal fluid, saliva, urine, and aqueous humor.4 Some studies have also reported the presence of LPA in conditions of nerve injury, in the serum of systemic sclerosis (SSc), in sepsis, in ascites related to pancreatic cancer and in the plasma of patients with chronic liver injury and obesity.4,5 LPA concentration significantly differs in the plasma and the serum. While the concentration of LPA in the plasma ranges from 0.7 to 80 nM, in serum it can reach 10 µM.4,21,28
Lysophosphatidic acid signaling and receptors
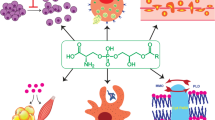
The first evidence for a role of LPA as a bioactive molecule dates from the 1960s with the identification of its effects on blood pressure, platelet activation and intracellular calcium release during smooth muscle contraction.3,29,30 Nearly two decades later, in the mid-1980s, a variety of cellular mechanisms, including cell growth, cell morphology, neurite retraction, and the development of actin stress fibers had already been associated with LPA signaling.31 However, the molecular mechanistic description of the involved metabotropic receptor with different G protein responses was only elucidated after 1996, when the first LPA receptor was cloned.32 Years before, the endothelial differentiating gene (Edg) family had been described. Nevertheless, their ligands had not been identified yet. The first result correlating LPA signaling through these GPCRs showed that the upregulation of Edg2 in murine neuronal cultures could lead to morphological changes and adenylyl cyclase inhibition in response to LPA.4 Even before the year 2000, another eight genes encoding Edg receptors in the human genome were identified. Among them, three encoding LPA receptors, namely LPAR1/Edg2,32 LPAR2/Edg433 and LPAR3/Edg7,12,34 which, in turn, are part of the rhodopsin GPCR family alpha subclass.35 In 2003, beyond the classical Edg family LPA receptors, the first non-Edg LPA receptor, which was further called LPAR4, was identified during experiments to find a P2Y9/GPR23 ligand.36,37 This discovery opened up the possibility of investigating the existence of other LPA receptors and revealed the existence of two more LPA receptors, the LPAR5 (GPR92/93) and LPAR6 (P2Y5). Together, these three receptors constitute a new family of non-Edg LPA receptors. Such receptors are part of the rhodopsin delta subclass and have stronger homology to P2Y purine receptors (P2YRs) (see Tables 1, 2, and Fig. 2).3,4,5,35,38,39

LPA receptors and signaling intermediates. LPA signals through six different LPA transmembrane G protein-coupled receptors (GPCRs and LPAR1–6) and other GPCRs (such as P2Y10 and GRP87). These receptors activate different intracellular signaling mediators to elicit different/several cellular responses (such as cytoskeleton reorganization, migration, proliferation, survival, and cell–cell communication). Finally, LPA was also recently described as a direct PPARγ agonist and in this sense intracellular LPA could also induce signaling through this receptor. Scheme created using Biorender.com
LPAR1 and LPAR2 signals through Gαi, Gαq, and Gα12/13, which trigger several downstream signaling cascades through PLC, mitogen-activated protein kinases (MAPK), protein kinase B (PKB/Akt) and rho-kinase pathways when LPAR1 is activated; and Ras GTPases, Rac GTPase, phosphatidylinositol 3-kinase (PI3K), MAPK, phospholipase C gamma (PLCγ), diacylglycerol, and Rho in the case of LPAR2 activation. Interestingly, LPAR3 couples only with Gαi and Gαq to induce calcium mobilization, adenylyl cyclase inhibition and activation, PLC activation and MAPK activation.4 Due to these distinct downstream pathways that can be activated, LPA can, therefore, induce a wide range of different cellular responses.
LPAR1 is widely expressed in several organs, including brain, uterus, testis, lung, small intestine, heart, stomach, kidney, spleen, thymus, placenta, and skeletal muscle; and its activation by LPA promotes cell proliferation and survival, cell–cell contact, cell migration, cytoskeletal changes, Ca2+ mobilization, and adenylyl cyclase inhibition.4,31,40,41 Moreover, recently it was demonstrated that LPAR1 can be considered a novel neural stem/progenitor cells (NSPC) marker as it has an interesting expression pattern in the adult dentate gyrus.42 LPAR2 signaling is also involved in cell survival and migration, immune function, myelination, and, like LPAR1, LPAR2 contributes to several aspects of nervous system development, function, and injuries.4,43 LPAR3 mediates Ca2+ mobilization, adenylyl cyclase signaling, MAPK activation, and presents strong expression in human heart, testis, prostate, pancreas, while it is less expressed in human lung, brain, heart, stomach, placenta, spleen, and thymus.4,5,40
In 2012, a new intracellular pathway activated by LPA was dissected. In an attempt to find the components of fetal bovine serum capable of inducing the nuclear localization of yes-associated protein (YAP), the effector of the Hippo pathway, the results pointed out that these components were bioactive lipids.44,45 The authors demonstrated that both sphingosine-1-phosphate (S1P) and LPA are capable of promoting YAP dephosphorylation in Ser127 which leads to YAP translocation to the nucleus, inducing the expression of several target genes of the Hippo pathway such as Cyr61 and connective-tissue growth factor (TGF). LPA was able to induce inactivation of large tumor suppressor kinase 1 (Lats 1) activity, an upstream component of the hippo pathway that marks YAP for degradation, while another upstream component, MST 1 showed no change of its kinase activity after treatment with LPA. Using specific chemical inhibitors or silencing RNAs, it was concluded that the actions of S1P occurs through its binding with the S1P2 receptor and not with S1PR1 or S1PR3 while the action of LPA occurs preferably through LPAR1 and LPAR3 that lead to activation of Rho GTPases inducing cell proliferation or cell migration.44,46,47 Interestingly, LPA is also able to bind to an intracellular receptor, regardless of its interaction with its receptors LPAR1–6. McIntyre and colleagues demonstrated that LPA is able to bind directly to the Peroxisome proliferator-activated receptor-gama (PPARγ) transcription factor and activates expression of a PPAR-responsive element reporter.48
Several interesting works pointed to the involvement of LPA receptors in cell motility. Different actions of LPAR1, LPAR2, and LPAR3 have been demonstrated, for example, in endothelial cells during chemotherapy using cisplatin or doxorubicin. One study reported that chemotherapy itself inhibited the expression of those LPA receptors. In addition, the motility was lower in the treated than in the untreated cells. Noteworthy, such effect was abolished when pancreatic cancer cells (PANC-1) were cocultured with treated endothelial cells, therefore suggesting that this effect was due to the expression of LPA receptors in endothelial cells.49 This work aimed to assess the LPA receptors’ roles in cellular responses during chemotherapy and to identify molecular targets involved in proceeding an effective antiangiogenic treatment in combination with chemotherapy.49 Surprisingly, it has been found that LPAR1 knockdown on endothelial cells increases cell mobility, while knocking down LPAR2 and LPAR3 inhibits it, suggesting that LPAR1 negatively regulates cell mobility and LPAR2 and LPAR3 positively regulate it in endothelial cells.49 This result demonstrated that LPA signaling is an important mechanism involved in modulating cell migration not only during development but also during pathological conditions and in the responses to treatment. Hence LPA signaling might be considered a promising target for cancer treatment, further studies still being necessary to confirm this hypothesis.
LPAR4, the first receptor described with similarity to P2Y purinergic receptors, can couple with the different G protein, Gαs, Gαi, Gαq, and Gα12/13.4,39,50,51 This receptor is strongly expressed in the human ovary, while it is less expressed in other organs, such as thymus, pancreas, colon, brain, heart, small intestine, testis, prostate, and spleen.4 Interestingly, LPA-induced cell migration through LPAR1 is inhibited when LPAR4 is activated, which shows a negative regulation loop of cell mobility induced by LPA. LPAR4-deficient mouse embryonic fibroblasts showed higher response to LPA-induced cell migration and reconstitution of LPAR4 on LPA-negative cells showed a less motile phenotype.37 Moreover, in B103 neuroblastoma cells without endogenous LPA receptors, the co-expression of ectopic LPAR4 with LPAR1 impaired LPAR1-driven migration and invasion.37 Another study also demonstrated that ectopic expression of LPAR4 in SQ-20B cells, a head and squamous cell carcinoma, expressing normal level of endogenous LPAR4, abolished LPA-induced motility as well as by Ki16425, Rac1, or Y-27632 LPA receptors inhibitors.52 More recent, it was shown that the LPAR4 knockdown in human pancreatic PANC-1 cells, PANC-sh4, the cell motility were enhanced accompanied by markedly stimulation on invasive activities.53 The same effect occurred on LPAR5 receptor knockdown PANC-1 cells, but not on LPAR6 receptor knockdown PANC-1 cells, which shows the distinct roles of LPA receptors in pancreatic cancer cells.53 In addition to other well-described functions of LPAR4 activation, such as cell aggregation, cell adhesion,54 vascular development,55,56 and osteogenesis regulation,57 it has recently been demonstrated the molecular mechanism of hypertensive response mediated by LPA.58 Different LPA analogs acted as LPAR agonizts, leading to hypertensive responses, and pretreatment with a Rho-kinase inhibitor that blocked Gα12/13 signaling attenuated this hypertensive effect induced by LPA. Corroborating this finding, another study reported that this effect is decreased in LPAR4 knockout mice, a Gα12/13-coupling LPA receptor.58 That study suggested a correlation between circulating LPA, produced by ATX, and the increase in blood pressure through its receptors, particularly LPAR4.
On the other hand, LPAR5 binds to Gαq and Gα12/13, Gαi and Gαs.3,4,39,59 LPAR5, identified in 2006, is highly expressed in the spleen and less expressed in the heart, small intestine, placenta, colon, and liver.4 This LPA receptor also negatively regulates cell motility as LPAR4 and is implicated in the induction of chemokine release.3 Moreover, LPAR5 is also responsible for LPA-induced neurite retraction and cytoskeleton stress fiber formation through a signaling cascade downstream of Gα12/13.60
LPAR6 binds to Gα12/13 and is the most recently discovered LPA receptor. Mutations in this receptor have been identified in patients with autosomal recessive hypotrichosis. Such findings indicate that LPAR6 may be a potential therapeutic target for human autosomal recessive hypotrichosis (alopecia).61 LPAR6 continues to be investigated to better understand its functions.3 Interestingly, LPAR6 was also related with prostate cancer progression in a past work.62 Recently, the crystal structure of LPAR6 was determined with the purpose of elucidating the ligand recognition mechanism of the non-Edg family of LPA receptors. This study was important for the identification of LPAR6 antagonists, which might provide with new therapeutic drugs for cancer treatment.63
GPR87 is also known as a LPA receptor since it was deorphanized in 2007 by Tabata and collaborators. Using CHO cells transfected with pcDNA5 vector containing GPR87-Gα16 fusion gene, they examined the effects of P2Y and P2Y-related receptor ligands on intracellular Ca2+ ([Ca2+]i) movements of GPR87 cells and showed that LPA significantly stimulated a [Ca2+]i increase in a dose-dependent manner. They also demonstrated that this effect was not related with the endogenous LPA receptors comparing LPA effects on the wild type and GPR87-G16α expressing cells. As a result, the GPR87 cells showed a higher Ca2+ response to LPA than the wild type cells. Besides that, different P2Y and P2Y-related receptor agonist such as ATP, UDP-glucose, phosphoribosyl pyrophosphate, α-ketoglutarate, leukotrienes, and phospholipids were analyzed to intend its capacity on activate GPR87 but only LPA significantly increased GPR87-mediated [Ca2+]i levels.64 Furthermore, they used siRNA to knockdown the expression of GPR87 which completely inhibited the LPA-induced Ca2+ response. The amino acids sequence homology of GPR87 with LPAR1, LPAR2, and LPAR3 is insignificant while share 27 and 25% similarity with LPAR4 and LPAR5, respectively. By the other hand GPR87 showed 41–48% amino acid identity with P2Y12, P2Y13, and P2Y14 receptors.64 The GPR87 gene is expressed at high levels in testis and brain and in other tissues, such as placenta, ovary, prostate, and skeletal muscle in a lower level, but it’s not expressed in heart, lung, kidney, liver or intestine.64
In 2013, a study addressed the role of GPR87 in urothelial carcinoma of the bladder through its ligand LPA. They showed a substantial expression of GPR87 gene in five human bladder cancer cells lines (HT1197, J82, TT112, RT4, and TCCSUP) and its correlation with cellular viability following gene silencing experiment. After GPR87 mRNA level suppression with transfection of Ad-shGPR87 until 120 h, they observed a significant reduction on cellular viability in a dose-dependent manner. Moreover, 38 of 71 non-muscle-invasive bladder cancers (54%) was positive for GPR87 immunostaining, were the positive-staining ratio in high-grade tumors showed a tendency to be higher than that in lower-grade tumors. GPR87-positive tumors also showed a higher median Ki-67 index than that in GRP87-negative tumors, thus a strong correlation with cell proliferation.65 As they have also reported enhanced expression of ATX in prostate cancers correlated with tumor grade and capsular invasion, they suggest that GPR87 is a potential prognostic marker for the progression of non-muscle-invasive bladder cancers.65,66 Also, in 2013, the same research group used A431 cells, a human epidermoid cancer cell, which previously have been shown to express GPR87, to study whether GPR87 acted as an LPA receptor.67 They demonstrated that when the expression of GPR87 was decreased by treatment with gpr87-specific siRNA, LPA-induced colony dispersal was significantly diminished. Before, Kam and Quaranta showed that A431 cells treated with LPA rapidly and synchronically dissociate cadherin-based adherents junctions68. Thus, they predicted that the LPA-induced cell scattering was via activation of GPR87 in A431 cells. The signaling pathways underlying this process were addressed and Erk, JNK, p38, and Akt were significantly activated 10 min after addition of LPA. Further, A431 cells were pretreated with the signaling pathways inhibitors, LPA-induced A431 colony dispersal was blocked completely.68
Another orphan GPCR, P2Y10 receptor was identified as LPA and S1P receptor in 2008.69 LPA was capable to evoke intracellular Ca2+ increase in the CHO cells which were inhibited by LPA receptor antagonists. In order to corroborate with these findings, they performed an assay using a siRNA designed for P2Y10 receptor into the P2Y10-CHO cells and showed that LPA-induced Ca2+ increased was blocked. Reverse transcription polymerase chain reaction (PCR) analysis showed assed its expression in different mice tissues, as reproductive organs, brain, lung, and skeletal muscle suggesting physiological roles.69
The transient receptor potential vanilloid 1 (TRPV1) ion channel responds to various stimuli, including LPA, which is also associated with neuropathic pain. Structural affinity determines the activation of TRPV1 and among the natural LPA analogs, Morales-Lázaro et al. found that only LPA 18:1, alkylglycerophosphate 18:1, and cyclic PA 18:1, all with a monounsaturated C18 hydrocarbon chain, are capable to activate TRPV1.70 The involvement of LPA in pain was first demonstrated in bone cancer pain through to its capacity to potentiate TRPV1 in dorsal root ganglion (DRG) neurons.71 Immunohistochemistry staining showed high levels of co-localization of LPAR1 with TRPV1 in DRG neurons and TRPV1 current were potentiated by LPA in isolated DRG neurons.71 Recently, a study aimed to underlie the mechanism by which LPA activates peripheral sensory neurons involved in intractable and continuous itch sensations present in atopic dermatitis, neurogenic lesions, uremia, and cholestasis, since LPA is an itch mediator found in cholestatic itch patients.72 They used a cheek injection model and the experiments revealed that the LPA-induced itch was dependent of transient receptor potential ankyrin 1 (TRPA1) and TRPV1, which leads to conclude that TRPV1 is one of the potential targets for cholestatic itch future treatments.73
LPA and CNS development
LPA is undoubtedly very important during embryonic development, as this bioactive lipid signaling regulates processes, such as cell proliferation, survival, differentiation, adhesion, migration, and morphology.19,74,75,76,77,78,79,80 The concentration of LP was found to vary greatly from nanomolar to micromolar ranges among different embryonic tissues, such as the brain, choroid plexus, neural tube, blood vessels, meninges, CSF, and spinal cord.81,82 Acting through specific GPCRs in a paracrine or autocrine fashion, LPA influences the formation and maintenance of the central nervous system (CNS). It was already demonstrated that LPA influences neuron–glia interaction, driving events such as neural progenitor cells (NPCs) commitment and maturation, and oligodendrocyte differentiation.76,77,78,83 Moreover, it was also demonstrated that LPA within certain lower spectrum of concentrations may function as an extracellular signal inducing the proliferation of NSPCs and their differentiation to specific cholinergic-committed neurons and unspecific microtubule-associated protein-2 (MAP2)-positive neurons.84 Normal brain development depends on LPA signaling and LPAR modulation and it is well established that LPAR expression occurs in a shifting spatio-temporal arrangement.5 To date, six LPARs have been described (LPAR1–6 for human and Lpar1–Lpar6 for nonhuman), however, only Lpar1, Lpar2, Lpar4, Lpar5, and Lpar6 have been detected during brain development.4,85,86 In the developing brain Lpar1 and Lpar2 are principally expressed in the ventricular zone (VZ) of the cerebral cortex, which is correlated with initiation, progression, and decline of neurogenesis.32,87 Moreover, Lpar1-null mice exhibit usually 50% of perinatal lethality that is correlated with defective suckling behavior which may be due to olfactory bulb and cerebral cortex defects, although no obvious abnormalities in the cerebral cortex were observed.88 Another variant of Lpar1-null mice (called “Málaga variant” or maLPAR1-null mice) exhibited a reduced VZ, premature neural progenitor cell maturation, and an increase in cortical apoptosis that causes a defect on the cortical layers in adults.89 Recently, it was demonstrated that LPAR1 is also expressed in some distinct areas of the brain, such as the amygdala, where there is a possible crosstalk with the endocannabinoid receptor 1 (CB1), where the lack of CB1 receptor in the amygdala promotes the increase in LPAR1 in this area.90 Interestingly, it was demonstrated that exogenous LPA exposure of ex vivo cerebral cortex induces gyri-like folds and widening of the cerebral cortex wall due to an increase in terminal mitosis and a decrease in cell death of the NPCs within the VZ.75 Exposure of ex vivo Lpar1–Lpar2-null mice cerebral cortex to exogenous LPS did not show these same effects, demonstrating that it was receptor mediated.75 LPA/Lpar1 signaling was also demonstrated to be important for survival and development of dopaminergic neurons (DA).91 Furthermore, the absence of Lpar1 signaling reduces not only cortical neuronal development but also cortical oligodendrocyte function and myelination.83,89
When ATX, (ENPP2), major enzyme responsible for extracellular LPA production from the precursor LPC, is constitutively removed, it results in vascular and neural tube defects with embryonic death around E9.5.92,93,94 In zebrafish, it was demonstrated that Enpp2 coordinates oligodendrocyte differentiation, showing that the important role of LPA in the developing brain is evolutionarily conserved.95 It was also demonstrated that ATX regulates cell positioning and adhesion of neuronal progenitor cells located in the VZ of the cerebral cortex. Moreover, ATX depletion leads to proliferation defects and alterations in the cell cycle.96
In situ data have demonstrated that, besides Lpar1, Lpar2, Lpar4, Lpar5, and Lpar6 are also expressed in the early developing brain.85,97 Lpar4 expression was observed mainly in the prospective midbrain–hindbrain boundary and in the margins of the neural folds, from the level of the rostral hindbrain to the forebrain, at E8.5. Later, at E11.5, it is also expressed at the midbrain and hindbrain.85 Lpar5 was strongly expressed at E8.5 in the rostral midbrain, forebrain and margins of the neural folds, and exhibited a diffuse expression in the developing brain including the choroid plexus.85 Lpar6 was observed in the neural plate of Xenopus neurulae and its depletion results in forebrain and hindbrain defects.97
As cortical neurogenesis occurs in mice from E10 to E18, LPA signaling plays an important role in NPCs’ proper commitment and influences directly young postmitotic neurons.75,98 It was shown that LPA can affect in vitro and in vivo neuronal migration by altering the actin cytoskeleton and promoting microtubule rearrangement in neurons.99,100 Moreover, LPA also improved the commitment of neuroblasts to the neural lineage through the Lpar1–Gαi pathway, as the exposure of Lpar1–Lpar2–null neuroblasts to LPA failed to recapitulate the same result.98
Neuron–glia interaction plays a pivotal role in brain development and, quite expectedly, LPA is also involved in this interaction. In vitro, LPA-primed astrocytes induced neuronal commitment by activating Lpar1–Lpar2. These events were mediated by modulation of laminin and fibronectin organization by astrocytes. The activation of Lpar1–Lpar2 induced astrocytes to secrete epidermal growth factor ligands (EGF and TGFα) and the activation of the MAPK cascade and protein kinase A (PKA) signaling pathways in response to LPA.76,77,78 LPA also stimulates, via the LPAR1, neuronal differentiation of neural progenitors, cortical neuroblasts, and early cortical neurons.98 There is no doubt that LPA is an essential factor for cortical neurogenesis, as it is able to induce depolarization of mouse cortical neuroblasts and to activate electrical responses in neuroblasts via gamma-aminobutyric acid signaling.101,102
LPA and CNS angiogenesis
Brain endothelial cells lie at a strategic position and function as a barrier for the exchange between circulating lipids and the brain parenchyma. Interestingly, an increasing set of in vitro and in vivo data points to a direct effect of the blood-borne bioactive LPA ligand in CNS angiogenesis and endothelial cell permeability, highlighting the importance of LPA signaling in various CNS pathologic conditions where vascular integrity is compromised. During development, after the initial phase of angiogenic sprouting from the perineural vascular plexus, CNS endothelial cells acquire unique features such as expressing specific tight junction proteins and membrane transporters establishing a selective blood–brain barrier (BBB) to regulate ion and molecule transfer to the cerebral microenvironment and ultimately ensure proper neural function.103 A more consistent body of literature has addressed the role of LPA signaling in BBB integrity rather than a role on CNS angiogenic sprouting and regulation, although, genetic deletion of ATX results in embryonic lethality due to the severe yolk sac and embryonic vascular malformations,92 demonstrating that LPA signaling is essential for overall vascular development, likely through LPA receptor-coupled G proteinα13.104 Data from genetic deletion studies of Lpar1–2 indicate a mild vascular phenotype, with some incidence of perinatal frontal hematoma,40,43 while Lpar4-deficient mice display several developmental vascular defects leading to embryo lethality.55 In a recent study, Yasuda and collaborators demonstrated the synergistic role of the LPA receptors Lpar4 and Lpar6 during vascular development. Lpar4/Lpar6 double-knockout (DKO) embryos dye around E11.5, presenting enlarged aortae, reduced vascular tree in the cephalic region, as well as yolk sac vascular defects.105 Further dissection of the molecular mechanism shows that LPA through its endothelial receptors Lpar4/Lpar6 represses β-catenin- and Notch intracellular domain-mediated dll4 activation via Gα12/α13-dependent activation of YAP-Taz.105
High levels of LPA have been shown to be increased in several models of brain pathologies in which the BBB is disrupted, such as intracerebral hemorrhage, cerebral ischemia, and edema.106,107,108 Also, in glioblastoma (GBM), a highly vascularized brain tumor with associated BBB disruption, ATX is also strongly expressed.109 In addition, LPA modulates cerebral circulation, as shown by dose-dependent local application under a cranial window in piglets.106 Dissecting the mechanisms of LPA in brain endothelial cell biology and BBB maintenance will be critical to understand its role in angiogenesis-mediated brain diseases as well as a possible therapeutic target both to treat brain diseases and to enhance drug delivery.
LPA receptor mRNA profile in vitro studies determined that Lpar1–4 are expressed in human and bovine brain microvascular endothelial cells (BMECs)110 and Lpar6 is present in rat BMECs.108 While in vivo studies demonstrated that Lpar1–4 mRNA transcripts were present in whole brain homogenates, robust Lpar1, 4, and 5 mRNA expressions were detected in brain capillary-enriched fractions.110 The effects of LPA on BBB permeability were determined through transendothelial electrical resistance and fluorescence transfer across the endothelial monolayer studies which demonstrated that LPA disrupts BBB integrity in different mammalian BMECs, including human.108,110,111,112 LPA induced a disruption in the junctional expression pattern of several BBB characteristic tight junction proteins, such as occludin, zonula occludens-1, and claudin-5 in rat BMECs,108 likely due to cytoskeletal rearrangements shown by F-actin stress fiber and focal contacts in LPA-treated porcine BMECs.111 Fulminant hepatic failure leading to BBB breakdown and cerebral edema were correlated with a significant increase in ATX serum levels in mice.108 The effect of stereotaxic microinjections of LPA in the mouse striatum leads to disruption of BBB as determined by Evans blue dye extravasation and ultrastructural electron microscopy analysis.113 Co-injection with the Rho and ROCK (Rho-associated protein kinase) signaling pathway inhibitor Y26732 indicated that LPA-induced BBB breakdown likely occurs through matrix metallopeptidase 9 (MMP9) and urokinase-type plasminogen activator (uPA) proteolytic enzyme activities mediated by the Rho/Rock signaling pathways.113 The BBB permeability induced by LPA was further analyzed in mice in terms of onset, duration, and magnitude using magnetic resonance imaging (MRI) and near-infrared fluorescence imaging.110 LPA-induced BBB breakdown occurred within minutes as demonstrated by MRI studies in mice using a low molecular weight contrast agent. Twenty minutes after LPA administration, the BBB integrity was restored as a second injection with contrast agent alone did not lead to increased signal, indicating that BBB disruption is a rapid and transient phenomenon. Ex vivo brain slice analysis by near-infrared fluorescence imaging using a large macromolecule contrast agent showed LPA-induced penetration in brain tissue within 20 min. The authors further tested the potential use of LPA to enhance drug delivery to the brain parenchyma. By co-injecting LPA with radiolabeled methotrexate, they showed that radioactivity detection in the brain fraction vs. plasma was increased in LPA-treated animals.110 Overall, these studies focused on the role of LPA in BBB integrity have highlighted a direct role of this LP on CNS endothelial cells. Although details of the mechanisms of LPA through G-coupled receptor signaling on non-CNS endothelial cells have been elucidated,114 only recently a study has focused on the role of LPA in different steps of CNS angiogenesis.115 Analysis of inducible endothelial-specific Lpar4/Lpar6 DKO retinas revealed a clear phenotype on retinal vascular expansion, tip cell, and sprouting morphogenesis. In this study, a series of elegant in vivo and in vitro assays allowed the authors to identify LPA plays an important role in CNS angiogenesis through Lpar4/Lpar6-mediated suppression of the angiogenic regulator dll4 through Gα12/α13-dependent activation of YAP-Taz.115 Future studies will be important to evaluate a possible role of these pathways in BBB integrity both in physiological and pathological conditions.
LPA and neuropathic pain
Pain is considered as an important clinical symptom common to several diseases. Furthermore, it is a warning signal related to the presence of actual or potential tissue injury.116 Pain might be classified into acute and chronic, and an additional classification subdivides pain into two other categories (neuropathic and nociceptive pain).116,117 Neuropathic pain is defined as “pain that arises as a direct consequence of a lesion or disease that affects the somatosensory system” where a transduction process is no longer needed to trigger neuropathic pain.118 Epidemiological studies have estimated that chronic pain affects nearly 100 million adults only in the United States,119 and neuropathic pain affects around 7–8% of the general population. Therefore, chronic neuropathic pain produces huge impacts on the quality of life of the affected individuals.120 Interestingly, multiple and sequential peripheral and central mechanisms have been proposed to explain the development and maintenance of neuropathic pain.121 Among those, LP are considered important players in the pathophysiology underlying the establishment of this type of pain. Of particular interest is LPA, a lipid mediator released under conditions of tissue injury. Both ATX, LPA, and its precursor LPC have been proved to participate in the mechanisms of neuropathic pain.122
The synaptic reorganization promoted by LPA via LPAR1 is one of the most discussed mechanisms in neuropathic pathophysiology. This complex process has been characterized by the presence of C-fiber retraction along with A-fiber demyelination and subsequent sprouting of dorsal root fibers, in which both processes would contribute to the development of allodynia, that can be interpreted as the occurrence of pain in response to innocuous stimuli.122,123,124 Although the molecular basis of such synaptic reorganization induced by LPA have not yet been totally clarified, there is evidence that partial sciatic nerve ligation (pSNL), an experimental model of neuropathic pain, promotes demyelination of dorsal root fibers through a pathway that encompasses the activation of RhoA and ROCK.125 LPA signaling via Gα12/13 activates the GTPase RhoA, which is then translocated to the plasma membrane where it activates multiple effectors such as Rho-kinase and ROCK.126 In fact, there is also evidence providing a direct link between Rho-ROCK and the development of neuropathic pain.127,128,129,130 However, that is not the only process associated with LPA-induced synaptic reorganization. Previous studies showed that LPA can induce the silencing of myelin basic protein and its transcription factor Sox10 in cultured S16 Schwann cells,131 as well as demyelination and downregulation of myelin proteins in ex vivo cultures of dorsal root fibers.132 A further study demonstrated the occurrence of demyelination, downregulation of MAG, which correlates to the development of sprouting of sensory fibers, and damage of Schwann cell partitioning of C-fiber-containing Remak bundles in the dorsal root and sciatic nerve, following sciatic nerve injury.133,134 Interestingly, the same changes could not be demonstrated in the dorsal root of LPAR1 receptor-deficient (Lpar1−/−) mice. Nevertheless, ex vivo experiments with LPA revealed the presence of demyelination in the sciatic nerve, spinal nerve, and dorsal root. In addition, it was also demonstrated that dorsal root demyelination induced by nerve injury is significantly reduced in mice heterozygous for autotaxin (Atx+/−). Confirming those findings, the addition of LPC to ex vivo dorsal root fibers cultures failed to produce demyelination in the absence of recombinant ATX. However, pronounced demyelination occurred in the presence of ATX. Finally, the intrathecal injection of LPC resulted in potent dorsal root demyelination, which was lessened or even eliminated in Lpar1−/− or Atx+/− mice. Such results suggest that the mechanisms that mediate the conversion of LPC to LPA would be necessary to induce the demyelination associated with neuropathic pain.122
Based on the results from some studies, a feed-forward system, which would be responsible for an amplified LPA production following pSNL, has been proposed.122,124 According to this model, the production of LPA depends on the simultaneous stimulation with N-methyl-d-aspartate and substance P, causing a synergistic and strong activation of cytosolic and calcium-independent phospholipase A2 (c/iPLA2), which in turn converts phosphatidylcholine to LPC. Afterward, ATX converts LPC into LPA. LPA would then stimulate microglia, acting through LPAR3, leading to increased production and release of interleukin-1β (IL-1β), which would then promote an upregulation/activation of c/iPLA2 in the adjacent neurons. The final result is an amplified production of LPA, associated with peripheral nerve lesions.135,136 Such a model would explain the production of LPA following pSNL, in experimental models of neuropathic pain, which nonselectively stimulates different types of peripheral sensory fibers, a process that does not occur in experimental models of inflammatory pain, which preferentially stimulates C-fibers.122
It has also been reported that LPA also plays an important role in the early phase of neuropathic pain following peripheral nerve injury, since the LPAR1 antagonist Ki-16425 completely blocked LPA-induced neuropathic pain-like behaviors in a time-dependent manner. For instance, such blockage peaked at 3 h after the peripheral nerve injury. However, it was not maintained after this period. In addition, the administration of Ki-16425 at 3 h but not at 6 h after the peripheral lesion produced a reduction of substance P in the dorsal horn of the spinal cord and an upregulation in the expression of voltage-gated calcium channel alpha(2) delta-1 subunit in the DRG.137 Although those results indicate that LPA is mainly related to the early phases of neuropathic pain, it has been largely demonstrated that the production of LPA also stimulates microglia through LPAR3 mechanism, leading to a higher release of IL-1β, activation of c/iPLA2 in the adjacent neurons and consequent amplified LPA production.135,136 Moreover, acting through LPAR1, LPA mediates dorsal root demyelination, which has also been related to neuropathic pain-behaviors and LPAR1 signaling upregulates Ephrin B1 and the α2δ1 calcium channel subunit (CAVα2δ1) in the DRG.125,138,139 Interestingly, gabapentinoids (e.g., pregabalin and gabapentin), one of the most widely used groups of drugs used in neuropathic pain treatment, bind to CAVα2δ which makes the connection between LPA and CAVα2δ1 extremely useful and attractive for future pharmacological studies.
Distinct roles have been attributed to microglia and astrocytes in the mechanisms of LPA-induced neuropathic pain, being activated astrocytes responsible for the maintenance of neuropathic pain. With this respect, one study showed that the initial pSNL-induced LPA production in the dorsal horn is blocked by depleting microglia or inhibiting its activation (e.g., with minocycline or Mac1-saporin) but not by l-a-aminoadipate (l-AA), gliotoxin that promotes a pharmacological ablation of astrocytes.140 Such findings lead some authors to elaborate the hypothesis that the LPA produced through microglial activation might act on astrocytes, inducing the release of chemokines and therefore contributing to the chronic phase instead of the acute neuropathic pain.122
Confirming the findings from experimental studies, a recent clinical study evaluated the concentrations of 12 species of lysophosphatidic acids, LPC, ATX, and the phosphorylated neurofilament heavy subunit in the samples of CSF of neuropathic pain patients. Regarding the LPA species, LPA 16:0, 16:1, and 18:1 were correlated with the average pain intensity, while the other LPA species were not. Moreover, the total amount of ATX and LPC was not associated with the pain intensity, suggesting that those molecules would act more mediating the development of neuropathic pain through LPA production than directly inducing neuropathic pain. Noteworthy, the association between LPA 18:1 and the clinical pain (subjectively reported by the studied patients) has been proved to be responsible for LPA amplification through LPAR1/LPAR3 and microglial activation.141
In the search for novel therapies to treat neuropathic pain, LPA antagonists emerge as promising candidates. For instance, in a recent work, the novel LPAR5 antagonist AS2717638 proved to have high potency and selectivity and was able to cross the BBB and penetrate the CNS.142 The effects of AS2717638 on mechanical allodynia and also in thermal hyperalgesia suggest an important outcome of this LPA antagonist on both TRPV1 and non-TRPV1 expressing neurons. This potentially provides an advantage to duloxetine and pregabalin, two of the most common pharmacological agents used to treat neuropathic pain symptoms, and that preferentially act by suppressing the symptoms related to the activity of non-TRPV1-expressing neurons. In fact, the analgesic effects of the novel LPAR5 antagonist AS2717638 might encompass not only neuropathic but also inflammatory pain.142
Studies also linked LPA to the neuropathic component of osteoarthritis (OA).125 In addition, a very recent study showed gender differences in several features of LPA-induced joint neuropathy.143 For instance, females exhibited more pronounced mechanical allodynia and saphenous nerve demyelination than males following LPA injection. Interestingly, blockade of voltage-gated sodium channel Nav1.8 decreased the allodynia driven by LPA treatment. Such results suggest sex-specific effects on LPA-induced joint neuropathy and a possible role of voltage-gated sodium channels in such mechanisms with possible future application in the development of pharmacological agents to treat OA and more particularly its neuropathic component.143
LPA and neurodegenerative diseases
LPA signaling has been reported to influence synapse formation, synaptic transmission and survival of mature postmitotic neurons,144,145 playing an important role in cerebral cortex formation and function.76,77,78,79 However, the impairment of LPA signaling has also been implicated in several neurological disorders such as schizophrenia, Alzheimer’s, and Parkinson’s disease (PD).146,147
PD is a progressive neurodegenerative motor disorder characterized by the selective loss of DA in the substantia nigra, that results in a pronounced depletion of dopamine in the striatum, to which neurons from the substantia nigra send their projections.148
In 2014, it was demonstrated, using PD rat models induced with 6-OHDA (6-hydroxy dopamine), that LPA signaling plays a role in the degeneration of DA and in the differentiation of adult rat mesenchymal stem cells (MSCs) into DA neurons. MSCs treated with LPA developed characteristic neuronal morphology and expressed the neuronal marker, neuron-specific enolase, while the expression of glial markers was absent. In this study, although a decrease in the Lpar1 expression in the substantia nigra of 6-OHDA PD rats was reported, there was no significant change in the striatum. The downregulation of Lpar1 expression in the 6-OHDA PD model, along with the differentiation of MSCs into DA neurons following LPA treatment, suggests that LPA/Lpar1 signaling pathway plays a key role in the development and maintenance of DA neurons. In addition, the low expression of Lpar1 in the 6-OHDA PD model may result in the death of DA neurons and thus contribute to the pathogenesis of PD.91
In Alzheimer’s disease (AD), the dysfunction in LPA signaling is associated with an impairment in expression and activity of autotaxin.149 Both LPA and ATX are highly expressed in the CNS and have been implicated in AD pathogenesis.150,151 Decreased levels of antioxidant enzyme activity and increased lipid peroxidation were observed in AD patients in comparison to healthy non-demented control subjects.152,153 The increase in lipid peroxidation triggers the neurotoxic events described in AD.154 One of the most vulnerable lipids to peroxidation is LDL.149 The oxidized LDL, whose LPA is the major bioactive component, disrupts the BBB, contributing to the pathogenesis of AD.155 Besides, high levels of oxidized LDL are a key contributor to β amyloid (Aβ) production and deposition in the brain parenchyma, which is the main pathophysiological mechanism in AD.156 The Aβ accumulation results in hyperphosphorylation of tau protein, which leads to synaptic failure and consequently, neuronal death.157
LPA signaling is present in other diseases that are a prominent risk factor for the development of AD, such as brain trauma.149 In traumatic brain injury (TBI), the increase in LPA activity derives from the upregulated expression of LPAR1, LPAR2, and LPAR3.4 Autopsy findings, from patients who died after brain trauma, showed diffuse Aβ plaques in the adjacent areas to the lesion site.158 The mechanisms responsible for the Aβ accumulation are most likely to be related with the BBB disruption, leading to ischemic insult and increasing the activity of β and γ secretases.159 In TBI, the increase of LPA and its metabolites seems to be involved in the early pathologic processes after the trauma, such as neurite retraction, reactive gliosis, inflammation, and cell death.160 Using a rat model of neurotrauma, Mcdonald and coworkers demonstrated that LPA increases diffusely after the injury, whereas LPA metabolites increases in white matter, in the hemorrhagic pericontused gray matter regions both LPA and its metabolites were increased, these events results in axonal injury and cell death, especially in the white matter.161
In patients, LPA levels increases in the blood and CSF within 24 h in TBI patients, indicating that LPA could possibly be one of the first mediators responding to the injury and suggesting it may be a biomarker for subsequent cellular pathology.161
The association between LPA and brain trauma provides indirect evidence that the impairment of LPA signaling can contribute and predispose to the pathogenesis of AD.162 In view of these evidences, ATX, LPA, and its receptors become promising therapeutic targets in neurological disorders.
LPA and cancer
Several studies have linked increased LPA signaling with the biological events related to carcinogenesis and tumor progression, as malignant transformation, increased cancer stem cell proliferation, enhanced invasion and metastasis, reprogramming of tumor and metastatic microenvironments, and development of therapy resistance.163,164
ATX and LPARs present numerous roles in tumor cells. Briefly, LPAR1 and LPAR4 have been mostly associated with cell motility and invasion.37,165,166,167 LPAR2 has been connected with cell survival and apoptosis inhibition.168 Both LPAR3 and LPAR6 have been implicated in the promotion of tumor proliferation, migration, invasiveness, aggressiveness, and progression of different cancers;169,170,171,172 besides, LPAR3 has also been related to the regulation of dendritic cell migration, possibly having a role in antitumor immunity,173 whereas LPAR5 has been involved with immune response inhibition and cancer progression.174,175,176,177 ATX and LPA are released by both tumor cells and cells of the tumor microenvironment (TME) increasing invasion, suppressing antitumor response and contributing to therapy resistance of cancers.176,177,178,179,180,181,182 LPA–LPAR signaling has been demonstrated to promote epithelial–mesenchymal transition (EMT), essential for cancer stem cells invasive and metastatic properties.183,184,185,186,187
Another important feature of LPA in TME is the induction of proinflammatory cytokines production, such as IL-6 and IL-8, impacting the progression and metastasis of tumors. Moreover, there is a positive loop in which the inflammatory milieu induces more LPA–LPAR signaling and chronic inflammation can lead to malignant transformation.188,189
LPA is a highly studied molecule in cancer. Based on cancers’ importance—regarding high incidence and mortality, investigation prevalence, and our group expertize, we decided to devote this section to discuss the publications on LPA and GBM, ovarian, breast, and colorectal cancers over the last 6 years.
Regarding colon cancer, it has been shown that LPA and WNT induce proliferation of these cells by activating β-catenin in a synergistic fashion and that Krüppel-like factor 5 (KLF5) acts as a cofactor in this process, facilitating the interaction between β-catenin and TCF4.190 Moreover, LPA has been linked to the induction of DNA synthesis and migration of colorectal cancer cells, probably in an EGFR-independent manner.191 Still, LPA has been correlated with S1P in colorectal cancer, since there were higher mRNA levels of LPAR2 and sphingosine kinase (SphK1), an oncogenic kinase that produces S1P, in tumor tissues when compared to normal tissue.192 Furthermore, it has been demonstrated that LPAR4 and LPAR6 negatively regulate the proliferation and migration induced by LPA on colon cancer cells.193 LPA also was shown to induce the proliferation of colon cancer cells via RhoA-ROCK and STAT-3 pathways associated with cell cycle progression.194
In breast cancer, it was shown that the high levels of ATX, which culminates in increased LPA production, are mainly originated from adipose-derived stem cells and adipocytes.195 An interesting recent study showed a direct mechanism involving LPA that links obesity and breast cancer progression, where it was revealed that in a breast cancer model under chronic diet-induced obesity LPA/PKD-1-CD36 signaling stimulated microvascular remodeling.196 LPA is also implicated in breast cancer metastasis to bone, as it induces the expression of IL-8, IL-11, and osteolytic cytokines in breast cancer cells.197 In another work, the proliferation and migration promoted by LPA and EGF in breast cancer cells were inhibited not only by n-3 fatty acid eicosapentaenoic acid but also by free fatty acid receptor (FFAR) agonizts, lighting up a possible anticancer role for FFAR.198 Concerning breast cancer cells motility, it was shown that LPA induces two different signaling pathways: Rho/ROCK, inducing a slow, directional, coherent, and consistent movement; and Gαi/o and Gα11/q dependent, promoting a lessen directionality and increase independent movement, favoring cell dispersal; so the balance between them defines the migratory reaction of cells, all depending on the cellular or microenvironmental context.199 Still, LPA was proven to induce breast cancer invasion via YB-1/EZH2/amphiregulin signaling axis200 and RhoA/ROCK/MMPs signaling pathway;201 further, curcumin was unprecedentedly shown to inhibit this effect by attenuating the RhoA/ROCK/MMPs pathway.201 In contrast, it has recently been reported that LPAR6 is downregulated in breast cancer, relating it to poor prognosis, which might implicate LPAR6 as a tumor suppressor gene in these cancers.202
LPA has a tremendously described influence on ovarian cancer. It acts on the EMT, glucose metabolism, cell migration, among other roles; and also functions as a biomarker. Recently, it has been shown that LPA can induce EMT through the activation of Wnt/β-catenin signaling183 as well as via Gαi2, Src, and HIF1α signaling nexus,183 and by repressing SIRT1.186 LPA also stimulates glycolytic metabolism through induction of HK2, promoting tumor cell proliferation203 and pseudohypoxic response, inducing aerobic glycolysis and has a pro-tumor role.204 It is also suggested that this glycolytic shift promoted by LPA is a priming event in the transition from normal to cancer-associated fibroblasts.205 LPA has been described as a biomarker of ovarian cancer regarding its high levels in plasma206,207 and the expression of its receptor in tissue,208 leading to the investigation of a biosensor to detect LPA in the serum of patients209 and even to the development of a device for fast detection of plasma LPA.210 Nevertheless, this plasma detection should be addressed carefully since it has been reported that sample processing and analysis could interfere in LPA levels.211 Still, it has been reported that LPA might also become a biomarker in vaginal fluid, but only for endometrioid ovarian cancer, being even more sensitive than the classical marker CA125.212 LPA promotes migration of ovarian cancer cells through different mechanisms/signaling pathways: RhoA/ROCK and Rac1/PAK,213 CXCL12‑CXCR4,214 uPA/uPAR,215 Gαi2/Src/β-pix/Rac,216 FOXM1,217 LPAR1,218 NHERF1/cpERMs,219 ETS-1,186 LPAR1/LPAR2/ERM,220 LPAR2/ERK, LPAR3/ERK, and LPAR6/AKT.221 LPA has also been implicated in the maintenance of cancer stem cell characteristics through ATX–LPA–LPAR1–AKT1 signaling axis in an autocrine loop.222 Moreover, LPA treatment leads to morphological change and alterations in the relative protein-to-lipid ratio on the surface of ovarian cancer cells.223 Interestingly, it has been shown that LPA in ascites is produced via PLA2 and ATX secreted by tumor-associated macrophages, where the 20:4 LPA species seems to have an important role in tumor recurrence.224 In fact, regarding the use of LPA signaling blockade as a possible therapy, it has been found that it increases cell death in response to chemotherapeutic agents.225
Lastly, GBM is the most common and deadliest primary malignant tumor of the CNS,226,227 and it has been a main focus of interest of our group. In this LPA context, ATX is highly upregulated and favors GBM migration/invasion.109,228,229,230 In GBM, LPAR1 signaling was shown to be important for tumor cell migration/invasiveness.109,229,231,232 Also, LPA signaling is implicated in tumor recurrence, and cancer stem cells have higher expression of LPAR1.231,233 Finally, our group recently reported that high LPAR1 and ATX correlates with glioma aggressiveness and patient prognosis.234 Also, we described that microglial cells, the major source of LPA in the CNS,230 secrete LPA in the TME, which induces GBM cell migration and proliferation via LPAR1.234
All these findings show that LPA signaling is an important pathway in carcinogenesis, tumor progression and establishment of the TME. In this sense, components of the ATX–LPA–LPARs axis can be important biomarkers or therapeutic targets for the treatment of different cancers,231,235 some of which will be reviewed in the next topic.
LPA signaling as a therapeutic target
Since the discovery that LPAR1 is overexpressed in samples from patients with idiopathic pulmonary fibrosis (IPF) and kidney fibrosis, several groups tried to target LPARs or ATX with small molecules to inhibit fibrosis. IFP is a chronic disease characterized by excessive collagen deposition in the lungs. ATX is known to be involved in the pathogenesis of IPF by increasing collagen production and migration of inflammatory cells to the lungs.236 In addition, LPAR1 knockout mice show a reduction in both renal and pulmonary fibrosis.237,238,239
Amira Pharmaceuticals developed the first highly selective LPAR1 inhibitors, AM966 and AM095. In vivo, oral administration of AM966 and AM095 reduced lung fibrosis, lung collagen deposition, vascular leakage, and inflammation.240,241 Moreover, AM095 inhibited kidney fibrosis induced by unilateral ureter obstruction and dermal fibrosis induced by bleomycin in murine models.238,240 Both inhibitors antagonize LPA-induced Ca+ release selectively in LPAR1 expressing cells by blocking the binding of guanosine 5′-O-[gamma-thio]triphosphate (GTPγS) to membranes induced by ligation of LPA to LPAR1.238,240,241
Other inhibitors of LPAR1 have already been tested in patients with IPF: BMS-986020 was tested in a phase 2 trial between April 2013 and February 2016. Patients had a minor improvement in disease control when compared to the placebo group, as stabilization of forced vital capacity (FVC) and improvement of lung fibrosis and inflammation biomarkers (C3M and adiponectin). Nevertheless, 80% of the patients in the treated group experienced hepatic adverse effects, and the trial was early terminated due to 3 cases of BMS-986020-related cholecystitis.242
Another phase 2 clinical trial was conducted with a small molecule antagonist of LPAR1 (SAR100842) for patients with diffuse cutaneous SSc. Overall, SAR100842 was safe and 80% of the patients in the treated group reported only minor adverse effects, compared to 71% in the placebo group.243
Ono Pharmaceuticals developed a selective ATX inhibitor, ONO-8430506. It is a tetrahydrocarboline derivative with a half-maximal inhibitory concentration (IC50) of approximately 10 nM.244 This compound was able to block the formation of LPA in the plasma of rats even after 24 h of treatment. The effect of ONO-8430506 endures for a longer period when compared to other ATX inhibitors.244 Since LPA induces the contraction of the urethra, the authors tested if ONO-8430506 could also inhibit this effect. The compound was able to decrease intraurethral pressure similarly to clinically used tamsulosin, an α1-adrenoceptor antagonist, without changing mean blood pressure.244 This inhibitor was also proven to have antitumor effects in preclinical metastatic breast and thyroid cancer models,188,245,246 showing the versatile potential of ATX inhibitors for the treatment of human diseases.
Recently, GLPG1690, a first in class drug that selectively inhibits ATX with an IC50 of approximately 100 nM247 was tested for the treatment of IPF. In a mouse model of bleomycin-induced pulmonary fibrosis, GLPG1690 showed efficacy in reducing extracellular matrix deposition in the lungs and LPA levels in plasma. GLPG1690 showed superior results compared to pirfenidone and was similar to nintedanib, a tyrosine kinase inhibitor, in reducing the Ashcroft fibrotic score and collagen levels.247 GLPG1690 was already tested in humans in order to test the safety, pharmacokinetics and pharmacodynamics of its doses,248 and was well tolerated in a Phase 2a study (FLORA study) involving 23 patients with IPF. Treated patients had reduced plasma LPA C18:2 levels and showed improvement of disease control, such as stabilized FVC after 12 weeks of treatment.249 Maher et al.249 started in November 2018 two identically designed phase 3 studies (Isabela I and II) to test GLPG1690 in 750 patients with IPF (see Table 3).
Finally, a new selective ATX inhibitor, BI-2545, was developed based on a previously published inhibitor by Pfizer (PF-8380). This improved inhibitor was able to reduce plasma LPA levels by 90% and with a higher in vivo potency compared to PF-8380.250
In the last 10 years, researchers have searched for multi-target drugs in order to treat complex diseases. IFP is also correlated to lung cancer which occurs as a late complication of the fibrotic tissue. EGFR inhibitors are effective in the treatment of non-small cell lung cancer (NSCLC). Thus, the discovery of a dual inhibitor for EGFR and ATX could be of great importance for treating not only IFP but also its late complications as NSCLC. Tetrahydropyrido[4,3-d]pyrimidine was found, by in vitro cytotoxic and enzymatic assays, to inhibit both ATX and EGFR with anticancer and antifibrotic activity superior to gefitinib, an EGFR inhibitor clinically used for the treatment of lung, breast and other cancers with overactive EGFR signaling. Tetrahydropyrido[4,3-d]pyrimidine also inhibits the expression of fibrotic markers, as TGF-β and tumor necrosis factor alfa (TNF-α).251
LPA and ATX play key roles in the progression of various cancers since one of the major hallmarks of cancer is cell migration. GBM is the most malignant brain tumor with high infiltration capacity in the cerebral parenchyma and both ATX and LPAR1 are highly expressed in several types of brain cancers. Motility of GBM cells seems to depend on ATX and LPAR1. Treatment of GBM cell line with LPAR1 antagonist, Ki16425, inhibits dramatically migration of these cells in vitro.110,234 Both α-bromomethylene phosphonate, a dual LPA receptor inhibitor/ATX inhibitor, and PF-8380, an ATX inhibitor, delayed in 20 days the growth of GL261 GBM cells injected in the hind limbs of nude mice.252,253
Since LPA induces secretion of proinflammatory cytokines and excessive scar formation, inhibition of the LPA pathway may also be an efficient strategy to improve brain and spinal cord damage. A mouse monoclonal antibody against LPA, called B3, was able to abolish the inhibitory effect of LPA on neurosphere formation and neuronal differentiation of human embryonic stem cells in vitro. Furthermore, B3 was also able to decrease astrogliogenesis, activation of microglia and neuronal apoptosis in a mouse model of spinal cord injury, reducing ERK phosphorylation and Rho-GTP levels.254 LPA is also highly secreted in TBI, both in humans and in mouse models as mentioned above. This antibody later renamed LpathomabTM reduced the size of hematoma and lesion 7 days after TBI, and also improved mice’ motor functions.162 In addition, LpathomabTM decreased IL-6 expression, an inflammatory cytokine correlated with poor outcome after TBI. This led the authors to conclude that anti-LPA antibody therapy could have neuroprotective effects following brain injury.162
In addition, it has been suggested that LPA and especially variations in the levels of LPA within different body fluids (e.g., serum, plasma, saliva, seminal plasma, and follicular fluid, among others) might be used as potential biomarkers to the determine the diagnosis and severity of diseases.255,256 In fact, LPA has been considered a biomarker in the diagnosis of ovarian cancer.257 Moreover, higher of LPA levels have been detected in the plasma of patients with renal failure258 as well as in the serum multiple myeloma patients.259 The production of LPA in the lung of asthmatic patients as well as the value of polyunsaturated LPA molecular species obtained from the bronchoalveolar lavage fluid have been also discussed as possible biomarkers not only to stablish the diagnosis but also to determine the severity of the disease.260
Conclusions
In summary, the current literature points to LPA regulating diverse important biological processes, and the cell response depends on the environment, cell type and developmental stage. Targeting LPA receptors, particularly using antagonist and agonist molecules or knockout mice, research groups have demonstrated several roles for LPARs in many disease models and also during embryo development.261 There is strong evidence regarding a chief role of LPA in cancer and neurodegenerative diseases as we explored here. There is no doubt that LPA and its receptors, LPAR1 and LPAR3, are crucial not only for the establishment but also for the chronification of neuropathic pain.122 Considering the huge literature supporting the role of LPA in so many diseases, it is important to explore new pharmacological agents capable of blocking the actions of different species of LPA without producing significant side effects.
References
Moolenaar, W. H., van Meeteren, L. A. & Giepmans, B. N. The ins and outs of lysophosphatidic acid signaling. Bioessays 26, 870–881 (2004).
Tokumura, A. Metabolic pathways and physiological and pathological significances of lysolipid phosphate mediators. J. Cell. Biochem. 92, 869–881 (2004).
Stoddard, N. C. & Chun, J. Promising pharmacological directions in the world of lysophosphatidic Acid signaling. Biomol. Ther. (Seoul.). 23, 1–11 (2015).
Yung, Y. C., Stoddard, N. C. & Chun, J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214 (2014).
Choi, J. W. et al. LPA Receptors: Subtypes and Biological Actions. Annu. Rev. Pharmacol. Toxicol. 50, 157–186 (2010).
van Meer, G., Voelker, D. R. & Feigenson, G. W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell. Biol. 9, 112–124 (2008).
Stefan, C., Jansen, S. & Bollen, M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem. Sci. 30, 542–550 (2005).
Okudaira, S., Yukiura, H. & Aoki, J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 92, 698–706 (2010).
Pagès, C., Simon, M. F., Valet, P. & Saulnier-Blache, J. S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat. 64, 1–10 (2001).
Brindley, D. N. & Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 50(Suppl), S225–S230 (2009).
Aoki, J., Inoue, A. & Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 1781, 513–518 (2008).
Bandoh, K. et al. Molecular cloning and characterization of a novel human G-protein- coupled receptor, EDG7, for lysophosphatidic acid. J. Biol. Chem. 274, 27776–27785 (1999).
Bandoh, K. et al. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 478, 159–165 (2000).
Yanagida, K. et al. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 284, 17731–17741 (2009).
Vancura, A. & Haldar, D. Regulation of mitochondrial and microsmal phospholipid synthesis by liver fatty acid-binding protein. J. Biol. Chem. 267, 14353–14359 (1992).
Zhukovsky, M. A., Filograna, A., Luini, A., Corda, D. & Valente, C. The Structure and Function of Acylglycerophosphate Acyltransferase 4/ Lysophosphatidic Acid Acyltransferase Delta (AGPAT4/LPAATδ). Front. Cell Dev. Biol. 7, 147 (2019).
Lin, M. E., Herr, D. R. & Chun, J. Lysophosphatidic acid (LPA) receptors: signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 91, 130–138 (2010).
Vancura, A., Carroll, M. A. & Haldar, D. A lysophosphatidic acid-binding cytosolic protein stimulates mitochondrial glycerophosphate acyltransferase. Biochem. Biophys. Res. Commun. 175, 339–343 (1991).
Sheng, X., Yung, Y. C., Chen, A. & Chun, J. Lysophosphatidic acid signalling in development. Development 142, 1390–1395 (2015).
Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 15, 477–489 (2004).
Valdés-Rives, S. A. & González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediators Inflamm. 2017, 9173090 (2017).
Hayashi, K. I. et al. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ. Res. 89, 251–258 (2001).
Baker, D. L., Umstot, E. S., Desiderio, D. M. & Tigyi, G. J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci. 905, 267–269 (2000).
Gerrard, J. M. & Robinson, P. Identification of the molecular species of lysophosphatidic acid produced when platelets are stimulated by thrombin. Biochim. Biophys. Acta, Lipids Lipid Metabol. 1001, 282–285 (1989).
Eichholtz, T., Jalink, K., Fahrenfort, I. & Moolenaar, W. H. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem. J. 291, 677–680 (1993).
Yoshida, K. et al. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation 108, 1746–1752 (2003).
Aikawa, S., Hashimoto, T., Kano, K. & Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 157, 81–89 (2015).
Gustin, C., Van Steenbrugge, M. & Raes, M. LPA modulates monocyte migration directly and via LPA-stimulated endothelial cells. Am. J. Physiol. Cell Physiol. 295, C905–C914 (2008).
Gerrard, J. M. et al. Lysophosphatidic acids. Influence on platelet aggregation and intracellular calcium flux. Am. J. Pathol. 96, 423–438 (1979).
Tokumura, A., Fukuzawa, K., Yamada, S. & Tsukatani, H. Stimulation effect of lysophosphatidic acids on uterine smooth muscles of non-pregnant rats. Arch. Int. Pharmacodyn. Ther. 245, 74–83 (1980).
Ishii, I., Fukushima, N., Ye, X. & Chun, J. Lysophospholipid Receptors: Signaling and Biology. Annu. Rev. Biochem. 73, 321–354 (2004).
Hecht, J. H., Weiner, J. A., Post, S. R. & Chun, J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 135, 1071–1083 (1996).
An, S., Bleu, T., Hallmark, O. G. & Goetzl, E. J. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J. Biol. Chem. 273, 7906–7910 (1998).
Im, D. S. et al. Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J. Biol. Chem. 275, 14281–14286 (2000).
Kihara, Y., Maceyka, M., Spiegel, S. & Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 171, 3575–3594 (2014).
Noguchi, K., Ishii, S. & Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 278, 25600–25606 (2003).
Lee, Z. et al. Role of LPA4/p2y9/GPR23 in negative regulation of cell motility. Mol. Biol. Cell. 19, 5435–5445 (2008).
Blaho, V. A. & Chun, J. ‘Crystal’ Clear? Lysophospholipid Receptor Structure Insights and Controversies. Trends Pharmacol. Sci. 39, 953–966 (2018).
Yanagida, K. & Ishii, S. Non-Edg family LPA receptors: the cutting edge of LPA research. J. Biochem. 150, 223–232 (2011).
Contos, J. J. A. et al. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl Acad. Sci. USA. 97, 13384–13389 (2000).
Fukushima, N. et al. Lysophospholipid receptors. Annu. Rev. Pharmacool. Toxicol. 41, 507–534 (2001).
Walker, T. L. et al. Lysophosphatidic Acid Receptor Is a Functional Marker of Adult Hippocampal Precursor Cells. Stem Cell Rep. 6, 552–565 (2016).
Contos, J. J. et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol. Cell Biol. 22, 6921–6929 (2002).
Yu, F. X. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012).
Miller, E. et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 19, 955–962 (2012).
Cai, H. & Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 11, 31 (2013).
Jeong, G. O. et al. TAZ mediates lysophosphatidic acid-induced migration and proliferation of epithelial ovarian cancer cells. Cell. Physiol. Biochem. 32, 253–263 (2013).
McIntyre, T. M. et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl Acad. Sci. USA. 100, 131–136 (2003).
Mori, S. et al. LPA signaling through LPA receptors regulates cellular functions of endothelial cells treated with anticancer drugs. Mol. Cell. Biochem. 408, 147–154 (2015).
Choi, J. W. et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl Acad. Sci. USA. 108, 751–756 (2011).
Lee, C. W., Rivera, R., Dubin, A. E. & Chun, J. LPA4/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing Gs-, Gq/Gi-mediated calcium signaling and G12/13-mediated Rho activation. J. Biol. Chem. 282, 4310–4317 (2007).
Matayoshi, S. et al. Lysophosphatidic acid receptor 4 signaling potentially modulates malignant behavior in human head and neck squamous cell carcinoma cells. Int. J. Oncol. 42, 1560–1568 (2013).
Ishii, S. et al. Diverse effects of LPA4, LPA5 and LPA6 on the activation of tumor progression in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 461, 59–64 (2015).
Yanagida, K. et al. LPA4/p2y9/GPR23 mediates rho-dependent morphological changes in a rat neuronal cell line. J. Biol. Chem. 282, 5814–5824 (2007).
Sumida, H. et al. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 116, 5060–5070 (2010).
Eino, D. et al. LPA4-mediated vascular network formation increases the efficacy of anti–PD-1 therapy against brain tumors. Cancer Res. 78, 6607–6620 (2018).
Liu, Y. B. et al. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J. Cell. Biochem. 109, 794–800 (2010).
Kano, K. et al. Molecular mechanism of lysophosphatidic acid-induced hypertensive response. Sci. Rep. 9, 2662 (2019).
Choi, J. W. & Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1831, 20–32 (2013).
Lee, C. W. et al. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 281, 23589–23597 (2006).
Nahum, S., Morice-Picard, F., Taieb, A. & Sprecher, E. A novel mutation in LPAR6 causes autosomal recessive hypotrichosis of the scalp. Clin. Exp. Dermatol. 36, 188–194 (2011).
Ketscher, A. et al. LSD1 controls metastasis of androgen-independent prostate cancer cells through PXN and LPAR6. Oncogenesis 3, e120 (2014).
Taniguchi, R. et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA 6. Nature 548, 356–360 (2017).
Tabata, K. et al. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 363, 861–866 (2007).
Okazoe, H. et al. Expression and role of GPR87 in urothelial carcinoma of the bladder. Int. J. Mol. Sci. 14, 12367–12379 (2013).
Nouh, M. A. A. M. et al. Expression of autotaxin and acylglycerol kinase in prostate cancer: Association with cancer development and progression. Cancer Science (2009).
Ochiai, S. et al. GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur. J. Pharmacol. 100, 1631–1638 (2013).
Kam, Y. & Quaranta, V. Cadherin-bound beta-catenin feeds into the Wnt pathway upon adherens junctions dissociation: Evidence for an intersection between beta-catenin pools. PLoS ONE. 4, e4580 (2009).
Murakami, M., Shiraishi, A., Tabata, K. & Fujita, N. Identification of the orphan GPCR, P2Y10 receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 371, 707–712 (2008).
Morales-Lázaro, S. L. et al. Structural determinants of the transient receptor potential 1 (TRPV1) channel activation by phospholipid analogs. J. Biol. Chem. 289, 24079–24090 (2014).
Pan, H. L., Zhang, Y. Q. & Zhao, Z. Q. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCε pathway in dorsal root ganglion neurons. Mol. Pain. 6, 85 (2010).
Kremer, A. E. et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 139, 1008–1018 (2010).
Kittaka, H., Uchida, K., Fukuta, N. & Tominaga, M. Lysophosphatidic acid-induced itch is mediated by signalling of LPA5 receptor, phospholipase D and TRPA1/TRPV1. J. Physiol. 595, 2681–2698 (2017).
Contos, J. J. A., Ishii, I. & Chun, J. Lysophosphatidic Acid Receptors. Mol. Cell. Biol. 58, 1188–1196 (2000).
Kingsbury, M. A. et al. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat. Neurosci. 6, 1292–1299 (2003).
Spohr, T. C. et al. Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. J. Biol. Chem. 283, 7470–7479 (2008).
Spohr, T. C. L., Dezonne, R. S., Rehen, S. K. & Gomes, F. C. A. Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. J. Neurochem. 119, 113–123 (2011).
Spohr, T. C. Ld. Se, Dezonne, Rm. S., Rehen, S. K. & Gomes, Fv. C. A. LPA-primed astrocytes induce axonal outgrowth of cortical progenitors by activating PKA signaling pathways and modulating extracellular matrix proteins. Front. Cell. Neurosc. 8, 1–9 (2014).
Weiner, J. A. et al. Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J. Neurosci. 21, 7069–7078 (2001).
Ye, X., Fukushima, N., Kingsbury, M. A. & Chun, J. Lysophosphatidic acid in neural signaling. Neuroreport 13, 2169–2175 (2002).
Baker, D. L. et al. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem. 292, 287–295 (2001).
Gardell, S. E., Dubin, A. E. & Chun, J. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 12, 65–75 (2006).
García-Díaz, B. et al. Loss of lysophosphatidic acid receptor LPA1 alters oligodendrocyte differentiation and myelination in the mouse cerebral cortex. Brain Struct. Funct. 220, 3701–3720 (2015).
Cui, H. L. & Qiao, J. T. Promotive action of lysophosphatidic acid on proliferation of rat embryonic neural stem cells and their differentiation to cholinergic neurons in vitro. Sheng Li Xue Bao. 58, 547–555 (2006).
Ohuchi, H. et al. Expression patterns of the lysophospholipid receptor genes during mouse early development. Dev. Dyn. 237, 3280–3294 (2008).
Suckau, O. et al. LPA 1, LPA 2, LPA 4, and LPA 6 receptor expression during mouse brain development. Dev. Dyn. 248, 375–395 (2019).
Moolenaar, W. H. Development of Our Current Understanding of Bioactive Lysophospholipids. Ann. N. Y. Acad. Sci. 905, 1–10 (2000).
Chun, J. et al. Neurobiology of receptor-mediated lysophospholipid signaling. From the first lysophospholipid receptor to roles in nervous system function and development. Ann. N. Y. Acad. Sci. 905, 110–117 (2000).
Estivill-Torrús, G. et al. Absence of LPA1 signaling results in defective cortical development. Cereb. Cortex. 18, 938–950 (2008).
González de San Román, E. et al. CB1 and LPA1 Receptors Relationship in the Mouse Central Nervous System. Front. Mol. Neurosci. 12, 1–12 (2019).
yun Yang, X. et al. LPA signaling is required for dopaminergic neuron development and is reduced through low expression of the LPA1 receptor in a 6-OHDA lesion model of Parkinson’s disease. Neurol. Sci. 36, 2027–2033 (2015).
van Meeteren, L. A. et al. Autotaxin, a Secreted Lysophospholipase D, Is Essential for Blood Vessel Formation during Development. Mol. Cell. Biol. 26, 5015–5022 (2006).
Perrakis, A. & Moolenaar, W. H. Autotaxin: structure-function and signaling. J. Lipid Res. 55, 1010–1018 (2014).
Tanaka, M. et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 281, 25822–25830 (2006).
Yuelling, L. W. et al. Autotaxin/ENPP2 regulates oligodendrocyte differentiation in vivo in the developing zebrafish hindbrain. Glia 60, 1605–1618 (2012).
Greenman, R. et al. Non-cell autonomous and non-catalytic activities of ATX in the developing brain. Front. Neurosci. 9, 1–17 (2015).
Geach, T. J. et al. An essential role for LPA signalling in telencephalon development. Development 141, 940–949 (2014).
Fukushima, N., Shano, S., Moriyama, R. & Chun, J. Lysophosphatidic acid stimulates neuronal differentiation of cortical neuroblasts through the LPA1-G(i/o) pathway. Neurochem. Int. 50, 302–307 (2007).
Fukushima, N. et al. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol. Cell. Neurosci. 20, 271–282 (2002).
Fukushima, N. & Morita, Y. Actomyosin-dependent microtubule rearrangement in lysophosphatidic acid-induced neurite remodeling of young cortical neurons. Brain Res. 1094, 65–75 (2006).
Dubin, A. E. et al. Lysophosphatidic acid stimulates neurotransmitter-like conductance changes that precede GABA and L-glutamate in early, presumptive cortical neuroblasts. J. Neurosci. 19, 1371–1381 (1999).
Kingsbury, M. A., Rehen, S. K., Ye, X. & Chun, J. Genetics and cell biology of lysophosphatidic acid receptor-mediated signaling during cortical neurogenesis. J. Cell. Biochem. 92, 1004–1012 (2004).
Daneman, R. & Prat, A. The Blood Brain Barrier (BBB). Cold Spring Harb. Perspect. Biol. 7, a020412 (2015).
Offermanns, S., Mancino, V., Revel, J. P. & Simon, M. I. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 275, 533–536 (1997).
Yasuda, D. et al. Lysophosphatidic acid-induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J. Clin. Invest. 129, 4332–4349 (2019).
Tigyi, G. et al. Lysophosphatidic acid alters cerebrovascular reactivity in piglets. Am. J. Physiol., Heart Circ. Physiol. 268, H2048–H2055 (1995).
Sun, G. Y., Lu, F. L., Lin, S. E. & Ko, M. R. Decapitation ischemia-induced release of free fatty acids in mouse brain - Relationship with diacylglycerols and lysophospholipids. Mol. Chem. Neuropathol. 17, 39–50 (1992).
Masago, K. et al. Lysophosphatidic acid receptor, LPA6, regulates endothelial blood-brain barrier function: Implication for hepatic encephalopathy. Biochem. Biophys. Res. Commun. 501, 1048–1054 (2018).
Kishi, Y. et al. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine TO lysophosphatidic acid. J. Biol. Chem. 281, 17492–17500 (2006).
On, N. H., Savant, S., Toews, M. & Miller, D. W. Rapid and reversible enhancement of blood-brain barrier permeability using lysophosphatidic acid. J. Cereb. Blood Flow. Metab. 33, 1944–1954 (2013).
Schulze, C., Smales, C., Rubin, L. L. & Staddon, J. M. Lysophosphatidic acid increases tight junction permeability in cultured brain endothelial cells. J. Neurochem. 68, 991–1000 (1997).
Nitz, T., Eisenblätter, T., Psathaki, K. & Galla, H. J. Serum-derived factors weaken the barrier properties of cultured porcine brain capillary endothelial cells in vitro. Brain Res. 981, 30–40 (2003).
Yu, Y. et al. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int. J. Mol. Med. 33, 661–669 (2014).
Avraham-Davidi, I., Grunspan, M. & Yaniv, K. Lipid signaling in the endothelium. Exp. Cell Res. 319, 1298–1305 (2013).
Tokumura, A. et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 277, 39436–39442 (2002).
Cohen, S. P. & Mao, J. Neuropathic pain: mechanisms and their clinical implications. BMJ 348, f7656 (2014).
Ballantyne, J. C., Fishman, S. M. & Rathmell, J. P. (eds) Bonica’s Management of Pain. (Lippincott Williams & Wilkins (LWW), 2010).
Treede, R. D. et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 70, 1630–1635 (2008).
Simon, L. S. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. J. Pain. Palliat. Care Pharmacother. 26, 197–198 (2012).
Bouhassira, D. et al. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 136, 380–387 (2008).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Ueda, H. Lysophosphatidic acid signaling is the definitive mechanism underlying neuropathic pain. Pain 158(Suppl 1), S55–S65 (2017).
Nakayama, T. et al. Astrocytogenesis of embryonic stem-cell-derived neural stem cells: Default differentiation. Neuroreport 17, 1519–1523 (2006).
Ueda, H. Lysophosphatidic acid as the initiator of neuropathic pain. Biol. Pharm. Bull. 34, 1154–1158 (2011).
McDougall, J. J. et al. Lysophosphatidic acid provides a missing link between osteoarthritis and joint neuropathic pain. Osteoarthr. Cartil. 25, 926–934 (2017).
Meyer zu Heringdorf, D. & Jakobs, K. H. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim. Biophys. Acta 1768, 923–940 (2007).
Ye, X., Inoue, M. & Ueda, H. Botulinum toxin C3 inhibits hyperalgesia in mice with partial sciatic nerve injury. Jpn J. Pharmacol. 83, 161–163 (2000).
Uehata, M. et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994 (1997).
Weiner, J. A. & Chun, J. Schwann cell survival mediated by the signaling phospholipid lysophosphatidic acid. Proc. Natl Acad. Sci. USA. 96, 5233–5238 (1999).
Mueller, B. K., Mack, H. & Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 4, 387–398 (2005).
Tsukahara, R. & Ueda, H. Myelin-related gene silencing mediated by LPA1 – Rho/ROCK signaling is correlated to acetylation of NFκB in S16 Schwann cells. J. Pharmacol. Sci. 132, 162–165 (2016).
Fujita, R., Kiguchi, N. & Ueda, H. LPA-mediated demyelination in ex vivo culture of dorsal root. Neurochem. Int. 50, 351–355 (2007).
Llorens, F., Gil, V. & Río, J. A. Emerging functions of myelin‐associated proteins during development, neuronal plasticity, and neurodegeneration. FASEB J. 25, 463–475 (2011).
Nagai, J. et al. Autotaxin and lysophosphatidic acid1receptor-mediated demyelination of dorsal root fibers by sciatic nerve injury and intrathecal lysophosphatidylcholine. Mol. Pain. 6, 78 (2010).
Ma, L., Nagai, J. & Ueda, H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J. Neurochem. 115, 643–653 (2010).
Yano, R., Ma, L., Nagai, J. & Ueda, H. Interleukin-1β plays key roles in LPA-induced amplification of LPA production in neuropathic pain model. Cell. Mol. Neurobiol. 33, 1033–1041 (2013).
Ma, L. et al. Evidence for lysophosphatidic acid 1 receptor signaling in the early phase of neuropathic pain mechanisms in experiments using Ki-16425, a lysophosphatidic acid 1 receptor antagonist. J. Neurochem. 109, 603–610 (2009).
Morioka, N. et al. Interleukin-1beta-induced substance P release from rat cultured primary afferent neurons driven by two phospholipase A2 enzymes: secretory type IIA and cytosolic type IV. J. Neurochem. 80, 989–997 (2002).
Uchida, H., Matsumoto, M. & Ueda, H. Profiling of BoNT/C3-reversible gene expression induced by lysophosphatidic acid: ephrinB1 gene up-regulation underlying neuropathic hyperalgesia and allodynia. Neurochem. Int. 54, 215–221 (2009).
Hagedorn, H. G., Bachmeier, B. E. & Nerlich, A. G. Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-beta in invasive carcinomas (Review). Int. J. Oncol. 18, 669–681 (2001).
Ma, L., Nagai, J., Chun, J. & Ueda, H. An LPA species (18:1 LPA) plays key roles in the self-amplification of spinal LPA production in the peripheral neuropathic pain model. Mol. Pain. 9, 29 (2013).
Murai, N. et al. Analgesic effects of novel lysophosphatidic acid receptor 5 antagonist AS2717638 in rodents. Neuropharmacology 126, 97–107 (2017).
O’Brien, M. S., Philpott, H. T. A. & McDougall, J. J. Targeting the Nav1.8 ion channel engenders sex-specific responses in lysophosphatidic acid-induced joint neuropathy. Pain 160, 269–278 (2019).
Noguchi, K., Herr, D., Mutoh, T. & Chun, J. Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 9, 15–23 (2009).
Fukushima, N., Weiner, J. A. & Chun, J. Lysophosphatidic acid (LPA) is a novel extracellular regular of cortical neuroblast morphology. Dev. Biol. 228, 6–18 (2000).
Roberts, C. et al. Neurochemical changes in LPA1 receptor deficient mice - A putative model of schizophrenia. Neurochem. Res. 30, 371–317 (2005).
Xiang, H., Lu, Y., Shao, M. & Wu, T. Lysophosphatidic Acid Receptors: Biochemical and Clinical Implications in Different Diseases. J. Cancer 11, 3519–3535 (2020).
Lotharius, J. & Brundin, P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 3, 932–942 (2002).
Ramesh, S. et al. Autotaxin–lysophosphatidic acid signaling in alzheimer’s disease. Int. J. Mol. Sci. 19, 1–25 (2018).
Aldred, S., Bennett, S. & Mecocci, P. Increased low-density lipoprotein oxidation, but not total plasma protein oxidation, in Alzheimer’s disease. Clin. Biochem. 43, 267–271 (2010).
Draczynska-Lusiak, B., Doung, A. & Sun, A. Y. Oxidized lipoproteins may play a role in neuronal cell death in alzheimer disease. Mol. Chem. Neuropathol. 33, 139–148 (1998).
Casado, Á., Encarnación López-Fernández, M., Concepción Casado, M. & De La Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and alzheimer dementias. Neurochem. Res. 33, 450–458 (2008).
Giasson, B. I., Ischiropoulos, H., Lee, V. M. Y. & Trojanowski, J. Q. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer’s and Parkinson’s diseases. Free Radic. Biol. Med. 32, 1264–1275 (2002).
Markesbery, W. R. & Carney, J. M. Oxidative Alterations in Alzheimer’s Disease. Brain Pathol. 9, 133–146 (1999).
Frisardi, V., Panza, F., Seripa, D., Farooqui, T. & Farooqui, A. A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 50, 313–330 (2011).
Sun, Y. X. et al. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 16, 136–144 (2003).
Rajmohan, R. & Reddy, P. H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 57, 975–999 (2017).
Perry, D. C. et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J. Neurosurg. 124, 511–526 (2016).
Pluta, R., Furmaga-Jabłońska, W., Maciejewski, R., Ułamek-Kozioł, M. & Jabłoński, M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 47, 425–434 (2013).
Adibhatla, R. M. & Hatcher, J. F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 49, 241–268 (2008).
McDonald, W. S. et al. Matrix-Assisted Laser Desorption Ionization Mapping of Lysophosphatidic Acid Changes after Traumatic Brain Injury and the Relationship to Cellular Pathology. Am. J. Pathol. 188, 1779–1793 (2018).
Crack, P. J. et al. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J. Neuroinflamm. 11, 37 (2014).
Tigyi, G. J. et al. Regulation of tumor cell - Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 71, 183–193 (2019).
Leblanc, R. & Peyruchaud, O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp. Cell Res. 333, 183–189 (2015).
Li, T. T. et al. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol. Cancer Res. 7, 1064–1077 (2009).
Shida, D. et al. Dual mode regulation of migration by lysophosphatidic acid in human gastric cancer cells. Exp. Cell Res. 301, 168–178 (2004).
Takahashi, K. et al. Lysophosphatidic acid (LPA) signaling via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochem. Biophys. Res. Commun. 483, 652–657 (2017).
Deng, W. et al. The Lysophosphatidic Acid Type 2 Receptor Is Required for Protection Against Radiation-Induced Intestinal Injury. Gastroenterology 132, 1834–1851 (2007).
Kato, K. et al. Opposite roles of LPA 1 and LPA 3 on cell motile and invasive activities of pancreatic cancer cells. Tumor Biol. 33, 1739–1744 (2012).
Altman, M. K. et al. Targeting melanoma growth and viability reveals dualistic functionality of the phosphonothionate analogue of carba cyclic phosphatidic acid. Mol. Cancer 9, 140 (2010).
Yu, S. et al. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J. Natl Cancer Inst. 100, 1630–1642 (2008).
Younis, S. & Rashid, S. Alpha conotoxin-BuIA globular isomer is a competitive antagonist for oleoyl-L-alpha-lysophosphatidic acid binding to LPAR6; A molecular dynamics study. PLoS ONE. 12, e0189154 (2017).
Chang, C. L. et al. Lysophosphatidic acid-induced interleukin-1 beta expression is mediated through Gi/Rho and the generation of reactive oxygen species in macrophages. J. Biomed. Sci. 15, 357–363 (2008).
Okabe, K. et al. Possible involvement of lysophosphatidic acid receptor-5 gene in the acquisition of growth advantage of rat tumor cells. Mol. Carcinog. 50, 635–642 (2011).
Tsujino, M. et al. Differential expressions and DNA methylation patterns of lysophosphatidic acid receptor genes in human colon cancer cells. Virchows Arch. 457, 669–676 (2010).
Oda, S. K. et al. Lysophosphatidic acid inhibits CD8 T cell activation and control of tumor progression. Cancer Immunol. Res. 1, 245–255 (2013).
Hu, J. et al. Lysophosphatidic Acid Receptor 5 Inhibits B Cell Antigen Receptor Signaling and Antibody Response. J. Immunol. 193, 85–95 (2014).
Lee, S. C. et al. Autotaxin and LPA1 and LPA5 receptors exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol. Cancer Res. 13, 174–185 (2015).
Jazaeri, A. A. et al. Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin. Cancer Res. 11, 6300–6310 (2005).
Gupta, P. B. et al. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 138, 645–659 (2009).
Shuyu, E. et al. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J. Biol. Chem. 284, 14558–14571 (2009).
Samadi, N., Gaetano, C., Goping, I. S. & Brindley, D. N. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene 28, 1028–1039 (2009).
Burkhalter, R. J., Westfall, S. D., Liu, Y. & Stack, M. S. Lysophosphatidic acid initiates epithelial to mesenchymal transition and Induces β-Catenin-mediated transcription in epithelial ovarian carcinoma. J. Biol. Chem. 290, 22143–22154 (2015).
Ha, J. H. et al. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget 7, 37664–37679 (2016).
Hashimoto, S. et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat. Commun. 7, 10656 (2016).
Ray, U., Roy, S. S. & Chowdhury, S. R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell. Physiol. Biochem. 41, 795–805 (2017).
Xu, M., Liu, Z., Wang, C., Yao, B. & Zheng, X. EDG2 enhanced the progression of hepatocellular carcinoma by LPA/PI3K/AKT/ mTOR signaling. Oncotarget 8, 66154–66168 (2017).
Benesch, M. G. K. et al. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr. Relat. Cancer 22, 593–607 (2015).
Benesch, M. G. K. et al. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 22, 593–607 (2015).
Guo, L., He, P., No, Y. R. & Yun, C. C. Krüppel-like factor 5 incorporates into the β-catenin/TCF complex in response to LPA in colon cancer cells. Cell. signal. 27, 961–968 (2015).
Tveteraas, I. H. et al. Lysophosphatidic acid induces both EGFR-dependent and EGFR-independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumor Biol. 37, 2519–2526 (2016).
Shida, D. et al. Sphingosine kinase 1 is upregulated with lysophosphatidic acid receptor 2 in human colorectal cancer. World J. Gastroenterol. 22, 2503–2511 (2016).
Takahashi, K. et al. Lysophosphatidic acid (LPA) signaling via LPA4and LPA6negatively regulates cell motile activities of colon cancer cells. Biochem. Biophys. Res. Commun. 483, 652–657 (2017).
Leve, F., Peres-Moreira, R. J., Binato, R., Abdelhay, E. & Morgado-Díaz, J. A. LPA induces colon cancer cell proliferation through a cooperation between the ROCK and STAT-3 pathways. PLoS ONE. 10, e0139094 (2015).
Schmid, R. et al. ADSCs and adipocytes are the main producers in the autotaxin–lysophosphatidic acid axis of breast cancer and healthy mammary tissue in vitro. BMC Cancer 18, 1273 (2018).
Dong, L. et al. Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling. Oncotarget 8, 22550–22562 (2017).
Nam, J. S. et al. Lysophosphatidic acid enhances breast cancer cells-mediated osteoclastogenesis. Korean J. Physiol. Pharmacol. 22, 503–511 (2018).
Hopkins, M., Zhang, Z., Liu, Z. & Meier, K. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J. Clin. Med. 5, 16 (2016).
Stuelten, C. H., Lee, R. M., Losert, W. & Parent, C. A. Lysophosphatidic acid regulates the motility of MCF10CA1a breast cancer cell sheets via two opposing signaling pathways. Cell. Signal. 45, 1–11 (2018).
Cho, K. H., Jeong, B. Y., Park, C. G. & Lee, H. Y. The YB-1/EZH2/amphiregulin signaling axis mediates LPA-induced breast cancer cell invasion. Arch. Pharm. Res. 42, 519–530 (2019).
Sun, K. et al. Curcumin inhibits LPA-induced invasion by attenuating RhoA/ROCK/MMPs pathway in MCF7 breast cancer cells. Clin. Exp. Med. 16, 37–47 (2015).
Tao, K. et al. Lysophosphatidic Acid Receptor 6 (LPAR6) Expression and Prospective Signaling Pathway Analysis in Breast Cancer. Mol. Diagn. Ther. 23, 127–138 (2019).
Mukherjee, A. et al. Lysophosphatidic acid up-regulates hexokinase II and glycolysis to promote proliferation of ovarian cancer cells. Neoplasia 17, 723–734 (2015).
Ha, J. H. et al. LPA induces metabolic reprogramming in ovarian cancer via a pseudohypoxic response. Cancer Res. 78, 1923–1934 (2018).
Radhakrishnan, R. et al. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 442, 464–474 (2019).
Cao, L. et al. Diagnostic value of plasma lysophosphatidic acid levels in ovarian cancer patients: A case-control study and updated meta-analysis. J. Obstet. Gynaecol. Res. 41, 1951–1958 (2015).
Li, Y. Y. et al. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: A meta-analysis. Lipids Health Dis. 14, 72 (2015).
Si, J., Su, Y., Wang, Y., Yan, Y. L. & Tang, Y. L. Expressions of lysophosphatidic acid receptors in the development of human ovarian carcinoma. Int. J. Clin. Exp. Med. 8, 17880–17890 (2015).
de la Franier, B. & Thompson, M. Detection of the ovarian cancer biomarker lysophosphatidic acid in serum. Biosensors 10, 13 (2020).
Wang, Y. et al. Synergistic Tailoring of Electrostatic and Hydrophobic Interactions for Rapid and Specific Recognition of Lysophosphatidic Acid, an Early-Stage Ovarian Cancer Biomarker. J. Am. Chem. Soc. 139, 11616–11621 (2017).
Yagi, T. et al. Challenges and inconsistencies in using lysophosphatidic acid as a biomarker for ovarian cancer. Cancers 11, 520 (2019).
Minis, E. et al. Evaluation of lysophosphatidic acid in vaginal fluid as a biomarker for ovarian cancer: A pilot study. Eur. J. Obstet. Gynecol. Reprod. Biol. X. 2, 100012 (2019).
Hwang, H., Kim, E. K., Park, J., Suh, P. G. & Cho, Y. K. RhoA and Rac1 play independent roles in lysophosphatidic acid-induced ovarian cancer chemotaxis. Integr. Biol. (Camb.). 6, 267–276 (2014).
Wang, H. et al. Effect of the LPA-mediated CXCL12-CXCR4 axis in the tumor proliferation, migration and invasion of ovarian cancer cell lines. Oncol. Lett. 7, 1581–1585 (2014).
Lizalek, J. et al. Lysophosphatidic acid stimulates urokinase receptor (uPAR/CD87) in ovarian epithelial cancer cells. Anticancer Res. 35, 5263–5270 (2015).
Ward, J. D., Ha, J. H., Jayaraman, M. & Dhanasekaran, D. N. LPA-mediated migration of ovarian cancer cells involves translocalization of Gαi2 to invadopodia and association with Src and β-pix. Cancer Lett. 356, 382–391 (2015).
Fan, Q., Cai, Q. & Xu, Y. FOXM1 is a downstream target of LPA and YAP oncogenic signaling pathways in high grade serous ovarian cancer. Oncotarget 6, 27688–27699 (2015).
Yu, X., Zhang, Y. & Chen, H. LPA receptor 1 mediates LPA-induced ovarian cancer metastasis: An in vitro and in vivo study. BMC Cancer 16, 846 (2016).
Oh, Y. S. et al. Dynamic relocalization of NHERF1 mediates chemotactic migration of ovarian cancer cells toward lysophosphatidic acid stimulation. Exp. Mol. Med. 49, e351 (2017).
Park, J. et al. LPA-induced migration of ovarian cancer cells requires activation of ERM proteins via LPA1 and LPA2. Cell. Signal. 44, 138–147 (2018).
Onallah, H., Davidson, B. & Reich, R. Diverse Effects of Lysophosphatidic Acid Receptors on Ovarian Cancer Signaling Pathways. J. Oncol. 2019, 7547469 (2019).
Seo, E. J. et al. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells 34, 551–564 (2016).
Bailey, K. A. et al. Chemical Analysis of Morphological Changes in Lysophosphatidic Acid-Treated Ovarian Cancer Cells. Sci. Rep. 7, 15295 (2017).
Reinartz, S. et al. Cell type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol. Oncol. 13, 185–201 (2019).
Rogers, L. A. C. et al. Blocking LPA-dependent signaling increases ovarian cancer cell death in response to chemotherapy. Redox Biol. 15, 380–386 (2018).
Geraldo, L. H. M. et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 5, 46–65 (2019).
Lima, F. R. S. et al. Glioblastoma: Therapeutic challenges, what lies ahead. Biochim. Biophys. Acta, Rev. Cancer 1826, 338–349 (2012).
Herr, D. R., Ong, J. H. J. & Ong, W. Y. Potential Therapeutic Applications for Inhibitors of Autotaxin, a Bioactive Lipid-Producing Lysophospholipase D, in Disorders Affecting the Nervous System. ACS Chem. Neurosci. 9, 398–400 (2018).
Herr, D. R., Chew, W. S., Satish, R. L. & Ong, W. Y. Pleotropic Roles of Autotaxin in the Nervous System Present Opportunities for the Development of Novel Therapeutics for Neurological Diseases. Mol. Neurobiol. 57, 372–392 (2020).
Hoelzinger, D. B. et al. Autotaxin: A secreted autocrine/paracrine factor that promotes glioma invasion. J. Neurooncol. 86, 297–309 (2008).
Benesch, M. G. K., Yang, Z., Tang, X., Meng, G. & Brindley, D. N. Lysophosphatidate Signaling: The Tumor Microenvironment’s New Nemesis. Trends Cancer 3, 748–752 (2017).
Valdés-Rives, S. A. et al. LPA1 receptor activation induces PKCα nuclear translocation in glioblastoma cells. Int. J. Biochem. Cell Biol. 110, 91–102 (2019).
Tabuchi, S. The autotaxin-lysophosphatidic acid-lysophosphatidic acid receptor cascade: proposal of a novel potential therapeutic target for treating glioblastoma multiforme. Lipids Health Dis. 14, 56 (2015).
Amaral, R. F. et al. Microglial Lysophosphatidic Acid Promotes Glioblastoma Proliferation and Migration via Lpa 1 Receptor. J. Neurochem. 3, https://doi.org/10.1111/jnc.15097 (2020). [Online ahead of print].
Barbayianni, E., Kaffe, E., Aidinis, V. & Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 58, 76–96 (2015).
Oikonomou, N. et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 47, 566–574 (2012).
Tager, A. M. et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 (2008).
Castelino, F. V. et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 63, 1405–1415 (2011).
Pradère, J. P. et al. Lysophosphatidic acid and renal fibrosis. Biochim. Biophys. Acta 1781, 582–587 (2008).
Swaney, J. S. et al. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J. Pharmacol. Exp. Ther. 336, 693–700 (2011).
Swaney, J. S. et al. A novel, orally active LPA 1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 160, 1699–1713 (2010).
Palmer, S. M. et al. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. Chest 154, 1061–1069 (2018).
Allanore, Y. et al. Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for Patients With Diffuse Cutaneous Systemic Sclerosis. Arthritis Rheumatol. 70, 1634–1643 (2018).
Saga, H. et al. A novel highly potent autotaxin/ENPP2 inhibitor produces prolonged decreases in plasma lysophosphatidic acid formation in vivo and regulates urethral tension. PLoS ONE. 9, e93230 (2014).
Benesch, M. G. K. et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 28, 2655–2666 (2014).
Iwaki, Y. et al. ONO-8430506: A Novel Autotaxin Inhibitor That Enhances the Antitumor Effect of Paclitaxel in a Breast Cancer Model. ACS Med. Chem. Lett. 11, 1335–1341 (2020).
Desroy, N. et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a First-in-Class Autotaxin Inhibitor Undergoing Clinical Evaluation. J. Med. Chem. 60, 3580–3590 (2017).
van der Aar, E. et al. Safety, Pharmacokinetics, and Pharmacodynamics of the Autotaxin Inhibitor GLPG1690 in Healthy Subjects: Phase 1 Randomized Trials. J. Clin. Pharmacol. 59, 1366–1378 (2019).
Maher, T. M. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 6, 627–635 (2018).
Kuttruff, C. A. et al. Discovery of BI-2545: A Novel Autotaxin Inhibitor That Significantly Reduces LPA Levels in Vivo. ACS Med. Chem. Lett. 8, 1252–1257 (2017).
Jing, T. et al. Discovery and optimization of tetrahydropyrido[4,3-d]pyrimidine derivatives as novel ATX and EGFR dual inhibitors. Bioorg. Med. Chem. 26, 1784–1796 (2018).
Schleicher, S. M. et al. Autotaxin and lpa receptors represent potential molecular targets for the radiosensitization of murine glioma through effects on tumor vasculature. PLoS ONE. 6, e22182 (2011).
Bhave, S. R. et al. Autotaxin inhibition with PF-8380 enhances the radiosensitivity of human and murine glioblastoma cell lines. Front. Oncol. 3, 236 (2013).
Goldshmit, Y. et al. Blockage of lysophosphatidic acid signaling improves spinal cord injury outcomes. Am. J. Pathol. 181, 978–992 (2012).
Shimizu, Y. & Tokumura, A. Physiological role of lysophosphatidic acid and its relevance to diseases. Seikagaku 83, 506–517 (2011).
Jesionowska, A., Cecerska, E. & Dolegowska, B. Methods for quantifying lysophosphatidic acid in body fluids: a review. Anal. Biochem. 453, 38–43 (2014).
Bese, T. et al. Comparison of total plasma lysophosphatidic acid and serum CA-125 as a tumor marker in the diagnosis and follow-up of patients with epithelial ovarian cancer. J. Gynecol. Oncol. 21, 248–254 (2010).
Sasagawa, T., Suzuki, K., Shiota, T., Kondo, T. & Okita, M. The significance of plasma lysophospholipids in patients with renal failure on hemodialysis. J. Nutr. Sci. Vitaminol. 44, 809–818 (1998).
Sasagawa, T., Okita, M., Murakami, J., Kato, T. & Watanabe, A. Abnormal serum lysophospholipids in multiple myeloma patients. Lipids 34, 17–21 (1999).
Ackerman, S. J. et al. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark. Med. 10, 123–135 (2016).
Ye, X. et al. LPA3-mediated lysophosphatidic acid signalling in implantation and embryo spacing. Nature 435, 104–108 (2005).
Acknowledgements
This study was supported by the Brazilian Funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Rio de Janeiro (FAPERJ), Instituto Nacional de Neurociência Translacional (INNT), and Programa de Pós-Graduação em Ciências Morfológicas (PCM), UFRJ. R.F.A. was financed by a fellowship of CNPq and L.H.M.G. was financed by fellowships of CAPES and of the European Society of Cardiology (ESC).
Author information
Authors and Affiliations
Contributions
Luiz Henrique M. Geraldo and Tânia C. L. de S. Spohr: conceptualization, writing-original draft preparation, writing, review and editing, and visualization; Rackele F. do Amaral, Anna C. C. da Fonseca, Celina Garcia, Fabio de A. Mendes, Catarina Freitas and Marcos F. dosSantos: writing, review, and editing; Flavia R. S. Lima: conceptualization, writing, review and editing, and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Geraldo, L.H.M., Spohr, T.C.L.d.S., Amaral, R.F.d. et al. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Sig Transduct Target Ther 6, 45 (2021). https://doi.org/10.1038/s41392-020-00367-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00367-5
- Springer Nature Limited
This article is cited by
-
EDI3 knockdown in ER-HER2+ breast cancer cells reduces tumor burden and improves survival in two mouse models of experimental metastasis
Breast Cancer Research (2024)
-
Structural basis of TRPV1 modulation by endogenous bioactive lipids
Nature Structural & Molecular Biology (2024)
-
Network analysis-guided drug repurposing strategies targeting LPAR receptor in the interplay of COVID, Alzheimer’s, and diabetes
Scientific Reports (2024)
-
Cholestatic Pruritus: Pathophysiology, Current Management Approach, and Emerging Therapies
Current Hepatology Reports (2024)
-
Lysophosphatidic Acid-Mediated Inflammation at the Heart of Heart Failure
Current Cardiology Reports (2024)