
Abstract
Neurogenic regions of mammalian brain produce many more neurons that will eventually survive and reach a mature stage. Developmental cell death affects both embryonically produced immature neurons and those immature neurons that are generated in regions of adult neurogenesis. Removal of substantial numbers of neurons that are not yet completely integrated into the local circuits helps to ensure that maturation and homeostatic function of neuronal networks in the brain proceed correctly. External signals from brain microenvironment together with intrinsic signaling pathways determine whether a particular neuron will die. To accommodate this signaling, immature neurons in the brain express a number of transmembrane factors as well as intracellular signaling molecules that will regulate the cell survival/death decision, and many of these factors cease being expressed upon neuronal maturation. Furthermore, pro-survival factors and intracellular responses depend on the type of neuron and region of the brain. Thus, in addition to some common neuronal pro-survival signaling, different types of neurons possess a variety of 'neuron type-specific' pro-survival constituents that might help them to adapt for survival in a certain brain region. This review focuses on how immature neurons survive during normal and impaired brain development, both in the embryonic/neonatal brain and in brain regions associated with adult neurogenesis, and emphasizes neuron type-specific mechanisms that help to survive for various types of immature neurons. Importantly, we mainly focus on in vivo data to describe neuronal survival specifically in the brain, without extrapolating data obtained in the PNS or spinal cord, and thus emphasize the influence of the complex brain environment on neuronal survival during development.
Similar content being viewed by others


Facts
-
During development neurons express a set of pro-survival/death molecules that are not present in adult brain.
-
Neuronal survival in the brain often relies on different external factors in comparison with the spinal cord and PNS.
-
Different types of neurons in the brain possess some common, but also distinct components of pro-survival signaling.
-
Immature neurons are more vulnerable to stress factors that trigger neuronal death than mature neurons.
Open questions
-
How abundant are distinct components of pro-survival signaling in different types of neurons that might adapt neuronal survival to the region of the brain, that is, neuron type-specific survival?
-
How do survival mechanisms of embryonically and adult-born neurons differ, that is, survival in immature versus mature brain?
-
During what period of brain development do the various types of neurons die?
-
What mechanisms account for higher vulnerability of immature neurons to stress factors?
During brain development, an excessive number of neurons is generated and, depending on the region and neuronal type, a varying number of neurons die before they mature.1, 2, 3, 4, 5 A high rate of neuronal death also occurs in the regions of adult neurogenesis.6, 7, 8, 9 The process of neuronal overproduction and elimination is necessary to optimize brain connectivity. Disturbances in regulating developmental neuronal death not only change cell composition and connectivity within local neuronal networks, but also alter global brain activity and, thus, cognition. Several types of brain disorders enhance the death of immature neurons (i.e., postmitotic neurons, but before complete maturation) during brain development that could lead to decline in cognitive abilities. After maturation, neurons become resistant to the signaling that was involved in the life/death decision at immature stages since, once neurogenesis is halted, it is advantageous to protect mature neurons that cannot be produced again (protection of immature and mature neurons is compared in Benn and Woolf10 and Kole et al.11).
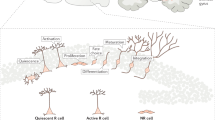
There are two distinct modes of neurogenesis – although the majority of neurons are generated during the embryonic period and their production is discontinued either in the embryonic brain or early postnatally (later referred to as embryonic neurogenesis),12 some populations of neurons are continuously generated throughout the life of an animal (later referred to as adult neurogenesis)13, 14 (see Figures 1a and b, respectively). The death of neurons that are born embryonically reaches a peak in the neonatal brain and affects neurons that are still immature,15, 16, 17 and the critical period for survival of adult-generated neurons is within 4 weeks after their birth; following this period of maturation, they become resistant to cell death.8, 9, 18

Neuronal death during embryonic and adult neurogenesis. (a) During embryonic brain development, neurons are born around the ventricles and migrate toward various brain regions. Cortical principal neurons and interneurons are born in the dorsal and ventral telencephalon, respectively. The majority of interneurons are born in the medial and caudal (data not shown) ganglionic eminences (MGE and CGE, respectively), whereas striatal medium spiny neurons are born in the lateral ganglionic eminence (LGE). Dopaminergic and cerebellar neurons are born in the ventricular zones of the mesencephalon and cerebellum, respectively. Red cells in each region depict dying immature neurons. Peak period of developmental cell death is given for each type of neurons. (b) The SGZ of the dentate gyrus in the hippocampus and the SVZ of the lateral ventricles continue to generate neurons throughout life. The SGZ generates neuroblasts that translocate within the SGZ and mature into excitatory granule cells. Neuroblasts that are generated in the SVZ migrate a long distance through the rostral migratory stream toward the olfactory bulb and mature into two major populations of inhibitory interneurons – granule and periglomerular cells. More than half of adult-generated neurons die by apoptosis. Red cells in each region depict dying immature neurons. Peak period of developmental cell death is given for each type of neurons. CB, cerebellum; CP, cortical plate; CX, cortex; DG, dentate gyrus; GE, ganglionic eminence; HP, hippocampus; LGE, lateral GE; LV, lateral ventricle; MB, midbrain; MGE, medial GE; RMS, rostral migratory stream; OB, olfactory bulb
Principles of neuronal survival are often generalized and data from different areas of the CNS are extrapolated to the CNS as a whole. Indeed, pro-survival signaling does converge on some common core components (Figure 2). However, data accumulated over the recent years show that different types of neurons in the brain might use different pro-survival mechanisms as there are a variety of routes by which core pro-survival components could be activated. Thus, we propose 'neuron type-specific' pro-survival mechanisms that will heavily rely upon (1) composition of extracellular pro-survival factors that are available in a certain brain area at a certain time period, (2) composition of transmembrane molecules (e.g. receptors or ion channels) that are expressed on distinct types of neurons and (3) composition of cytosolic molecules that could propagate pro-survival signaling from the cell membrane toward common core components (Figure 2).

Components of survival/death signaling in immature neurons. Extracellular pro-survival factors that are available in a certain brain area stimulate a variety of receptors and ion channels on neurons located in the area. Transcription factors involved in neuronal differentiation determine what combination of receptors and ion channels will be expressed on a particular neuron. Such neuron type-specific combination of receptors and channels propagates pro-survival signaling to intermediate components, some of which express broadly, whereas others have restricted expression only in one or few types of neurons. Finally, all pro-survival signaling converges on core components that inhibit neuronal death
It should be noted that not only pro-survival, but also pro-death pathways could be neuron type specific. In general terms, it is pro-survival signaling that blocks intrinsic pro-death signaling, and when there is a lack of pro-survival signaling, pro-death pathways are triggered. However, in a recent paper19 it was shown that survival of CNS neurons during development is regulated by 'dependence receptors' that activate pro-death signaling when not bound to their ligands (reviewed in Dekkers et al.20). Although the extent of expression and the number of dependence receptors still remain to be determined in the developing brain, the presence of such a mechanism indicates that neuron type-specific pro-death pathways do exist.
Neuron type-specific pro-survival mechanisms
As different types of neurons survive in different brain areas and at different periods of brain development, the transcriptome of the surviving neuron should 'prepare' the neuron to survive in a certain environment. The preparation is coordinated by distinct sets of transcription factors that are involved in differentiation of specific types of neurons. These transcription factors drive expression of transmembrane and intracellular molecules that are necessary to recognize and respond to the local environment. Neurons failing to differentiate properly are less likely to respond to signals from local brain environment and could be eliminated during maturation. Interestingly, the period of developmental cell death differs across types of neurons/brain areas. For instance, GABAergic interneurons of the cortex and medium spiny neurons exhibit one peak of cell death at P7-P111 and P2-P7,21 respectively, whereas two distinct peaks of developmental cell death have been observed for dopaminergic neurons, at P0-P6 and ~P14,2 and for Purkinje cells, at ~E15 and ~P3.22
The difference in survival mechanisms between embryonically and adult-born neurons illustrates the importance of time period of neuronal survival with regard to brain maturation, since embryonically born immature neurons must survive in immature brain, whereas adult-born immature neurons must survive in mature brain. Thus, there is high pressure for adult-born neurons to integrate into the pre-existing mature circuits, which is absent for embryonically born neurons. This is supported, for instance, by a higher vulnerability of adult-born neurons to impairment in NMDA receptor (NMDAR) expression, since ablation of NR1 or NR2B subunit markedly augments death of adult-born neurons during maturation,23, 24, 25 whereas studies of global or early postnatal knockout of these subunits do not report increase in apoptosis of embryonically produced neurons.26, 27, 28
The effect of brain maturation on neuronal survival might also be illustrated by a decrease in survival of small axonless neurons – a type of neurons that is generated both during embryonic and adult neurogenesis.29 The majority of these neurons survive in the deep cortical layers when circuits are still immature, and gradual maturation of the brain correlates with a decreased number of newly added neurons,29 although the number of these neurons could be increased by pathological conditions such as stroke.30
Support of neuronal survival by the local environment depends on whether a specific factor itself and its receptor are expressed in the region. Availability of pro-survival factors varies within the brain and even cortical layers,31, 32, 33 and response to different pro-survival factors markedly changes over a course of neuronal maturation.34 Moreover, certain intracellular pro-survival molecules are present only in some types of neurons, but not in others. For instance, BDNF promotes survival of dopaminergic neurons, medium spiny neurons and cerebellar granule cells,35, 36, 37 but it is dispensable for survival of GABAergic neurons in the cortex1 although the latter express TrkB receptor and BDNF is available in the surrounding environment.31, 38
In the following, we summarize the evidence for neuron type-specific pro-survival mechanisms during embryonic and adult neurogenesis (see overview in Table 1).
Embryonic neurogenesis: glutamatergic neurons
The most information regarding survival of glutamatergic neurons in the brain was obtained by studying cerebellar granule cells and principal neurons of the hippocampus and cortex (Figure 3a). The peak of cortical principal neuron cell death is at P4–P8,39 whereas the majority of immature cerebellar granule cells die at P5–P9.40 Although knockout of a single neurotrophic factor or its receptor does not have large effects on neuronal survival during brain development,41 double knockout of Ntrk2 and Ntrk3 (genes coding for TrkB and TrkC, respectively) results in the massive death of immature granule cells in the cerebellum and dentate gyrus.42 This could be explained either by redundancy of intracellular pro-survival pathways that are triggered by each of the receptors or by compensatory effects in knockout mice. Furthermore, often data obtained in vivo differs from in vitro experiments, highlighting importance of brain environment for action of a particular pro-survival factor. For instance, BDNF was shown to promote neuronal survival in the culture,43 but deletion of Bdnf in all postmitotic neurons in the brain did not have a large effect on their survival.44

Neuron type-specific pro-survival signaling in embryonically born neurons. (a) Signaling involved in survival and cell death of glutamatergic neurons exemplified by cerebellar granule cells and cortical projection neurons. (b) Pro-survival and apoptotic signaling in GABAergic neurons illustrated by Purkinje cells, medium spiny neurons and cortical interneurons. (c) Signaling regulating survival or cell death in dopaminergic neurons. Green arrows: activation of signaling; dashed green arrow: proposed activation of signaling; red blunt arrows: inhibition of signaling; black arrows: activation of receptors on immature neurons from the extracellular space; dashed black arrows: protein secretion to the extracellular space; red cross: lack of signaling; Pi: phosphorylation
Granule cells of the cerebellum represent a population of glutamatergic neurons that could be a target of pro-survival action of BDNF. Deleting Camk4 and Camkk2 genes in mice enhances apoptosis in immature granule cells in the cerebellum, which is associated with a decrease in levels of CREB1 and BDNF expression.37 It was proposed that Ca2+ entering immature granule cells triggers activation of the calmodulin/CaMKK2/CaMKIV cascade, which, in turn, activates CREB1 and transcription of Bdnf gene.37 Survival of granule cells is also promoted by IGF1 that enhances expression of Bcl-2 and Bcl-xL thus inhibiting caspase-3 activity.45
The existence of neuron type-specific pro-survival mechanisms in glutamatergic neurons was recently highlighted by the identification of a pro-survival pathway that was largely restricted to cortical principal neurons of layer V, which require trophic support from microglia to survive during early postnatal development.46 Microglia secrete IGF1, which binds to IGF1R on immature layer V neurons and activates the IRS1/PI3K/Akt1 cascade inhibiting caspase-3-dependent apoptosis.46 Microglia are activated via CX3CL1, which is released from layer V neurons and interacts with CX3CR1 on microglia.
Interestingly, caspase-3-dependent apoptosis of cortical excitatory, but not inhibitory, neurons was shown to be activated by Rho GTPase RhoA.47 Inhibiting RhoA signaling in the developing brain rescues up to 25% of cortical neurons from apoptosis.
Embryonic neurogenesis: GABAergic neurons
Only few studies have investigated developmental death of GABAergic neurons, and these were mainly focused on Purkinje cells of the cerebellum and medium spiny neurons of the striatum that exhibit a peak of cell death at ~E15 and ~P3,22 and at P2–P7,21 respectively (Figure 3b). Lhx1/Lhx5 transcription factors together with their co-activator Ldb1 promote survival of postmitotic Purkinje cells at E13.5–E15.5.48 Interestingly, two members of the EBF (early B-cell factor) family of transcription factors – EBF1 and EBF2 – are involved in survival of medium spiny49 and Purkinje neurons,50 respectively, during perinatal development. In Purkinje cells, EBF2 binds to Igf1 promoter and activates Igf1 expression that results in local IGF1 secretion and potentiation of Akt1-dependent pro-survival signaling.51 All the aforementioned transcription factors were also shown to be involved in differentiation and/or migration of medium spiny and Purkinje neurons, and thus immature neurons might die because they are not able to complete their differentiation programs.
Although, overall, neurotrophins do not have a large role in survival of immature GABAergic neurons, BDNF and NT-3 were shown to enhance survival of immature medium spiny neurons, as they are secreted by midbrain dopaminergic neurons during a critical period of striatal neuron survival and activate pro-survival signaling via TrkB and TrkC receptors.35
Recently, it was shown that around 40% of immature cortical GABAergic interneurons die during the first two postnatal weeks (with the peak at P7–P11).1 Their survival did not depend on TrkB expression, but was regulated by either cell-autonomous or population-autonomous mechanisms that activated pro-apoptotic Bax signaling.
Embryonic neurogenesis: dopaminergic neurons
Apoptosis of immature dopaminergic neurons occurs at two developmental stages – at P0–P6 and ~P14.2 Three main transcription factors involved in specification dopaminergic neurons – Nurr1, Pitx3 and En1 – also regulate their survival.52, 53, 54, 55 Both Nurr1 and Pitx3 were shown to activate expression of BDNF,56, 57 which promotes survival of a subpopulation of dopaminergic neurons from E16 onward36 via TrkB receptors58, 59 (Figure 3c).
Another BDNF receptor, low-affinity neurotrophin receptor p75NTR, promotes cell death of immature dopaminergic neurons.60 Expression of p75NTR is repressed by En1/2,60 and as En1 was also proposed to co-activate expression of Nurr1-dependent genes,61 En1 could enhance survival of immature dopaminergic neurons via two pathways – enhancing BDNF expression (via Nurr1) and repressing p75NTR expression. Pro-death signaling from p75NTR suppresses ERK1/2 activity and likely inhibits anti-apoptotic activity of Bcl-2 family members,60 thus activating a classical apoptosis pathway via Bax, caspase-3 and caspase-9.62 Caspase-3/-9 activation is inhibited by dual-specificity tyrosine-phosphorylation regulated kinase 1A (Dyrk1a), a Down syndrome-associated gene.63
Involvement of neuron type-specific signaling in survival of dopaminergic neurons is highlighted by inhibition of developmental apoptosis by TGFβ-Smad-Hipk2 pathway.64 Interestingly, although transforming growth factor (TGF) β1 and β2 had little effect on modulation of survival of immature dopaminergic neurons, stimulation by TGFβ3 led to activation of Smad2/3 that directly interacted with Hipk2 and inhibited caspase-3-dependent apoptosis.
Adult neurogenesis: subventricular zone (SVZ)
Survival of postnatally born neurons in the olfactory bulb is regulated by neuronal activity (Figure 4a). Ablation or enhancement of olfactory activity onto maturing granule cells decreases or increases their survival, respectively.65, 66 However, similar enhancement does not affect periglomerular neurons,9, 66 which could be explained by neuron type-specific pro-survival mechanisms. Furthermore, stimulation of periglomerular neurons by a single odorant decreases their survival in the region that is activated by the odorant.9 Apoptosis is stimulated by connective tissue growth factor (CTGF) that, in combination with TGFβ2, activate TGFβRs and Smads in immature periglomerular neurons.9

Neuron type-specific pro-survival signaling in adult-born neurons. (a) Signaling involved in survival and cell death of immature neurons that are born during adult neurogenesis in the SVZ. (b) Signaling involved in survival and cell death of immature neurons that are born during adult neurogenesis in the SGZ. Green arrows: activation of signaling; red blunt arrows: inhibition of signaling; black arrows: activation of receptors on immature neurons from the extracellular space; Pi: phosphorylation
Few neurotransmitter receptors on newborn SVZ neurons mediate pro-survival effects of neuronal activation. Glutamate NMDAR activity is required for survival of neuroblasts during their migration from the SVZ through the RMS and when maturing in the olfactory bulb.23, 67 This pro-survival effect likely depends on Ca2+ that enters into neuroblasts via NMDAR. When already in the olfactory bulb, expression of nicotinic acetylcholine receptor (nAChR) subunit β2 regulates apoptosis in newborn granule cells.68 Knockout of the subunit results in 50% increase in survival of immature neurons, and stimulation of nAChR could be considered as another 'negative' regulator of immature neuronal survival in postnatal neurogenesis, similar to CTGF.
Phosphorylation of CREB1 was shown to promote survival of SVZ-derived neuroblasts,69, 70 where CREB1 might be activated by Ca2+ signaling via calmodulin and CaMKIV.71, 72 As NMDAR are involved in survival of SVZ neuroblasts,23, 67 and upon opening they allow Ca2+ entry into neuroblasts,67 it is likely that Ca2+ entry via NMDAR triggers CREB1-dependent pro-survival cascade (although other receptors on neuroblasts could also mediate Ca2+ entry).72, 73 Knockout of Creb1 was shown to decrease expression of the polysialylated isoform of the neural cell adhesion molecule (PSA-NCAM),70 which, in turn, could promote survival of immature olfactory bulb neurons by inhibiting p75NTR expression.74 Among p75NTR activating neurotrophins only the role of BDNF in postnatal SVZ neurogenesis has been studied, and Ntrk2 knockout decreases the survival of dopaminergic periglomerular neurons, but not any other cells.75, 76
Mammalian target of rapamycin (mTOR) pathway promotes the survival of SVZ neuroblasts via hypoxia-inducible factor 1a (HIF1A).77 Tuberous sclerosis proteins 1 and 2 (TSC1/2) inhibit mTOR, and HIF1A is strongly upregulated in Tsc1−/− neuroblasts, thereby increasing their survival.77 mTOR is most likely activated by PI3K/Akt1 signaling as many components of this pathway were shown to be present in SVZ neuroblasts.72, 78
Finally, pro-survival signaling in newborn SVZ neurons converges on Bcl-2 family members and caspase−3/−9.7, 79
Adult neurogenesis: subgranular zone (SGZ)
Less is known regarding neuronal survival in the SGZ in comparison with the SVZ. Activation of NMDAR on newborn SGZ neurons enhances their survival,24 and it is likely that the pro-survival effect depends on Bcl-2 stimulation (Figure 4b).80 Protection of newborn dentate gyrus neurons by Bcl-2 signaling was also shown in transgenic mice that overexpress Bcl-2.81 Bcl-2 activity might be stimulated by Akt1 signaling, which was shown to enhance neuronal survival in the SGZ.82 Cyclin-dependent kinase-like 5 (CDKL5) activates Akt1 and also inhibits Gsk-3β thus activating CREB1-dependent gene expression. Similar to the SVZ, apoptosis in newborn SGZ neurons converges on Bcl-2/Bax activity.6
Two growth factors promote survival of granule cells in the SGZ – TGFβ1 and IGF1.83, 84 Importantly, both factors have little (if any) contribution to survival of adult-born neurons in the olfactory bulb,9, 85 indicating neuron type-specific role of TGFβ1 and IGF1 in survival of adult-born neurons.
Common signaling that regulates neuronal survival in the brain
Many neuron type-specific pro-survival pathways eventually converge on pro-apoptotic and pro-survival members of Bcl-2 family and caspase-3/caspase-9 (Figure 2). Neuronal apoptosis in the brain is inhibited by Bcl-2 and Bcl-xL pro-survival proteins,86, 87, 88, 89 whereas pro-apoptotic proteins, mainly Bax and Bak, promote neuronal death.87, 88 Massive death of immature neurons in the brain of Bcl2l1−/− (gene name for Bcl-xL) mice suggests that Bcl-xL is the major neuronal pro-survival protein of Bcl-2 family,86, 87 and it becomes important for survival only at the stage of postmitotic neurons, but not before.88 Another anti-apoptotic member of the Bcl-2 family, myeloid cell leukemia 1 (Mcl-1), was also shown to be critical for survival of immature neurons during embryonic development.90
Several transcription factors promote neuronal survival, most likely by activating transcription of pro-survival genes and/or inhibiting pro-apoptotic genes. A family of myocyte enhancer factor 2 (MEF2) transcription factors, MEF2A, 2C and 2D, are expressed in the mouse brain during development and are critical for the survival of immature neurons.91 Widespread loss of neurons was also reported for knockout of another transcription factor – p73 (a member of p53 family proteins).92 The loss of neurons started to be visible during second postnatal week, and was attributed to the anti-apoptotic role of the truncated form of p73, ΔNp73, which antagonizes p53 function and inhibits Bax and caspase-3/-9-dependent apoptosis.93 Finally, members of the CREB family of transcription factors, CREB1 and CREM, activate pro-survival signaling in postmitotic neurons around the time of perinatal development (E16.5-P0).94
Activity-dependent survival of immature neurons via action of GABA and/or glutamate neurotransmitters was proposed for many neuronal subtypes.95 For instance, deletion of syntaxin-binding protein 1 (Stxbp1) that is required for synaptogenesis and neurotransmission results in widespread neuronal death during brain development.96 Furthermore, pharmacological inhibition of NMDAR leads to a pronounced decrease in survival of neurons during postnatal brain development.97, 98, 99 However, as discussed above, knockouts of genes coding for NMDAR subunits show marked increase in neuronal death only during adult neurogenesis.26, 27, 28, 100
Neuronal activity also generates reactive oxygen species (ROS) that could damage maturing neurons and trigger apoptosis. Protection from ROS is particularly important for immature neurons since they are often easier to excite than mature ones.101, 102 It was recently shown that knockout of the gene coding for the antioxidant protein lanthionine synthetase C-like protein 1 (LanCL1) causes massive neuronal death in the brain due to reduced glutathione-mediated antioxidant defense and via Bax activation.103
Survival of neurons in injured brain
Immature neurons are more vulnerable to stress factors than mature neurons, as it is easier for external stimuli to trigger neuronal death during development than in adult brain.11 Although the exact mechanisms of such vulnerability are unknown, it is likely that neurons over maturation devise a highly protective strategy against any external stress. Furthermore, expression of some pro-death molecules, for example, dependence receptors,19, 20 could be limited to immature neurons. Therefore, similar stress factors might be more potent enhancers of neuronal death during development than in adult brain.
In addition to common stress factors that stimulate neuronal death both during development and in adult, few factors are specific for the developing brain – for instance, misplacement of neurons could trigger their death due to impairment in neuronal connectivity. Certain types of immature neurons are more strongly affected by the stress than the others highlighting neuron type-specific mechanisms of survival. Below we discuss factors that affect survival of neurons during abnormal brain development.
Oxidative stress
Oxidative stress contributes to severe neurodevelopmental deficits in the developing mammalian brain caused by chronic exposure to either reduced (hypoxia–ischemia) or elevated (hyperoxia) levels of oxygen (Figure 5).

Cell signaling under hypoxic (purple arrows) and hyperoxic (blue arrows) conditions in immature neurons in vivo. Arrows: activation of signaling; blunt arrows: inhibition of signaling; vertical small arrow: elevated expression level
Perinatal hypoxia–ischemia or neonatal stroke is the main cause of neurodevelopmental deficits in newborns. It is accompanied by an overall decrease in cortical and hippocampal volumes due to neuronal death and atrophy. One of the major causes of neuronal death is excitotoxicity due to overactivation of NMDAR on immature neurons by the release of glutamate.104, 105 Pathological influx of Ca2+ via NMDAR is followed by aberrant production of free radicals and mitochondrial dysfunction, which leads to the release of cytochrome C and, consequently, neuronal death.106, 107 Importantly, interneurons were shown to be less susceptible to hypoxic cell death – although neonatal hypoxia slows maturation of interneurons, it does not affect their survival.108
A glutamate-independent mechanism contributing to hypoxia–ischemia-induced neuronal death reveals transient receptor potential melastatin 7 (TRPM7) as a key factor.109 As early as 24 h after neonatal ischemic insult, TRPM7 protein levels were upregulated, which might lead to increase in caspase-3-dependent apoptosis by inhibiting Akt1 and promoting Bax versus Bcl-2 expression.
Overexposure to oxygen could cause hyperoxia in the brain, which was shown to affect preterm born neonates receiving oxygen supplementation.110 Hyperoxia mainly affects cortical areas and in mice the effect on neuronal survival is most pronounced between P3 and P7.111 Apoptosis is caspase-3 dependent and could be enhanced because of decreased pro-survival signaling from Akt1 and Erk1/2.112 Importantly, the effect is limited to immature neurons, as hyperoxia at later ages does not affect neuronal survival. Hyperoxia also triggers an inflammatory response that could further promote neuronal death via increased levels of several interleukins - IL-1β, IL-18 and IL-18 receptor α (IL-18Rα).113
Fetal alcohol spectrum disorders (FASDs)
FASDs are triggered by gestational alcohol exposure and lead to impaired brain development accompanied by deficits in cognitive functions.114 Data from animal models of prenatal alcohol exposure suggest that neuronal cell death is one of the major effects contributing to the disease phenotype (Figure 6).115

Cell signaling upon alcohol exposure of immature neurons in vivo. Green arrows: activation of signaling; red blunt arrows: inhibition of signaling; vertical small arrow: elevated expression level
Early postnatal (P7) exposure of rats to EtOH induces widespread apoptosis, indicated by increased activation of caspase-3 as early as 8 h and neurodegeneration within less than 24 h after EtOH treatment.116 Differential susceptibility of immature neurons to alcohol-induced stress is underlined by variability of the extent of neuronal death in different brain regions. Thus, the retrosplenial cortex and hippocampus were most affected, whereas the olfactory bulb and piriform cortex exhibited much less apoptosis.116 In another study, the overall architecture of mouse brains exposed to alcohol at P7 appeared to be unaltered, but the number of calretinin-positive and parvalbumin-positive GABAergic neurons was strongly reduced, indicating that they are more prone to alcohol-induced cell death when immature.117 Misplacing GABAergic neurons could contribute to their death since low doses of prenatal alcohol increase ambient GABA levels in the extracellular space and upregulate GABAA receptors on neuroblasts that lead to aberrant neuroblast migration.118
Ethanol possesses NMDA antagonist and GABAA agonist activities and both activities could induce apoptosis during brain development.97, 119 Thus, apoptotic effects of ethanol exposure are closely related to those observed with either disrupted NMDA or elevated GABA signaling. The former has been extensively studied in immature neurons using NMDAR inhibitors causing rapid neuronal death of both excitatory and inhibitory neurons associated with decreased Bcl-2, Erk1/2 and CREB1 and increased activated caspase-3 levels.120, 121, 122
Embryonically administered EtOH was also shown to decrease activation of pro-survival PI3K/Akt1 signaling and increase activation of glycogen synthase kinase-3β (GSK-3β).123 The latter could stimulate neuronal death by activating Bax and, thus, caspase-3-dependent apoptosis.124
Neuronal cell loss as a consequence of alcohol exposure in models of FASD can be attributed in part to oxidative stress. Analysis of the cerebella of P1 rats chronically exposed to ethanol from E6 shows a decrease in mRNA levels of mitochondrial respiration complex genes in granule cells, combined with increased expression of pro-apoptotic p53 and oxidative stress markers.125 EtOH also inhibits nuclear translocation of nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor that is responsible for expression of those genes that protect against oxidative stress and reduce production of ROS.126 In the cerebellum, ROS can activate c-jun N-terminal kinase (JNK) at P4, but not at P7 rats, highlighting a time window in immature granule cells when they are most vulnerable to the oxidative stress.127, 128 JNK, in turn, removes pro-survival 14-3-3 protein from its dimer with Bax, thus making it possible for cytosolic Bax to translocate into the mitochondria leading to mitochondrial dysfunction and neuronal apoptosis via release of cytochrome C.
Traumatic brain injury (TBI)
Although brain injury due to physical trauma is common in both developing and adult brains, the effect of such injury on the immature brain is much more devastating.129 Strikingly, in a rat model of the disorder, the extent of neuronal apoptosis is age-related, with the P3–P7 brains being most vulnerable.130 Apoptosis of immature neurons was associated with enhanced expression of c-Jun and reduced expression of Bcl-2 and Bcl-xL leading to the release of cytochrome C and neuronal cell death.130, 131 Caspase-1 was shown to promote neuronal death by activating two proinflammatory cytokines, IL-1β and IL-18, acting via IL-18 R on neurons.113, 132 Interestingly, immature neurons are also the most affected by TBI population in the regions of adult neurogenesis in mice.133, 134
Other diseases
Neuronal death contributes to phenotypic effects observed in several other neurodevelopmental disorders. Defects in microtubules because of mutations in tubulin alpha or beta genes are often associated with cortical malformations (e.g., lissencephaly or polymicrogyria) because of neuronal misplacement and subsequent death of misplaced neurons.135 For instance, deletion of Tubb2 gene during brain development causes aberrant neuronal migration and arrest of cells near the ventricles that eventually leads to enhanced neuronal apoptosis.135, 136
Although apoptosis was proposed to be augmented in a variety of psychiatric disorders, including schizophrenia and autism spectrum disorders (ASDs), the data were often obtained by analyzing adult brains. Experimental evidence in younger brains is rather limited to gene expression measurements using western blot or PCR.137 Furthermore, it remains to be investigated whether a reduction in the number of GABAergic neurons that was reported in postmortem brains of patients with schizophrenia, bipolar disorder and ASDs138, 139 occurs before neuronal maturation is finished. In addition, it might be that the strength of marker expression rather than the number of neurons is affected.140 Although knockout/knockdown of genes that are associated with psychiatric disorders has been reported to decrease the number of immature neurons in mice,141 other studies showed that maturation rather than survival of immature neurons is affected.142, 143, 144
Conclusions
The mammalian brain is the most complex tissue and includes many more neuronal subtypes than other parts of the nervous system. During perinatal development and in the regions of adult neurogenesis, neurons in the brain are overproduced and multitudes of immature neurons die before they reach maturity. Although there are certain core components of survival/apoptotic machinery in immature neurons, it seems that various types of neurons also exploit pro-survival pathways that are specific only for one or few type(s) and not utilized in others. Such neuron type-specific components of pro-survival signaling could indicate adaptation toward an optimal survival rate of overproduced neurons according to type of neuron and brain region. The number, type and position of neurons that survived should affect both local neuronal circuits and higher brain activities, for example, oscillations. As there is increasing evidence that some types of neurons are more susceptible to certain injuries in the developing brain, more targeted therapeutic strategies might be needed to treat such brain disorders. The advantage of targeting neuron type-specific pro-survival pathways is to avoid side effects of the therapy on other neuron/cell types that are not affected in the disorder. Future studies will determine the extent to which neuron type-specific pro-survival signaling is utilized in normal brain and in pathological conditions and how it contributes to brain information processing.
References
Southwell DG, Paredes MF, Galvao RP, Jones DL, Froemke RC, Sebe JY et al. Intrinsically determined cell death of developing cortical interneurons. Nature 2012; 491: 109–113.
Oo TF, Burke RE . The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Brain Res Dev Brain Res 1997; 98: 191–196.
Burek MJ, Oppenheim RW . Programmed cell death in the developing nervous system. Brain Pathol 1996; 6: 427–446.
White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD . Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci 1998; 18: 1428–1439.
Lossi L, Merighi A . In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian CNS. Prog Neurobiol 2003; 69: 287–312.
Sun W, Winseck A, Vinsant S, Park OH, Kim H, Oppenheim RW . Programmed cell death of adult-generated hippocampal neurons is mediated by the proapoptotic gene Bax. J Neurosci 2004; 24: 11205–11213.
Kim WR, Kim Y, Eun B, Park OH, Kim H, Kim K et al. Impaired migration in the rostral migratory stream but spared olfactory function after the elimination of programmed cell death in Bax knock-out mice. J Neurosci 2007; 27: 14392–14403.
Mouret A, Gheusi G, Gabellec MM, de Chaumont F, Olivo-Marin JC, Lledo PM . Learning and survival of newly generated neurons: when time matters. J Neurosci 2008; 28: 11511–11516.
Khodosevich K, Lazarini F, von Engelhardt J, Kaneko H, Lledo PM, Monyer H . Connective tissue growth factor regulates interneuron survival and information processing in the olfactory bulb. Neuron 2013; 79: 1136–1151.
Benn SC, Woolf CJ . Adult neuron survival strategies—slamming on the brakes. Nat Rev Neurosci 2004; 5: 686–700.
Kole AJ, Annis RP, Deshmukh M . Mature neurons: equipped for survival. Cell Death Dis 2013; 4: e689.
Buss RR, Sun W, Oppenheim RW . Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci 2006; 29: 1–35.
Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH . Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev 2014; 94: 991–1026.
Khodosevich K, Alfonso J, Monyer H . Dynamic changes in the transcriptional profile of subventricular zone-derived postnatally born neuroblasts. Mech Dev 2013; 130: 424–432.
Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M . Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience 1990; 39: 451–458.
Ferrer I, Soriano E, del Rio JA, Alcantara S, Auladell C . Cell death and removal in the cerebral cortex during development. Prog Neurobiol 1992; 39: 1–43.
Finlay BL, Slattery M . Local differences in the amount of early cell death in neocortex predict adult local specializations. Science 1983; 219: 1349–1351.
van Praag H, Kempermann G, Gage FH . Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 1999; 2: 266–270.
Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L et al. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature 2010; 467: 59–63.
Dekkers MP, Nikoletopoulou V, Barde YA . Cell biology in neuroscience: death of developing neurons: new insights and implications for connectivity. J Cell Biol 2013; 203: 385–393.
Fishell G, van der Kooy D . Pattern formation in the striatum: neurons with early projections to the substantia nigra survive the cell death period. J Comp Neurol 1991; 312: 33–42.
Dusart I, Guenet JL, Sotelo C . Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum 2006; 5: 163–173.
Lin CW, Sim S, Ainsworth A, Okada M, Kelsch W, Lois C . Genetically increased cell-intrinsic excitability enhances neuronal integration into adult brain circuits. Neuron 2010; 65: 32–39.
Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH . NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature 2006; 442: 929–933.
Kelsch W, Li Z, Eliava M, Goengrich C, Monyer H . GluN2B-containing NMDA receptors promote wiring of adult-born neurons into olfactory bulb circuits. J Neurosci 2012; 32: 12603–12611.
Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M et al. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 1994; 13: 325–338.
Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E et al. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 1996; 16: 333–344.
von Engelhardt J, Doganci B, Jensen V, Hvalby O, Gongrich C, Taylor A et al. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron 2008; 60: 846–860.
Le Magueresse C, Alfonso J, Khodosevich K, Arroyo Martin AA, Bark C, Monyer H . "Small axonless neurons": postnatally generated neocortical interneurons with delayed functional maturation. J Neurosci 2011; 31: 16731–16747.
Kreuzberg M, Kanov E, Timofeev O, Schwaninger M, Monyer H, Khodosevich K . Increased subventricular zone-derived cortical neurogenesis after ischemic lesion. Exp Neurol 2010; 226: 90–99.
Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 1999; 98: 739–755.
Katoh-Semba R, Takeuchi IK, Semba R, Kato K . Distribution of brain-derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem 1997; 69: 34–42.
Patz S, Wahle P . Developmental changes of neurotrophin mRNA expression in the layers of rat visual cortex. Eur J Neurosci 2006; 24: 2453–2460.
Catapano LA, Arnold MW, Perez FA, Macklis JD . Specific neurotrophic factors support the survival of cortical projection neurons at distinct stages of development. J Neurosci 2001; 21: 8863–8872.
Baydyuk M, Xie Y, Tessarollo L, Xu B . Midbrain-derived neurotrophins support survival of immature striatal projection neurons. J Neurosci 2013; 33: 3363–3369.
Baquet ZC, Bickford PC, Jones KR . Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci 2005; 25: 6251–6259.
Kokubo M, Nishio M, Ribar TJ, Anderson KA, West AE, Means AR . BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J Neurosci 2009; 29: 8901–8913.
Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A . Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 2002; 129: 3147–3160.
Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS Jr. . Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci 2000; 22: 125–138.
Wood KA, Dipasquale B, Youle RJ . In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron 1993; 11: 621–632.
Henderson CE . Role of neurotrophic factors in neuronal development. Curr Opin Neurobiol 1996; 6: 64–70.
Minichiello L, Klein R . TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev 1996; 10: 2849–2858.
Murase S, Owens DF, McKay RD . In the newborn hippocampus, neurotrophin-dependent survival requires spontaneous activity and integrin signaling. J Neurosci 2011; 31: 7791–7800.
Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci 2010; 30: 1739–1749.
Chrysis D, Calikoglu AS, Ye P, D'Ercole AJ . Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci 2001; 21: 1481–1489.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 2013; 16: 543–551.
Sanno H, Shen X, Kuru N, Bormuth I, Bobsin K, Gardner HA et al. Control of postnatal apoptosis in the neocortex by RhoA-subfamily GTPases determines neuronal density. J Neurosci 2010; 30: 4221–4231.
Zhao Y, Kwan KM, Mailloux CM, Lee WK, Grinberg A, Wurst W et al. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc Natl Acad Sci USA 2007; 104: 13182–13186.
Garel S, Marin F, Grosschedl R, Charnay P . Ebf1 controls early cell differentiation in the embryonic striatum. Development 1999; 126: 5285–5294.
Croci L, Chung SH, Masserdotti G, Gianola S, Bizzoca A, Gennarini G et al. A key role for the HLH transcription factor EBF2COE2,O/E-3 in Purkinje neuron migration and cerebellar cortical topography. Development 2006; 133: 2719–2729.
Croci L, Barili V, Chia D, Massimino L, van Vugt R, Masserdotti G et al. Local insulin-like growth factor I expression is essential for Purkinje neuron survival at birth. Cell Death Differ 2011; 18: 48–59.
Arenas E, Denham M, Villaescusa JC . How to make a midbrain dopaminergic neuron. Development 2015; 142: 1918–1936.
Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci 2009; 29: 15923–15932.
Sonnier L, Le Pen G, Hartmann A, Bizot JC, Trovero F, Krebs MO et al. Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J Neurosci 2007; 27: 1063–1071.
van den Munckhof P, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF et al. Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development 2003; 130: 2535–2542.
Peng C, Aron L, Klein R, Li M, Wurst W, Prakash N et al. Pitx3 is a critical mediator of GDNF-induced BDNF expression in nigrostriatal dopaminergic neurons. J Neurosci 2011; 31: 12802–12815.
Volpicelli F, Caiazzo M, Greco D, Consales C, Leone L, Perrone-Capano C et al. Bdnf gene is a downstream target of Nurr1 transcription factor in rat midbrain neurons in vitro. J Neurochem 2007; 102: 441–453.
Checa N, Canals JM, Gratacos E, Alberch J . TrkB and TrkC are differentially regulated by excitotoxicity during development of the basal ganglia. Exp Neurol 2001; 172: 282–292.
Zaman V, Nelson ME, Gerhardt GA, Rohrer B . Neurodegenerative alterations in the nigrostriatal system of trkB hypomorphic mice. Exp Neurol 2004; 190: 337–346.
Alavian KN, Sgado P, Alberi L, Subramaniam S, Simon HH . Elevated P75NTR expression causes death of engrailed-deficient midbrain dopaminergic neurons by Erk1/2 suppression. Neural Dev 2009; 4: 11.
Veenvliet JV, Dos Santos MT, Kouwenhoven WM, von Oerthel L, Lim JL, van der Linden AJ et al. Specification of dopaminergic subsets involves interplay of En1 and Pitx3. Development 2013; 140: 3373–3384.
Yamaguchi Y, Miura M . Programmed cell death in neurodevelopment. Dev Cell 2015; 32: 478–490.
Barallobre MJ, Perier C, Bove J, Laguna A, Delabar JM, Vila M et al. DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson's disease. Cell Death Dis 2014; 5: e1289.
Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J et al. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat Neurosci 2007; 10: 77–86.
Petreanu L, Alvarez-Buylla A . Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J Neurosci 2002; 22: 6106–6113.
Rey NL, Sacquet J, Veyrac A, Jourdan F, Didier A . Behavioral and cellular markers of olfactory aging and their response to enrichment. Neurobiol Aging 2012; 33: 626 e629–626 e623.
Platel JC, Dave KA, Gordon V, Lacar B, Rubio ME, Bordey A . NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron 2010; 65: 859–872.
Mechawar N, Saghatelyan A, Grailhe R, Scoriels L, Gheusi G, Gabellec MM et al. Nicotinic receptors regulate the survival of newborn neurons in the adult olfactory bulb. Proc Natl Acad Sci USA 2004; 101: 9822–9826.
Giachino C, De Marchis S, Giampietro C, Parlato R, Perroteau I, Schutz G et al. cAMP response element-binding protein regulates differentiation and survival of newborn neurons in the olfactory bulb. J Neurosci 2005; 25: 10105–10118.
Herold S, Jagasia R, Merz K, Wassmer K, Lie DC . CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol Cell Neurosci 2011; 46: 79–88.
Khodosevich K, Monyer H . Signaling in migrating neurons: from molecules to networks. Front Neurosci 2011; 5: 28.
Khodosevich K, Seeburg PH, Monyer H . Major signaling pathways in migrating neuroblasts. Front Mol Neurosci 2009; 2: 7.
Khodosevich K, Zuccotti A, Kreuzberg MM, Le Magueresse C, Frank M, Willecke K et al. Connexin45 modulates the proliferation of transit-amplifying precursor cells in the mouse subventricular zone. Proc Natl Acad Sci USA 2012; 109: 20107–20112.
Gascon E, Vutskits L, Jenny B, Durbec P, Kiss JZ . PSA-NCAM in postnatally generated immature neurons of the olfactory bulb: a crucial role in regulating p75 expression and cell survival. Development 2007; 134: 1181–1190.
Bergami M, Vignoli B, Motori E, Pifferi S, Zuccaro E, Menini A et al. TrkB signaling directs the incorporation of newly generated periglomerular cells in the adult olfactory bulb. J Neurosci 2013; 33: 11464–11478.
Galvao RP, Garcia-Verdugo JM, Alvarez-Buylla A . Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J Neurosci 2008; 28: 13368–13383.
Feliciano DM, Zhang S, Quon JL, Bordey A . Hypoxia-inducible factor 1a is a Tsc1-regulated survival factor in newborn neurons in tuberous sclerosis complex. Hum Mol Genet 2013; 22: 1725–1734.
Khodosevich K, Monyer H . Signaling involved in neurite outgrowth of postnatally born subventricular zone neurons in vitro. BMC Neurosci 2010; 11: 18.
Miwa N, Storm DR . Odorant-induced activation of extracellular signal-regulated kinase/mitogen-activated protein kinase in the olfactory bulb promotes survival of newly formed granule cells. J Neurosci 2005; 25: 5404–5412.
Mu Y, Zhao C, Toni N, Yao J, Gage FH . Distinct roles of NMDA receptors at different stages of granule cell development in the adult brain. Elife 2015; 4: e07871.
Kuhn HG, Biebl M, Wilhelm D, Li M, Friedlander RM, Winkler J . Increased generation of granule cells in adult Bcl-2-overexpressing mice: a role for cell death during continued hippocampal neurogenesis. Eur J Neurosci 2005; 22: 1907–1915.
Fuchs C, Trazzi S, Torricella R, Viggiano R, De Franceschi M, Amendola E et al. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol Dis 2014; 70: 53–68.
Kandasamy M, Lehner B, Kraus S, Sander PR, Marschallinger J, Rivera FJ et al. TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J Cell Mol Med 2014; 18: 1444–1459.
Lichtenwalner RJ, Forbes ME, Sonntag WE, Riddle DR . Adult-onset deficiency in growth hormone and insulin-like growth factor-I decreases survival of dentate granule neurons: insights into the regulation of adult hippocampal neurogenesis. J Neurosci Res 2006; 83: 199–210.
Hurtado-Chong A, Yusta-Boyo MJ, Vergano-Vera E, Bulfone A, de Pablo F, Vicario-Abejon C . IGF-I promotes neuronal migration and positioning in the olfactory bulb and the exit of neuroblasts from the subventricular zone. Eur J Neurosci 2009; 30: 742–755.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995; 267: 1506–1510.
Shindler KS, Latham CB, Roth KA . Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci 1997; 17: 3112–3119.
Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S et al. Bcl-xL is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci 2016; 36: 5448–5461.
Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM . Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci 2005; 25: 6721–6728.
Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci 2008; 28: 6068–6078.
Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA et al. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS ONE 2012; 7: e34863.
Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD . p73 is required for survival and maintenance of CNS neurons. J Neurosci 2002; 22: 9800–9809.
Jacobs WB, Walsh GS, Miller FD . Neuronal survival and p73/p63/p53: a family affair. Neuroscientist 2004; 10: 443–455.
Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 2002; 31: 47–54.
Luhmann HJ, Sinning A, Yang JW, Reyes-Puerta V, Stuttgen MC, Kirischuk S et al. Spontaneous neuronal activity in developing neocortical networks: from single cells to large-scale interactions. Front Neural Circuits 2016; 10: 40.
Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000; 287: 864–869.
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999; 283: 70–74.
Heck N, Golbs A, Riedemann T, Sun JJ, Lessmann V, Luhmann HJ . Activity-dependent regulation of neuronal apoptosis in neonatal mouse cerebral cortex. Cereb Cortex 2008; 18: 1335–1349.
Wagner-Golbs A, Luhmann HJ . Activity-dependent survival of developing neocortical neurons depends on PI3K signalling. J Neurochem 2012; 120: 495–501.
Maskos U, McKay RD . Neural cells without functional N-Methyl-D-Aspartate (NMDA) receptors contribute extensively to normal postnatal brain development in efficiently generated chimaeric NMDA R1 -/- <—> +/+ mice. Dev Biol 2003; 262: 119–136.
Schmidt-Hieber C, Jonas P, Bischofberger J . Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 2004; 429: 184–187.
Wang XQ, Deriy LV, Foss S, Huang P, Lamb FS, Kaetzel MA et al. CLC-3 channels modulate excitatory synaptic transmission in hippocampal neurons. Neuron 2006; 52: 321–333.
Huang C, Chen M, Pang D, Bi D, Zou Y, Xia X et al. Developmental and activity-dependent expression of LanCL1 confers antioxidant activity required for neuronal survival. Dev Cell 2014; 30: 479–487.
Gucuyener K, Atalay Y, Aral YZ, Hasanoglu A, Turkyilmaz C, Biberoglu G . Excitatory amino acids and taurine levels in cerebrospinal fluid of hypoxic ischemic encephalopathy in newborn. Clin Neurol Neurosurg 1999; 101: 171–174.
Pu Y, Li QF, Zeng CM, Gao J, Qi J, Luo DX et al. Increased detectability of alpha brain glutamate/glutamine in neonatal hypoxic-ischemic encephalopathy. AJNR Am J Neuroradiol 2000; 21: 203–212.
Fiskum G, Murphy AN, Beal MF . Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab 1999; 19: 351–369.
Kumar A, Mittal R, Khanna HD, Basu S . Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008; 122: e722–e727.
Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML et al. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci 2013; 33: 13375–13387.
Chen W, Xu B, Xiao A, Liu L, Fang X, Liu R et al. TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Mol Brain 2015; 8: 11.
Deuber C, Terhaar M . Hyperoxia in very preterm infants a systematic review of the literature. J Perinat Neonat Nur 2011; 25: 268–274.
Ikonomidou C, Kaindl AM . Neuronal death and oxidative stress in the developing brain. Antioxid Redox Signal 2011; 14: 1535–1550.
Kaindl AM, Sifringer M, Zabel C, Nebrich G, Wacker MA, Felderhoff-Mueser U et al. Acute and long-term proteome changes induced by oxidative stress in the developing brain. Cell Death Differ 2006; 13: 1097–1109.
Felderhoff-Mueser U, Sifringer M, Polley O, Dzietko M, Leineweber B, Mahler L et al. Caspase-1-processed interleukins in hyperoxia-induced cell death in the developing brain. Ann Neurol 2005; 57: 50–59.
Riley EP, Infante MA, Warren KR . Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev 2011; 21: 73–80.
Goodlett CR, Horn KH, Zhou FC . Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 2005; 230: 394–406.
Wilson DA, Peterson J, Basavaraj BS, Saito M . Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol Clin Exp Res 2011; 35: 1974–1984.
Smiley JF, Saito M, Bleiwas C, Masiello K, Ardekani B, Guilfoyle DN et al. Selective reduction of cerebral cortex GABA neurons in a late gestation model of fetal alcohol spectrum disorder. Alcohol 2015; 49: 571–580.
Cuzon VC, Yeh PW, Yanagawa Y, Obata K, Yeh HH . Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. J Neurosci 2008; 28: 1854–1864.
Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000; 287: 1056–1060.
Coleman LG Jr, Jarskog LF, Moy SS, Crews FT . Deficits in adult prefrontal cortex neurons and behavior following early post-natal NMDA antagonist treatment. Pharmacol Biochem Behav 2009; 93: 322–330.
Hansen HH, Briem T, Dzietko M, Sifringer M, Voss A, Rzeski W et al. Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol Dis 2004; 16: 440–453.
Lema Tome CM, Nottingham CU, Smith CM, Beauchamp AS, Leung PW, Turner CP . Neonatal exposure to MK801 induces structural reorganization of the central nervous system. Neuroreport 2006; 17: 779–783.
de la Monte SM, Wands JR . Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell Mol Life Sci 2002; 59: 882–893.
Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z et al. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J Neurosci Res 2009; 87: 2793–2802.
Chu J, Tong M, de la Monte SM . Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol 2007; 113: 659–673.
Kumar A, Singh CK, Lavoie HA, Dipette DJ, Singh US . Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol Pharmacol 2011; 80: 446–457.
Heaton MB, Paiva M, Kubovec S . Differential effects of ethanol on bid, tBid, and Bax:tBid interactions in postnatal day 4 and postnatal day 7 rat cerebellum. Alcohol Clin Exp Res 2015; 39: 55–63.
Heaton MB, Paiva M, Kubovic S, Kotler A, Rogozinski J, Swanson E et al. Differential effects of ethanol on c-jun N-terminal kinase, 14-3-3 proteins, and Bax in postnatal day 4 and postnatal day 7 rat cerebellum. Brain Res 2012; 1432: 15–27.
Giza CC, Prins ML . Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci-Basel 2006; 28: 364–379.
Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H et al. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol 1999; 45: 724–735.
Felderhoff-Mueser U, Sifringer M, Pesditschek S, Kuckuck H, Moysich A, Bittigau P et al. Pathways leading to apoptotic neurodegeneration following trauma to the developing rat brain. Neurobiol Dis 2002; 11: 231–245.
Sifringer M, Stefovska V, Endesfelder S, Stahel PF, Genz K, Dzietko M et al. Activation of caspase-1 dependent interleukins in developmental brain trauma. Neurobiol Dis 2007; 25: 614–622.
Kim DH, Ko IG, Kim BK, Kim TW, Kim SE, Shin MS et al. Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol Behav 2010; 101: 660–665.
Zhou H, Chen L, Gao X, Luo B, Chen J . Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol 2012; 71: 348–359.
Jaglin XH, Poirier K, Saillour Y, Buhler E, Tian G, Bahi-Buisson N et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet 2009; 41: 746–752.
Stottmann RW, Donlin M, Hafner A, Bernard A, Sinclair DA, Beier DR . A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Hum Mol Genet 2013; 22: 4053–4063.
Wei H, Alberts I, Li X . The apoptotic perspective of autism. Int J Dev Neurosci 2014; 36: 13–18.
Pantazopoulos H, Wiseman JT, Markota M, Ehrenfeld L, Berretta S . Decreased numbers of somatostatin-expressing neurons in the amygdala of subjects with bipolar disorder or schizophrenia: relationship to circadian rhythms. Biol Psychiatry 2016; 81: 536–547.
Hashemi E, Ariza J, Rogers H, Noctor SC, Martinez-Cerdeno V . The number of parvalbumin-expressing interneurons is decreased in the medial prefrontal cortex in autism. Cereb Cortex 2016 (doi:10.1093/cercor/bhw021).
Chung DW, Fish KN, Lewis DA . Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am J Psychiatry 2016; 173: 1131–1139.
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011; 147: 235–246.
Kim JY, Liu CY, Zhang F, Duan X, Wen Z, Song J et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 2012; 148: 1051–1064.
Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 2014; 511: 236–240.
Watanabe Y, Khodosevich K, Monyer H . Dendrite development regulated by the schizophrenia-associated gene FEZ1 involves the ubiquitin proteasome system. Cell Rep 2014; 7: 552–564.
Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008; 59: 399–412.
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E . Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci 2005; 25: 1089–1094.
Frielingsdorf H, Simpson DR, Thal LJ, Pizzo DP . Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol Dis 2007; 26: 47–55.
Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO et al. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci 1995; 15: 2888–2905.
Muller M, Triaca V, Besusso D, Costanzi M, Horn JM, Koudelka J et al. Loss of NGF-TrkA signaling from the CNS is not sufficient to induce cognitive impairments in young adult or intermediate-aged mice. J Neurosci 2012; 32: 14885–14898.
Scardigli R, Capelli P, Vignone D, Brandi R, Ceci M, La Regina F et al. Neutralization of nerve growth factor impairs proliferation and differentiation of adult neural progenitors in the subventricular zone. Stem Cells 2014; 32: 2516–2528.
Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 1994; 368: 246–249.
Bartkowska K, Paquin A, Gauthier AS, Kaplan DR, Miller FD . Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 2007; 134: 4369–4380.
Ma L, Harada T, Harada C, Romero M, Hebert JM, McConnell SK et al. Neurotrophin-3 is required for appropriate establishment of thalamocortical connections. Neuron 2002; 36: 623–634.
Shimazu K, Zhao M, Sakata K, Akbarian S, Bates B, Jaenisch R et al. NT-3 facilitates hippocampal plasticity and learning and memory by regulating neurogenesis. Learn Mem 2006; 13: 307–315.
Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M . Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci 1997; 9: 2045–2056.
Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F . Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 1995; 14: 717–730.
Hodge RD, D'Ercole AJ, O'Kusky JR . Insulin-like growth factor-I (IGF-I) inhibits neuronal apoptosis in the developing cerebral cortex in vivo. Int J Dev Neurosci 2007; 25: 233–241.
Harrist A, Beech RD, King SL, Zanardi A, Cleary MA, Caldarone BJ et al. Alteration of hippocampal cell proliferation in mice lacking the beta 2 subunit of the neuronal nicotinic acetylcholine receptor. Synapse 2004; 54: 200–206.
Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N et al. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 1995; 374: 65–67.
Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP . Increased neurodegeneration during ageing in mice lacking high-affinity nicotine receptors. EMBO J 1999; 18: 1235–1244.
Brionne TC, Tesseur I, Masliah E, Wyss-Coray T . Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003; 40: 1133–1145.
Stritt C, Stern S, Harting K, Manke T, Sinske D, Schwarz H et al. Paracrine control of oligodendrocyte differentiation by SRF-directed neuronal gene expression. Nat Neurosci 2009; 12: 418–427.
Bernabeu RO, Longo FM . The p75 neurotrophin receptor is expressed by adult mouse dentate progenitor cells and regulates neuronal and non-neuronal cell genesis. BMC Neurosci 2010; 11: 136.
McQuillen PS, DeFreitas MF, Zada G, Shatz CJ . A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci 2002; 22: 3580–3593.
Lotta LT, Conrad K, Cory-Slechta D, Schor NF . Cerebellar Purkinje cell p75 neurotrophin receptor and autistic behavior. Transl Psychiatry 2014; 4: e416.
Zanin JP, Abercrombie E, Friedman WJ . Proneurotrophin-3 promotes cell cycle withdrawal of developing cerebellar granule cell progenitors via the p75 neurotrophin receptor. Elife 2016: 5.
Catts VS, Al-Menhali N, Burne TH, Colditz MJ, Coulson EJ . The p75 neurotrophin receptor regulates hippocampal neurogenesis and related behaviours. Eur J Neurosci 2008; 28: 883–892.
Adams SM . de Rivero Vaccari JC, Corriveau RA. Pronounced cell death in the absence of NMDA receptors in the developing somatosensory thalamus. J Neurosci 2004; 24: 9441–9450.
Maskos U, Brustle O, McKay RD . Long-term survival, migration, and differentiation of neural cells without functional NMDA receptors in vivo. Dev Biol 2001; 231: 103–112.
Acknowledgements
Work in the Khodosevich lab is supported by the Novo Nordisk Foundation (Hallas-Møller Investigator, NNF16OC0019920) and Agnes og Poul Friis Fond.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pfisterer, U., Khodosevich, K. Neuronal survival in the brain: neuron type-specific mechanisms. Cell Death Dis 8, e2643 (2017). https://doi.org/10.1038/cddis.2017.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.64
- Springer Nature Limited
This article is cited by
-
Neurodevelopmental disorders—high-resolution rethinking of disease modeling
Molecular Psychiatry (2023)
-
CNS Delivery of Nucleic Acid Therapeutics: Beyond the Blood–Brain Barrier and Towards Specific Cellular Targeting
Pharmaceutical Research (2023)
-
Single-cell transcriptomic analysis reveals the adverse effects of cadmium on the trajectory of neuronal maturation
Cell Biology and Toxicology (2023)
-
Pleiotropic predisposition to Alzheimer’s disease and educational attainment: insights from the summary statistics analysis
GeroScience (2022)
-
Small extracellular vesicles encapsulating CCL2 from activated astrocytes induce microglial activation and neuronal apoptosis after traumatic spinal cord injury
Journal of Neuroinflammation (2021)