
Abstract
We report in this work coaxial electrospun fibers with potential applications in the treatment of Parkinson’s disease. The fibers comprise a fixed dose combination (FDC) containing the active ingredients levodopa and carbidopa, loaded in a fast dissolving polyvinylpyrrolidone (PVP) shell and an insoluble but swellable Eudragit® RLPO core. Under appropriate processing conditions we are able to prepare fibers with distinct core/shell architectures and diameters of approximately 400 nm. X-ray diffraction and differential scanning calorimetry analyses revealed that the drugs are dispersed on the molecular level within the polymer carriers, and IR spectroscopy indicated the presence of intermolecular interactions. At pH 1, the composite fibers yields extended release over more than 8 h, with an initial loading dose being freed from the PVP shell and then a sustained release phase following from the insoluble core. This is markedly extended over the release period of the commercial FDC product, and thus the fibers generated here have the potential to be used to reduce the required dosing frequency.
Graphic Abstract

Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a disabling disorder that significantly affects a patient’s quality of life [1]. Treatments are available, but only 10% of PD patients fully adhere to their treatment regimens [2]. Non-adherence is a serious problem linked to worsening symptoms and increased motor fluctuations [3,4,5]. Levodopa (LD) remains the most effective treatment for PD [6]. However, a single dosing treatment is only effective in the early phase of the disease. In the severe phase, 4–5 years after diagnosis, the therapeutic window of dopamine becomes narrow, and due to its short half-life of between 0.7 and 1.4 h dosing of LD will be required every 2 h [7,8,9]. One way to ameliorate this issue is to co-administer LD with carbidopa (CD), which can increase LD’s oral bioavailability to 40–70% [10].
A major goal of PD pharmacotherapy has been the development of an oral formulation able to keep LD in the therapeutic plasma concentration range [11]. The timely intake of LD is of crucial importance to avoid complications arising [12]. There exists an approved combination formulation of LD and CD, available in a dose range between 100/25 and 200/50 mg (Sinemet CR®; Merck). This takes the form of sustained-release (SR) tablets which release their ingredients over a 4 to 6 h period [11, 13]. The SR tablets are advantageous over an immediate release (IR) formulation of LD alone, because SR can reduce fluctuations of dose in the systemic circulation [5]. However, LD absorption might be delayed if the SR tablet is used alone, so effective therapy often requires co-administration of an IR dose of LD/CD, particularly for the first dose of the day [11]. In this work, we seek to combine both immediate and sustained release of LD and CD into a single formulation, with the ultimate aim of improving patient compliance in PD treatment.
The formulation approach taken here is to use electrospinning to produce drug-loaded polymer fibers. Electrospinning first involves the preparation of a mixed solution of a polymer and drug in a volatile solvent, followed by ejection of this solution through a narrow-bore needle and the application of an electrical field to solidify the product. It is beneficial over other techniques such as spray drying and hot-melt extrusion because it does not require the application of heat, and thus thermally labile active ingredients can be processed [14]. Further, electrospinning allows multiple different fluids to be processed simultaneously, leading to fiber products with highly tunable properties [15, 16]. Electrospun fibers have been widely explored to generate drug delivery systems, and there are reports in the literature of fast-dissolving [17,18,19,20], extended release [21,22,23], and pH responsive systems [24, 25], among many others. Fixed-dose combination formulations have also been prepared by electrospinning [26, 27], but there are few studies where multiple drugs have been combined into a single fiber formulation.
The most simple electrospinning experiment involves a single polymer/drug solution, and results in a monolithic product with the drug uniformly dispersed throughout the carrier in most cases. Coaxial electrospinning, dispensing two fluids simultaneously to prepare core/shell fibers, is a more complex process but can create formulations with a number of benefits over monolithic systems from single-needle electrospinning [28, 29]. For instance, while a burst of drug release is typically observed from monolithic fibers (because of the presence of drug at the surface), this can be avoided in a core/shell system by having a drug-free shell layer [30]. Obviating this burst release is particularly important for hydrophilic small molecule drugs [31]. Other benefits of coaxial spinning lie in the development of drug delivery systems with multi-phasic release, as is desired here. Because coaxial electrospinning allows the combination of two polymers with very different properties (e.g. in terms of solubility/dissolution rate), it can result in products giving release at two different rates. This has been shown to be the case when polyvinylpyrrolidone (PVP) and zein were used to construct core/shell fibers [32].
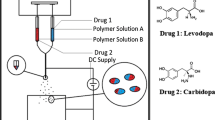
In this work, for the first time, coaxial electrospinning was used to generate FDCs in the form of core/shell fibers (see Fig. 1a) designed for the treatment of Parkinson’s disease. The FDC formulations are prepared using two base polymer solutions containing PVP [shell] and Eudragit® RL PO (E-RLPO) [core] (Fig. 1b). The aim of the work is to fabricate oral FDCs giving an initial burst release to provide a loading dose of the drug, followed by extended release. An immediate burst of drug release from the PVP shell is expected, since PVP is a well-known hydrophilic polymer and will dissolve rapidly upon contact with gastric fluids. This should be followed by extended release from the E-RLPO core, since the latter is insoluble in water but undergoes pH-independent swelling in the digestive fluid to provide time-controlled release. The FDC formulation parameters were first optimised, before two sets of fibers were produced. These were characterised in detail using electron microscopy, X-ray diffraction, differential scanning calorimetry, and IR spectroscopy. The drug loading and encapsulation efficiency were calculated, and the fibers’ drug release profiles explored.

A schematic diagram of the coaxial electrospinning process showing a the experimental setup and b the core/shell FDCs prepared in this work
Experimental Section
Materials
Levodopa (LD), carbidopa (CD), and PVP (MW 360,000 Da) were purchased from Sigma-Aldrich, UK. Eudragit® RLPO (E-RLPO; MW 150,000 Da) was a gift from Evonik (Germany). Analytical grade ethanol, acetonitrile, methanol, and N,N-dimethylacetamide (DMAc) were procured from Sigma-Aldrich (UK). Deionised water was used where needed, and all other chemicals used were of analytical grade.
Solution Preparation
Two base polymer solutions prepared in ethanol were used in this work. These comprised 10% w/v PVP and 20% w/v Eudragit RLPO (E-RLPO). Each solution was stirred overnight to ensure complete dissolution. The required volume of each polymer solution was then mixed with a solution of the active pharmaceutical ingredients (APIs) dissolved in 1 M aqueous HCl. Full details of the formulations are given in Table 1. In order to protect LD and CD from photolytic degradation, the samples were stored in dark amber glass containers. The combined drug/polymer solutions were mixed at room temperature for 6 h to obtain homogeneous solutions.
Electrospinning
The setup of the coaxial electrospinning apparatus is shown in Fig. 1a. For each formulation, the polymer solutions were loaded into separate plastic syringes (BD plastic, MediSupplies, UK) with care taken to avoid any air bubbles. Each syringe was fitted on a syringe pump (KDS100, Cole Parmer, UK) and connected with plastic tubing to infuse the polymer solution into a concentric spinneret comprising two nested stainless steel needles (core: 18G, 0.83 mm inner diameter; shell: 13G, 1.83 mm inner diameter). A positive voltage was applied to the spinneret using an HCP 35-35000 power supply (FuG Elektronik GmbH, Germany). To collect the fibers, a flat metal collector (5 × 5) cm covered with aluminium foil was situated below the spinneret tip. The electrospinning process took place in ambient conditions of 19 to 26 °C, and 31 to 49% relative humidity.
Optimisation of the electrospinning parameters was first performed to find the most appropriate flow rate, voltage, and spinneret to collector distance. The range of parameters explored and optimal processing conditions identified are detailed in Table 1.
Characterisation
Scanning electron microscopy (SEM) images were obtained on a Quanta 200 FEG SEM (FEI, USA). Prior to examination, samples were sputter-coated with gold under argon to make them electrically conductive. Images were taken at an excitation voltage of 5 kV. The average diameter of the fiber formulations was determined from the SEM images by making manual measurements at 100 different points using the ImageJ software (National Institutes of Health, USA).
The electrospun fibers were also examined by transmission electron microscopy (TEM). For this, samples were electrospun directly onto a lacy carbon coated copper grid (TAAB, UK). TEM images were taken on a Philips/FEI CM 120 Bio-Twin instrument (Netherlands).
X-ray diffraction (XRD) patterns were obtained on a MiniFlex 600 diffractometer (Rigaku, Japan) supplied with Cu Kα radiation (λ = 1.5418 Å) at a voltage of 40 kV and current of 15 mA. Samples were fixed on an aluminium holder and data recorded over the 2θ range between 3° and 43° at a scan speed of 5° min−1. Differential scanning calorimetry (DSC) studies were conducted using a Q2000 calorimeter (TA Instruments, USA). Non-hermetically sealed samples in aluminium pans (Tzero premium pan/lid, TA instruments) were heated from 30 °C to an appropriate temperature (ideally above the melting point of the active ingredient of interest but below the degradation point of the polymer). Experiments were undertaken at a rate of 10 °C min−1 and under a nitrogen flow of 50 mL min−1. Data were analysed using the TA instruments Universal Analysis software.
Samples from each formulation were analysed in attenuated total reflectance (ATR) mode IR spectroscopy on a Spectrum 100 instrument (PerkinElmer, USA). Samples were studied over the range 4000 to 650 cm−1, with the spectral resolution set at 1 cm−1. Four scans per sample were recorded.
Functional Performance Assessment
A high performance liquid chromatography (HPLC) assay for simultaneous analysis of both LD and CD was devised based on the method for Co-Careldopa detailed in the British Pharmacopeia [33]. The analysis was performed on a 1200 Infinity Series instrument (Agilent Technologies, USA). The stationary phase was a C18(2) column (00G-4252-E0, Phenomenex Luna, UK) held at 25 °C. Analysis was carried out at a flow rate of 1.5 mL min−1, and an injection volume of 20 μL was used. A UV detector was used for quantification at 282 nm, where LD can be detected at t = 3.7 min and CD at t = 6.2 min. The mobile phase comprised 20:80 v/v methanol: phosphate buffered saline (PBS; pH 2.8, 0.01 mol/L). Adjustment of the PBS pH was undertaken using 1 M orthophosphoric acid (85%, HPLC grade, Fisher Scientific, UK). The mobile phase solutions were mixed and filtered through a 0.22 μm membrane filter (Millex syringe filter, Merck Millipore, Germany) before use.
The loading of each drug in the electrospun fibers was evaluated by taking 20 ± 1 mg of each sample and dissolving this in 100 mL of 1 M HCl under stirring. The vessels used all comprised darkened glass to protect the samples from light degradation. A calibration curve for LD and CD was prepared from stock solutions stored in the dark and freshly diluted on the day of the experiment.
Drug release experiments were carried out using USP apparatus type 1, with a rotating basket (ST7 dissolution tester, Caleva, UK). Fiber samples of 30 ± 1 mg were loaded in hard gelatin capsules (size 0). Each capsule was placed in the rotating basket at 50 rpm, then immersed in a vessel containing 750 mL of 0.1 M HCl, at a temperature of 37 °C. At predetermined time points, 1 mL samples were withdrawn and replaced with an equal amount of fresh pre-heated medium. The release of each API was quantified by HPLC as described above. Three independent experiments were carried out, and cumulative release percentages are reported as mean ± S.D.
Results and Discussion
Fiber Morphology
SEM images of the raw APIs and the coaxial electrospun fibers F1 and F2, together with the diameter distributions of the latter, are displayed in Fig. 2. The SEM images of the raw materials (Fig. 2) show LD to exist as large particles with sharp edges and CD as elongated particles with irregular edges. This is consistent with both being crystalline materials. The production of fibers was successful for both formulations, as shown in Fig. 2. The images of the fibers do not show any evidence for particles of LD or CD, indicating a solid dispersion was generated. In general the fibers have cylindrical shapes with a smooth surfaces, although for F2 some flattened fibers can also be observed. The fiber diameters are 380 ± 120 nm and 1300 ± 330 nm for F1 and F2 respectively. This difference can be attributed to the greater amount of solid material dispensed per unit time with F2. Similar results have been reported by Pornsopone et al. [34, 35], who produced fibers sized ~ 1300 nm from 20% w/v E-RLPO, and fibers sized ~ 2200 nm from 25% w/v E-RLPO.

SEM images of LD, CD, F1 (19.0/4.8% w/w LD/CD), and F2 (8.9/2.2% w/w LD/CD), with fiber diameter distributions
TEM images of the fibers are shown in Fig. 3. Samples were collected at different times after electrospinning was commenced, in order to investigate whether core/shell fibers were continually produced. The images of F1 (Fig. 3a, b) reveal the formation of core/shell fibers in both samples collected, with the core around 154–160 nm in diameter and the shell at 226–295 nm. The images of F2 (Fig. 3c, d) do not confirm the formation of core/shell fibers, however. It appears that here monolithic fibers have been produced. It is not clear why this is, but the solutions used for F2 fabrication have higher polymer concentrations than those used for F1. This will result in them having higher viscosities and different drawing and drying behaviour under the electric field. As a result of these factors, it appears that mixing of the fluids occurred during spinning.

TEM images of the fibers, showing F1 after dispensing a 3 mL and b 6 mL of liquid, and F2 after dispensing c 6 mL and d 12 mL
Physical Form and Component Compatibility
The physical form of the drugs in the fibers was assessed with XRD and DSC. The XRD patterns are given in Fig. 4. The patterns of the API raw materials show numerous sharp Bragg reflections for LD (e.g. at 18.36°, 21.40°, 22.98°, and 25.08°) and CD (e.g. at 9.66°, 18.66°, and 19.56°). This illustrates that both drugs existed in the crystalline form before electrospinning. In contrast, the XRD patterns for the polymers show broad haloes without any Bragg reflections, which indicates the amorphous nature of both polymers. In the patterns of the fiber composites, only a broad halo is seen, without the presence of any characteristic reflections from LD or CD. This confirmed that both drugs are present as amorphous solid dispersions within the fibers. This result was expected since it is known that electrospinning involves a very rapid drying process, which does not allow sufficient time for recrystallisation. Coaxial fibers prepared from PVP and other Eudragit polymers have been found to have similar amorphous structures [36, 37].

XRD diffraction patterns of the raw APIs (LD and CD); the pure polymers (PVP and E-RLPO) and the coaxial fibre formulations (F1 and F2)
The DSC data for the raw API materials (Fig. 5) show endothermic peaks corresponding to melting, with onset for LD at 284 °C and for CD at 196 °C. These results are consistent with values reported in the literature for LD (284–286 °C; [38]) and CD (203–208 °C; [33, 39]). This observation indicates that both drugs were initially in the crystalline form before processing, and agrees with the findings from XRD. In contrast, the data for the polymer raw materials (Fig. 5) show no melting events, consistent with the XRD patterns and confirming them to be amorphous materials. Instead, PVP shows a broad endothermic peak at around 30 to 150 °C due to the evaporation of adsorbed water, and Tg appears at 176 °C, close to the literature value of 166 °C [40]. E-RLPO shows a Tg at 53 °C, followed by a broad dehydration endotherm between 67 and 140 °C. The Tg here is similar to a value previously reported for E-RLPO [41] (53.1 °C). The events observed at 166 °C for E-RLPO have been described by Parikh et al. as polymer degradation [42].

DSC data for the raw materials and fibers
The thermograms of the coaxial fibers (Fig. 5) show they undergo dehydration, in the form of a broad endotherm between room temperature and 140 °C for F1 or 150 °C for F2. These endotherms are very shallow, indicating minimal solvent inclusion in the systems. No melting endotherms from either LD or CD are visible for the fibers, which agrees with the findings from XRD and demonstrates the fibers to comprise amorphous solid dispersions. The thermogram of F2 shows Tgs at 56 °C from E-RLPO, and at 179 °C from PVP. In F1 these glass transitions cannot be clearly observed, but the noise at ca. 166 °C could be related to E-RLPO degradation.
The chemical structures of the raw materials are presented in Fig. 6a. The FTIR spectra of the API raw materials (Fig. 6b) present characteristic bands for each material. In the spectrum of LD, characteristic peaks appear from O–H stretching at 3200, 3063, and 2982 cm−1, from phenyl group C = C vibrations at 1458, 1404, 1352, 817, and 674 cm−1, and from C—H stretches at 2929 and 2982 cm−1. NH2 stretches are visible at 1562 cm−1, and C=O stretching at 1651 cm−1. In the spectrum of CD, O–H stretching bands arise at 3519, 3322, 3286 cm−1, phenyl group C=C vibrations are present at 1457, 1402, 1262, 879, and 830 cm−1, and C–H stretches can be seen at 3101, 3059, and 1371 cm−1. N–H and NH2 stretches arise at 1527 cm−1 and 1122 cm−1, and C=O stretching at 1626 cm−1. The spectra of the raw polymer powders (Fig. 6b) show that PVP has a broad band between 3400 to 2950 cm−1 corresponding to O–H stretches from adsorbed water, C=O vibrations at 1654 cm−1, and a C–N stretch at 1285 cm−1. The spectrum of E-RLPO displays C–H stretching at 2951 cm−1 and 1448 cm−1, a C=O stretch at 1723 cm−1, and C–O–C vibrations at 1237 and 1143 cm−1.

a Chemical structures of the APIs and polymers, together with b FTIR spectra
In the FTIR spectra of the coaxial fiber mats (Fig. 6b), formulation F1 shows E-RLPO bands at 2951 cm−1 (C–H stretching), 1728 cm−1 (C=O stretching), and 1148 cm−1 (C–O–C vibrations). Bands from PVP can be seen between 3400 and 2950 cm−1 (O–H and C-H stretches), at 1652 cm−1 from C=O vibrations, and at 1289 cm−1 from C–N stretching. Similarly, for formulation F2, peaks from E-RLPO can be seen at 2951 cm−1, 1726 cm−1, and at 1150 cm−1, and vibrations from PVP are visible at 3400 to 2984 cm−1, 2950 cm−1, 1651 cm−1, and 1289 cm−1. In comparison to the spectra of pure LD and CD, in the fibers many of the drugs’ characteristic peaks have shifted positions or merged with the peaks from the carrier polymers, indicating that the APIs were successfully loaded in the fiber mat with significant intermolecular interactions between the drug and the polymer.
Drug Loading and Encapsulation Efficiency
The drug loading and encapsulation efficiency were determined by HPLC, and the results are listed in Table 2. The loadings are in most cases very close to 100% of the theoretical amount. While the calculated loadings come out at a little over 100% in places, the error of the measurements is such that these values are essentially identical to the theoretical content. The deviations seem can be ascribed to inhomogeneities in the density of the fiber mat. The exception is for CD in the F2 formulation where the encapsulation efficiency (EE) is ca. 80%. This decrease in CD content could be a result of degradation during either the electrospinning or HPLC sample preparation processes. The observation of close to 100% EE in most cases supports the DSC data in indicating that there is very little solvent present in the fibers. Given the loadings calculated in Table 2, to deliver a therapeutic dose of 100 mg LD/25 mg CD, around 500 mg of the F1 fibers would be required. This is an eminently feasible size for a dosage form.
Drug Release Studies
In the British Pharmacopoeia (BP), it is recommended to perform dissolution testing of the LD/CD combination SR tablet in 0.1 M HCl [43]. This protocol was followed here. The SR tablet contains LD/CD at 200/50 mg. This is relatively high, and thus the high drug-loaded formulation F1 seemed more appropriate for study here. Further, the TEM images of F2 did not confirm the formation of core/shell fibers (Fig. 3). The F1 fibers were manually loaded into a hard gelatin capsule (size 0) for release studies, since the intention is to deliver the formulation orally. The in vitro drug release results are given in Fig. 7a.

a In vitro dissolution profiles showing release of LD and CD from Sinemet CR® 200/50 mg tablets and F1 at pH 1. Data are reported as mean ± S.D. (n = 3). b A representative HPLC chromatogram, taken after 24 h of immersion of F1 at pH 1
In Fig. 7a, the release of both drugs from the Sinemet CR tablet show similar trends, with ca. 50% of the loaded drug released in the first hour, and the remaining 50% in the second hour. To allow a direct comparison with the SR tablet, drug release from the formulations was explored in HCl. This will allow the drug molecules to be released and dissolve into the acidic medium of the stomach, avoiding degradation. Since the fibers are expected to have mucoadhesive properties, they could be developed into gastro-retentive systems to this end. The in vitro drug release data are presented in Fig. 7a. The release of both drugs was similar, showing a multiphasic release profile. In the first 2 h, over 25% of the loaded drug was rapidly released, followed by another 25% release over the next 6 h. Half the loading of each drug is present with hydrophilic PVP in the shell layer, and it is expected that it is this half of the dose which was freed in the first stages of release. A similar rapid release pattern from PVP has been reported from coaxial fibers made of a PVP shell and ethyl cellulose core [29].
Drug release from the E-RLPO core can be seen to take place over 8 to 24 h. After 24 h, 84 ± 1% of the LD and 79 ± 3% of the CD loading has been freed into solution. There is no evidence for degradation products in the HPLC data (Fig. 7b), and thus the residual LD and CD must be retained inside the fibers even after 24 h. As described earlier, E-RLPO is a water-insoluble polymer with low permeability, but it undergoes pH-independent swelling allowing it to slowly release a drug cargo [41]. The slow release of LD and CD observed from E-RLPO arises due to diffusion of the loaded drug through and out of the polymer matrix [44]. This extended release from the core layer is a comparable to results reported by Pornsopone et al. who showed E-RLPO fibers loaded with 10% w/w indomethacin to reach only 18% cumulative release after 24 h [35]. Overall, it is clear that the F1 formulation provides the desired release profile. The results here are similar to others reported for coaxial fibers, for instance those of Yu et al. where core/shell systems were prepared with the non-steroidal anti-inflammatory agent ketoprofen, PVP as the shell polymer and either zein [32] or ethyl cellulose [29] as the core matrix. Other authors have employed a cellulose acetate shell and a core containing sodium hyaluronate and naproxen-loaded liposomes to achieve a biphasic release profile [45]. In this work however, we make an advance on these previous studies by incorporating multiple active ingredients into each compartment of the fiber architecture.
Conclusions
This study aimed to employ coaxial electrospinning to fabricate a core/shell fiber fixed dose formulation. Two polymers, PVP and E-RLPO, were used to build the core/shell fibers. In each layer, two model drugs (LD and CD) were loaded, resulting in an FDC which could be used for the treatment of PD. We were able to develop fibers with theoretical drug loadings of LD/CD at 19.0/4.8% (F1) and 8.9/2.2% w/w (F2). SEM images showed the production of cylindrical fibers with smooth surfaces. TEM images confirmed the formation of coaxial structure in the F1 fibers, but not for F2. Physical form characterisation studies with XRD and DSC revealed that both formulations comprised amorphous solid dispersions. Drug-polymer interactions between both drugs and the polymers were confirmed with FTIR spectroscopy. Encapsulation efficiencies were close to 100% in most cases. An in vitro dissolution study revealed the ability of the core/shell fibers to provide extended release profiles, providing an immediate burst release (giving a loading dose) followed by extended release for longer than was observed with commercial controlled release tablets. The F1 fibers should thus be able to help maintain a constant LD plasma concentration, reducing the dosing frequency and undesirable side effects from overuse of the conventional treatments in PD.
References
Grosset D, Taurah L, Burn DJ, MacMahon D, Forbes A, Turner K, et al. A multicentre longitudinal observational study of changes in self reported health status in people with Parkinson’s disease left untreated at diagnosis. J Neurol Neurosurg Psychiatry2007, 78, 465.
Leopold NA, Polansky M, Hurka MR. Drug adherence in Parkinson’s disease. Mov Disord2004, 19, 513–7.
Kulkarni AS, Balkrishnan R, Anderson RT, Edin HM, Kirsch J, Stacy MA. Medication adherence and associated outcomes in medicare health maintenance organization-enrolled older adults with Parkinson’s disease. Mov Disord2008, 23, 359–65.
Fernandez HH. Updates in the medical management of Parkinson disease. Cleve Clin J Med2012, 79, 28–35.
Ngwuluka N, Pillay V, Du Toit LC, Ndesendo V, Choonara Y, Modi G et al. Levodopa delivery systems: Advancements in delivery of the gold standard. Expert Opin Drug Deliv2010, 7, 203–24.
https://www.nice.org.uk/guidance/ng71/chapter/recommendations.
Contin M, Riva R, Martinelli P, Procaccianti G, Cortelli P, Avoni P, et al. Response to a standard oral levodopa test in Parkinsonian patients with and without motor fluctuations. Clin Neuropharmacol1990, 13, 1–28.
Seeberger LC, Hauser RA. Optimizing bioavailability in the treatment of Parkinson’s disease. Neuropharmacology2007, 53, 791–800.
Grosset D, Antonini A, Canesi M, Pezzoli G, Lees A, Shaw K, et al. Adherence to anti-Parkinson medication in a multicenter European study. Mov Disord2009, 24, 826–32.
Contin M, Martinelli P. Pharmacokinetics of levodopa. J Neurol2010, 257, S253–61.
Hsu A, Yao HM, Gupta S, Modi NB. Comparison of the pharmacokinetics of an oral extended-release capsule formulation of carbidopa-levodopa (Ipx066) with immediate-release carbidopa-levodopa (Sinemet((R))), sustained-release carbidopa-levodopa (Sinemet((R)) CR), and carbidopa-levodopa-entacapone (Stalevo((R))). J Clin Pharmacol2015, 55, 995–1003.
Stocchi F. The hypothesis of the genesis of motor complications and continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Parkinsonism Relat Disord2009, 15 Suppl 1, S9–S15.
Williams GR, Raimi-Abraham BT, Luo CJ. Nanofibres in drug delivery. London: UCL Press; 2018.
Yu DG, Li JJ, Williams GR, Zhao M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J Control Release2018, 292, 91–110.
Yu D-G, Wang M, Li X, Liu X, Zhu L-M. Annie Bligh SW. Multifluid electrospinning for the generation of complex nanostructures. Wiley Interdiscip Rev Nanomed Nanobiotechnol2019. https://doi.org/10.1002/wnan.1592.
Illangakoon UE, Gill H, Shearman GC, Parhizkar M, Mahalingam S, Chatterton NP et al. Fast dissolving paracetamol/caffeine nanofibers prepared by electrospinning. Int J Pharm2014, 477, 369–79.
Tort S, Yıldız A, Tuğcu-Demiröz F, Akca G, Kuzukıran Ö, Acartürk F. Development and characterization of rapid dissolving ornidazole loaded PVP electrospun fibers. Pharm Dev Tech2019, 24, 864–73.
Giram PS, Shitole A, Nande SS, Sharma N, Garnaik B. Fast dissolving moxifloxacin hydrochloride antibiotic drug from electrospun Eudragit l-100 nonwoven nanofibrous mats. Mater Sci Eng C2018, 92, 526–39.
Kajdič S, Vrečer F, Kocbek P. Preparation of poloxamer-based nanofibers for enhanced dissolution of carvedilol. Eur J Pharm Sci2018, 117, 331–40.
Feng K, Li C, Wei Y-S, Zong M-H, Wu H, Han S-Y. Development of a polysaccharide based multi-unit nanofiber mat for colon-targeted sustained release of salmon calcitonin. J Colloid Interface Sci2019, 552, 186–95.
Dias AM, da Silva FG, Monteiro APdF, Pinzón-García AD, Sinisterra RD, Cortés ME. Polycaprolactone nanofibers loaded oxytetracycline hydrochloride and zinc oxide for treatment of periodontal disease. Mater Sci Eng C2019, 103, 109798.
Angkawinitwong U, Awwad S, Khaw PT, Brocchini S, Williams GR. Electrospun formulations of bevacizumab for sustained release in the eye. Acta Biomater2017, 64, 126–36.
Jin M, Yu DG, Geraldes CF, Williams GR, Bligh SW. Theranostic fibers for simultaneous imaging and drug delivery. Mol Pharm2016, 13, 2457–65.
Shen X, Yu D, Zhu L, Branford-White C, White K, Chatterton NP. Electrospun diclofenac sodium loaded Eudragit(r) l 100-55 nanofibers for colon-targeted drug delivery. Int J Pharm2011, 408, 200–7.
Bukhary H, Williams GR, Orlu M. Electrospun fixed dose formulations of amlodipine besylate and valsartan. Int J Pharm2018, 549, 446–55.
Li H, Zhu J, Chen S, Jia L, Ma Y. Fabrication of aqueous-based dual drug loaded silk fibroin electrospun nanofibers embedded with curcumin-loaded RSF nanospheres for drugs controlled release. RSC Adv2017, 7, 56550–8.
Zhang Y, Huang Z-M, Xu X, Lim CT, Ramakrishna S. Preparation of core–shell structured PCL-r-gelatin bi-component nanofibers by coaxial electrospinning. Chem Mater2004, 16, 3406–9.
Yu DG, Wang X, Li XY, Chian W, Li Y, Liao YZ. Electrospun biphasic drug release polyvinylpyrrolidone/ethyl cellulose core/sheath nanofibers. Acta Biomater2013, 9, 5665–72.
Wang C, Yan K-W, Lin Y-D, Hsieh PCH. Biodegradable core/shell fibers by coaxial electrospinning: Processing, fiber characterization, and its application in sustained drug release. Macromolecules2010, 43, 6389–97.
Chou SF, Carson D, Woodrow KA. Current strategies for sustaining drug release from electrospun nanofibers. J Control Release2015, 220, 584–91.
Jiang YN, Mo HY, Yu DG. Electrospun drug-loaded core-sheath PVP/zein nanofibers for biphasic drug release. Int J Pharm2012, 438, 232–9.
Commission BP. British pharmacopoei. Carbidopa assay standard. London: Stationery Office; 2013.
Pornsopone V, Supaphol P, Rangkupan R, Tantayanon S. Electrospinning of methacrylate-based copolymers: Effects of solution concentration and applied electrical potential on morphological appearance of as-spun fibers. Polym Eng Sci2005, 45, 1073–80
Pornsopone V, Supaphol P, Rangkupan R, Tantayanon S. Electrospun methacrylate-based copolymer/indomethacin fibers and their release characteristics of indomethacin. J Polym Res2007, 14, 53–9
Illangakoon UE, Yu DG, Ahmad BS, Chatterton NP, Williams GR. 5-fluorouracil loaded Eudragit fibers prepared by electrospinning. Int J Pharm2015, 495, 895–902.
Yang Y-Y, Zhang M, Wang K, Yu D-G. pH-sensitive polymer nanocoating on hydrophilic composites fabricated using modified coaxial electrospraying. Mater Lett2018, 227, 93–6.
O’Neil MJ, Smith A, Heckelman PE, Budavari S. The Merck index-an encyclopedia of chemicals, dugs, and biologicals. Whitehouse Station: Merck and Inc. Co; 2001. p. 4342.
http://polymerdatabase.com/polymers/polyvinylpyrrolidone.html.
Dave VS, Fahmy RM, Bensley D, Hoag SW. Eudragit((R)) RSPO/RLPO as rate-controlling matrix-formers via roller compaction: Influence of formulation and process variables on functional attributes of granules and tablets. Drug Dev Ind Pharm2012, 38, 1240–53.
Parikh T, Singh Gupta SS, Meena A, Serajuddin ATM. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion: iii—Polymethacrylates and polymethacrylic acid based polymers. J Excip Food Chem2014, 5, 56–64.
Commission BP. British pharmacopoeia. Co-careldopa tablets, Appendix XII B. Annex: Recommendations on dissolution testing. London, England: Stationary Office; 2016.
Yadav SK, Mishra S, Mishra B. Eudragit-based nanosuspension of poorly water-soluble drug: Formulation and in vitro-in vivo evaluation. AAPS PharmSciTech2012, 13, 1031–44.
Li Z, Kang H, Che N, Liu Z, Li P, Li W et al. Controlled release of liposome-encapsulated naproxen from core-sheath electrospun nanofibers. Carbohydrate Polymers2014, 111, 18–24.
Acknowledgements
The authors thank the Umm AlQura University (Kingdom of Saudi Arabia) for provision of a scholarship to HB. We also gratefully acknowledge Dr Andrew Weston and Mrs Kate Keen for assistance with obtaining SEM and TEM images.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bukhary, H., Williams, G.R. & Orlu, M. Fabrication of Electrospun Levodopa-Carbidopa Fixed-Dose Combinations. Adv. Fiber Mater. 2, 194–203 (2020). https://doi.org/10.1007/s42765-020-00031-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42765-020-00031-1