
Abstract
Introduction
Time-to-event data from clinical trials are routinely extrapolated using parametric models to estimate the cost effectiveness of novel therapies, but how this approach performs in the presence of heterogeneous populations remains unknown.
Methods
We performed a simulation study of seven scenarios with varying exponential distributions modelling treatment and prognostic effects across subgroup and complement populations, with follow-up typical of clinical trials used to appraise the cost effectiveness of therapies by agencies such as the UK National Institute for Health and Care Excellence (NICE). We compared established and emerging methods of estimating population life-years (LYs) using parametric models. We also proved analytically that an exponential model fitted to censored heterogeneous survival times sampled from two distinct exponential distributions will produce a biased estimate of the hazard rate and LYs.
Results
LYs are underestimated by the methods in the presence of heterogeneity, resulting in either under- or overestimation of the incremental benefit. In scenarios where the overestimation of benefit is likely, which is of interest to the healthcare provider, the method of taking the average LYs from all plausible models has the least bias. LY estimates from complete Kaplan–Meier curves have high variation, suggesting mature data may not be a reliable solution. We explore the effect of increasing trial sample size and accounting for detected treatment–subgroup interactions.
Conclusions
The bias associated with heterogeneous populations suggests that NICE may need to be more cautious when appraising therapies and to consider model averaging or the separate modelling of subgroups when heterogeneity is suspected or detected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Heterogeneity in time-to-event data may not be identified in current health technology appraisals and may result in biased estimates of treatment benefit that would affect prices and patient access to therapy. |
Heterogeneity should be considered. Methods such as averaging across plausible models, encouraging larger trial populations, and accounting for detectable heterogeneity may reduce the bias associated with heterogeneity compared with current methods used in health technology appraisals undertaken by the UK National Institute for Care and Excellence. |
1 Introduction
Health technology assessment (HTA) agencies, such as the National Institute for Health and Care Excellence (NICE) in England and Wales, assess the clinical and cost effectiveness of health technologies based on the appraisal of supporting clinical evidence, usually from at least one clinical trial, which is then incorporated alongside a series of assumptions into an economic model.
Clinical trials often demonstrate heterogeneity in treatment efficacy among patients, with some patients receiving less or even no clinical benefit [1, 2]. This heterogeneity may increase when converting clinical benefits into quality-adjusted life-years (QALYs), which are used in an attempt to present a level playing field on which the effectiveness of all treatments for all diseases can be judged. QALYs are usually obtained by estimating the expected number of life-years (LYs) and multiplying by a health utility value that captures the expected quality of health a patient is expected to experience whilst they remain alive, which may vary as patients pass through different stages of disease, though other methods are possible.
For severe, terminal diseases such as advanced cancers, the goals of treatments are to delay disease progression and/or extend survival as the prospect of being cured is unlikely. Treatments for such diseases usually report clinical outcomes based on their relative efficacy using a hazard ratio, whereas the relative benefit will be measured using the gain in QALYs for cost-effectiveness assessments. A hazard ratio uses only observed data, whereas LYs often involve extrapolations.
This use of differing scales between clinical and cost effectiveness assessments means that heterogeneous treatment effects are even harder to identify. A treatment could appear more clinically effective for a subgroup of patients compared with the complement in terms of a hazard ratio yet offer less benefit in the subgroup when examining the LY/QALY benefit because of the influence of prognostic factors. For example, a subgroup and complement may have hazard of 0.5 and 0.25, with average LYs of 2 and 4, respectively. A treatment with a hazard ratio of 0.7 in the subgroup and 0.8 in the complement might suggest the treatment has a stronger effect in the subgroup; however, the LYs are 2.86 and 5 when the subgroup and complement are treated, meaning the complement population gains 1 LY and the subgroup gains 0.86. However, the reverse could also be true, with different clinical responses in the subgroup and its complement resulting in equivalent LY benefits. Factors that may clinically be prognostic, such as age, could become treatment-effect modifiers when appraising a therapy from a health economic perspective.
Given the increasing pressure on healthcare budgets, it is vital that the implications of current methods are fully understood to assist decision makers and ensure fair access to health technologies.
Our aims are to demonstrate the relationship between hazard ratios and LY efficacy estimates and to explore the ability of current methodology to accurately estimate LYs when the population includes a subgroup with heterogeneity in overall survival and treatment effect compared with its complement.
2 Method Overview
We undertook a series of simulations capturing seven distinct scenarios, each replicating follow-up for a time-to-event outcome from a phase III clinical trial at the point of appraisal by an HTA agency. Each scenario contained a different combination of prognosis and treatment effect for a subgroup and complement population, with half the trial population featuring in the subgroup. Five methods of estimating LYs were implemented, all based on a set of candidate parametric models.
2.1 Simulation Method
An overview of the simulation is provided in Table 1. The survival times for each subgroup/complement and treatment/control group were sampled from different exponential distributions reflecting plausible hazard ratios of treatment and prognostic effect.
The seven scenarios considered (Table 2) were as follows:
-
Scenario 0 serves as a reference point and features no difference in prognosis or treatment effect between the subgroup and complement.
-
Scenario 1 models no difference in prognosis between the subgroup and complement, with a treatment effect only in the subgroup.
-
Scenario 2 features a treatment effect only in the subgroup, but the subgroup has a worse prognosis than the complement.
-
Scenario 3 models a subgroup with a worse prognosis, but the treatment has an equal hazard ratio of effect across the subgroup and complement.
-
Scenario 4 features a subgroup with a worse prognosis, but the treatment only has an effect in the complement.
-
Scenario 5 models a subgroup with a worse prognosis, and the hazard ratio of treatment effect is slightly stronger in the subgroup than in the complement.
-
Scenario 6 features a subgroup with a worse prognosis, whereas the treatment has a positive effect in the subgroup and a slight negative effect in the complement.
Our sample size for each scenario was based on assumptions of an overall hazard ratio of 0.75, 90% power, and a 5% alpha and did not consider treatment effect interactions. The probability of an event in the follow-up period was 0.60, and probability for withdrawing was 0.05, giving a sample size of 896 rounded up to the nearest multiple of 8 to allow for consistently sized subgroups in every simulation for each scenario, using Stata’s ‘power cox’ command.
We replicated trial follow-up by generating censoring times using a Gompertz distribution (shape = 3.5, rate = 0.00005). This gave an average censoring time of 3 years, with very few patients censored before 2 years or beyond 4 years of follow-up (see Fig. A1 in the electronic supplementary material [ESM] for an example). Our scenarios had varying power, with the hazard rates used suggesting mortality rates of 41–78% at 3 years. All survival data were generated and survival models fitted using the ‘flexsurv’ package in R [3], with post-simulation analysis conducted in Stata 16.
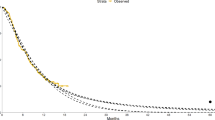
We fixed the proportion of the subgroup at 0.5 of the whole population but anticipated that our results would generalise to subgroups of all proportions. Figure 1 demonstrates the pooling of subgroup and complement survival curves, whilst Fig. 2 shows parametric curves fitted to a heterogeneous population. The true expected LY for each heterogenous population was calculated using the LY of the respective component population restricted to the first 30 years, weighted by their prevalence. For example, LYs for one arm =
where \({\lambda }_{1}\) and \({\lambda }_{2}\) are the hazard rates in the subgroup and complement, respectively, and \(p\) and \((1-p)\) are the respective prevalences.

Example Kaplan–Meier plot of subgroup, complement and combined data for where the treatment is only effective in the subgroup and subgroup has a worse prognosis (scenario 2)

Example of parametric curves failing to predict true survival for a heterogeneous population. Here λ1 = 0.5 and λ2 = 0.25
2.2 Analytical Method
We fitted eight parametric models (exponential, Weibull, log-normal, log-logistic, gamma, generalised gamma, Gompertz, and generalised F) independently to each arm for each set of simulated trial follow-up data, ignoring subgroup effects, and estimated LYs from the extrapolation of these models. We removed implausible models by assessing each model’s prediction of survival at 5 and 10 years. Estimates were considered implausible if they fell outside a ± 7.5 percentage unit window around the true 5-year survival percentage or a ± 5% window around the 10-year value when survival rates are expected to be much lower. These windows were consistent with the variation in predictions made by clinical experts in NICE technology appraisals from the authors’ experiencev.
We considered three distinct approaches to obtaining a LY estimate. First, we chose the single best-fitting models for each arm independently according to Akaike information criterion (AIC) and Bayesian information criterion (BIC), despite there being limitations with this approach [4]. This means different parametric models could be chosen for each arm, which is only encouraged by the NICE technical support document (TSD)-14 when justified by “clinical expert judgement, biological plausibility, and robust statistical analysis” [5]. Second, in keeping with NICE TSD 14, we selected the model with the combined lowest AIC/BIC for both arms by adding AIC/BIC across both arms, the approach we believe to be most consistent with current practice [6, 7]. Finally, we calculated the mean average of the LY estimates from all plausible models, as presented by Gallacher et al. [8], which generally outperformed information criteria-based weights. For reference, we measured the area under the Kaplan–Meier curve for each simulation, estimating the LYs as they would have been had follow-up been complete without any censoring.
We fitted two Cox models in each simulation [9], the first only estimating a treatment effect, and the second estimating treatment and subgroup effects and a treatment-by-subgroup interaction effect.
Finally, we explored the impact of doubling the trial sample size and of fitting separate parametric models to the subgroup and complement populations of the treatment and control arms whenever a significant interaction term was detected by a Cox model at the 0.05 significance level threshold.
The code for this paper can be accessed online (https://github.com/daniel-g-92/heterogeneity).
3 Results
3.1 Main Scenarios
Scenario 0 served as a reference point, demonstrating the performance of the different approaches when there is no heterogeneity within either arm. There is little to distinguish between the methods of single model selection, with each showing almost no bias (Table 3). Even in the absence of heterogeneity, few estimates of LYs from the fitted models were within 10% of the true LYs, with the highest being 27%. LY estimates from complete Kaplan–Meier follow-up were within this range for 39% of simulations.
Scenario 1 applied the hazard ratio only in the subgroup, with no prognostic differences. The methods tended to underestimate incremental LYs (Table 3) because of the benefit of the intervention was underestimated (Fig. 3). In just over one-half of simulations, neither a significant treatment effect nor a significant treatment subgroup interaction were detected (46 and 43%, respectively; Table 3).

Violin plot of difference in life-year estimation for each method, by arm where estimates for the intervention are on the left of each violin, and controls are on the right. Dashed line indicates the mean, occasionally distinguishable from the solid median line. AIC Akaike information criterion, BIC Bayesian information criterion, LY life-year, sig. significant
Scenario 2 applied the hazard ratio only to the subgroup, which also had a worse prognosis than the complement. The methods underestimated LYs in both arms but overestimated incremental LYs. BIC-based selection was associated with the highest bias. Significant hazard ratios and interaction terms were detected in just over one-half of simulations (56.5 and 54.7%).
Scenario 3 applied the hazard ratio to the whole population, but the subgroup had a worse prognosis. The LYs for the intervention were generally underestimated, leading to underestimation of the incremental benefit. A significant treatment effect was detected in almost all simulations (97.8%), but a significant interaction term was rare (5.2%).
Scenario 4 featured a hazard ratio in the complement, whereas the subgroup population had a worse prognosis. This scenario was analogous to scenario 2, and the results were consistent with the switch in treatment efficacy. Incremental efficacy was underestimated, with the BIC-based methods being the most severe. A significant treatment effect was not detected in the majority of simulations (43.7%), but a significant interaction was (52.9%).
Scenario 5 applied a hazard ratio to both the subgroup and the complement, but this was stronger in the subgroup, which had a worse prognosis. LYs were underestimated for both arms by all methods, but these largely cancelled out to provide unbiased estimates of incremental benefit. A significant treatment effect was detected in most simulations (91.4%), but a significant interaction effect was not (11.8%).
Scenario 6 applied a hazard ratio of positive treatment effect in the subgroup, which also had a worse prognosis, along with a negative treatment effect in the complement. Methods tended to underestimate LYs for both arms, but this was more considerable in the control arm, leading to overestimation of the incremental benefit. The majority of simulations for this scenario did not detect a significant treatment effect in the whole population (35.6%) but did detect a significant treatment subgroup interaction (75.8%).
The optimal method varied by scenario, and there was little to distinguish between model averaging and the AIC-based methods in terms of bias and accuracy. Examination of the distributions of the results (Fig. 3) suggested that estimates coming from model averaging were less skewed and so may be more reliable.
LY estimates for all methods were most accurate when there was little or no heterogeneity within either arm (scenarios 0, 3, and 5), with a noticeably higher percentage of estimates falling within 10% of the true incremental LYs; however, they were all outperformed by complete Kaplan–Meier follow-up. The LY estimates from complete follow-up still had high variability, with the percentage of LY estimates that fell within ± 10% of the true LYs varying across scenarios from 9 to 38%.
Estimates of all approaches were most accurate (least biased and highest percentage within 10% of true value) in scenarios when little or no heterogeneity was present (scenarios 0, 3, and 5).
The ESM contains results of LY estimates from each of the parametric models (Tables A2–A3, Fig. A3). Fitting to the censored follow-up of combined populations of two heterogeneous exponential groups, the exponential, Weibull, and gamma models on average underestimated survival, whereas the generalised F, log-normal, and log-logistic overestimated, though scenario 0 suggests this may be due to poor fit rather than heterogeneity. The generalised gamma and Gompertz had a lot of variation but were generally unbiased.
3.2 Additional Analyses
Table 4 and Fig. 4 contain the results of exploratory analyses examining (1) the effects of increasing the sample size to 896 per arm, (2) fitting separate parametric models to the subgroup and complement population when a significant treatment subgroup interaction was detected, and (3) both (1) and (2) simultaneously. Scenario 2 was chosen as it modelled a simple interaction that was already often detected in the original scenario. However, the results will generalise to all scenarios of heterogeneous effects.

Violin plot of difference in life-year estimation for each method for variations of scenario 2. Estimates for the intervention are on the left of each violin, and controls are on the right. Dashed line indicates the mean, occasionally distinguishable from the solid median line. AIC Akaike information criterion, BIC Bayesian information criterion, LY life-year, sig. significant
Increasing the sample size increased the detection of both significant treatment effects and treatment subgroup interactions. It also slightly reduced the bias for the methods of model selection.
Fitting separate models when significant interactions were detected reduced the bias from all methods. When combined with the larger sample size, all methods produced unbiased LY estimates. This approach relies on correct identification of subgroup interactions, may increase variance where interactions are falsely identified, and cannot be applied when subgroups are not identified.
4 Discussion
Through simulation, we demonstrated the performance of current methodology used in HTA in estimating treatment benefits. We assessed the bias of this methodology when heterogeneity was present in censored follow-up. Across every scenario, we showed that the methods had problems accurately predicting LYs, underestimating where heterogeneity was present. When estimates of LYs in two treatment groups are used to estimate incremental LYs, this can result in either under- or overestimation of the true benefit, varying by scenario. This issue of biased estimation is therefore a concern to both healthcare providers/decision makers and pharmaceutical manufacturers.
These simulations are supported by an analytical result that when fitting a single exponential model to immature follow-up of a heterogenous population made up of two components, the survival times of which come from two distinct exponential distributions, the fitted model will always overestimate the true hazard rate, thus underestimating the mean survival time. Defining the true average hazard rate as
where \(y\) is the hazard rate in the subgroup, of size \(np\), and \(z\) is the hazard rate in the complement, of size \((1-p)n\). Assuming all patients begin follow-up at the same time, and so those that remain event free are all censored at the same point, we denote the estimated hazard rate at time \(t\) as
It can be shown that \(\widehat{\lambda }\left(t\right)>\lambda\) for all \(t\), so that an estimate of the LY based on this will be an underestimate. This result can be generalised to show that the hazard is overestimated for any distribution of censoring times, relaxing the assumption on recruitment and censoring times. A detailed proof is presented in the ESM.
It is common when almost all patients have died to estimate LYs from the Kaplan–Meier curves instead of parametric models [6, 7]. This approach avoids debate on the choice of preferred extrapolation. We showed that, across all six scenarios, complete follow-up without any censoring yielded an estimate of mean survival that deviated at least 10% from the true value in the majority of simulations (up to 91%). This raises the question of whether mature follow-up from clinical trials is sufficiently reliable for decision makers, especially when sample sizes are small. We recommend that the uncertainty in the Kaplan–Meier estimates is considered, perhaps through the 95% confidence interval curves.
As access to therapies is ascertained not just on clinical efficacy but also on cost effectiveness, greater consideration of the economic assessment should be accounted for in the trial design and data collection. Cost-effectiveness analysis protocols should be established during the trial development stage to promote transparency. This may be a challenge to pharmaceutical manufacturers, as methods of assessing cost effectiveness vary by country. Consideration should be given not only to powering trials to detect clinically meaningful differences at key follow-up milestones but also to accurately capturing patient survival for a single arm [10], leading to increased confidence in the output from extended follow-up. Our simulations demonstrated the challenge of estimating cost effectiveness from a study powered for a clinical outcome. In cases where it may be appropriate to make treatment available for only a subgroup of patients, it is critical that the correct group are identified. When such discrimination is not appropriate, it remains imperative that these groups are identified to accurately estimate the treatment benefit in a heterogeneous population.
Heterogeneity could also be more prevalent in routine care than in clinical trials, for example where populations tend to be underrepresented in research [11], leading to differences between actual and predicted benefits.
Scenarios 3 and 5 featured varying treatment benefits on the LY scale between the subgroup and complement that was not reflected on the hazard ratio scale. Scenario 5 was more complex in that the hazard ratio suggested a stronger benefit in the subgroup, but the better prognosis of the complement meant that the complement gained more LYs. This scenario could cause confusion if attempting to prioritise patient access.
The often worse performance of the BIC-based methods was perhaps due to their preference for models with the fewest parameters, which may have been the worst at capturing the heterogeneity.
In four scenarios, the analytical methods underestimated the incremental LYs. This means the healthcare provider obtains better value for money than was anticipated at the point of appraisal and that the pharmaceutical manufacturer does not maximise their potential reimbursement. It is likely that pharmaceutical manufacturers already watch for these potential conditions and take steps to minimise their occurrence. It is not necessarily the priority of the healthcare provider to reduce the bias in these scenarios. However, in scenarios 2 and 6, the incremental LY was overestimated, quite considerably by some methods, which should be of concern to the healthcare provider. Consequently, the avoidance of these scenarios is less of a priority to the pharmaceutical manufacturer and so are potentially more likely to occur. These scenarios both featured a treatment effect only in a prognostically worse subgroup. In both, the bias was reduced when LY estimates were taken by either using arm-independent AIC selection or taking the average of all plausible models, compared with obtaining LYs through one of the other methods. Given the skewed nature of the independent AIC selection in these scenarios, taking the average of all plausible models appears to be more reliable, also featuring a higher percentage of LY estimates within 10% of the true range. Hence, we recommend that decision makers such as NICE encourage the presentation of analyses using the average of all plausible models where the treatment effect may interact with a prognostic factor, or model subgroup populations separately if a significant interaction is detected.
Such an approach is not without risk since phase III trials are not usually designed with the power to detect efficacy among known prognostic or potential treatment-modifying subgroups. Any observed difference in treatment effects could occur by chance and may lead to unnecessary restrictions being applied, resulting in unfair pricing and unfair access to interventions.
To make strides towards personalised medicine, NICE could consider offering greater incentivisation for treatments where the developer has identified novel patient subgroups, which will likely incur additional costs compared with developing a non-stratified therapy. This would ensure patients receive the best therapy for them and avoid treatment prices being based on potentially biased estimates [12].
If heterogeneity is suspected, but not detected or attributable to any known covariate, fitting separate models for different subgroups is not an option. It is possible that flexible parametric approaches [13] or mixture cure models [14] might better capture the heterogeneity than would traditional parametric approaches; however, these were beyond the scope of this study, and further investigation is needed. Data appearing to follow a complex hazard rate may be a consequence of a heterogeneous population containing subgroups that each have a much simpler underlying hazard rate.
A major strength of our study was that it captured a range of interesting scenarios of varying subgroup and complement treatment efficacies representative of clinical trial follow-up used for appraising the cost effectiveness of therapies by agencies such as NICE. These scenarios could potentially feature in any and every technology appraisal. However, our study did have limitations. It assumed that the clinical predictions of efficacy were unbiased, whereas this may not be the case in practice. The size of the bias is certainly affected by sample size, subgroup prevalence, and length of follow-up, which were not explored in detail in this study.
Our source distributions were all exponential, which often led to the exclusion of the log-normal and log-logistic curves when plausibility was assessed. We anticipate our results to be generalisable to scenarios beyond those based on the exponential distribution, wherever heterogeneous populations exist, regardless of the underlying distribution. Our results are relevant to not only technology appraisals but also published cost-effectiveness studies, where methods of extrapolating survival are similar [15, 16]. Spline models were not included in this simulation because of their additional manual specification when selecting knot frequency and location but have been shown to fit well to trial and registry data where sample sizes are larger than in typical clinical trials [17, 18]. Similarly, cure models incorporating external data have been shown to perform well but could not be applied in this simulation [19].
Our study demonstrated that the relationship between clinical benefit and LY benefit is not always linear, which can raise challenges when valuing treatments. Our results were consistent with and may explain the observations of Ouwens et al. [20], who reported that parametric models fitted to trial follow-up underestimated mean survival compared with more mature follow-up. More work exploring further ways of discovering and accounting for this heterogeneity is necessary to increase the likelihood of conducting a balanced assessment of cost effectiveness.
5 Conclusion
Our study presented simulated trial follow-up for seven scenarios of varying combinations of treatment effects and prognoses where these differ in different parts of the population, where the information is typical of that used to assess the cost effectiveness of therapies.
We demonstrated how existing methods cope poorly with censored data containing heterogeneous treatment effects, which can either under- or overestimate the incremental LYs. Taking the average LY estimate from all plausible models performs well in scenarios where the incremental LYs are likely to be overestimated, and we encourage decision makers to consider this approach in future appraisals.
The high variability of estimates present in observed follow-up suggests that mature follow-up may not be reliable for estimating mean survival, particularly when sample sizes are small. We demonstrated the improved LY estimation obtained by increasing the sample size and modelling subgroup data separately when significant interactions were detected.
References
Raman G, Balk EM, Lai L, Shi J, Chan J, Lutz JS, et al. Evaluation of person-level heterogeneity of treatment effects in published multiperson N-of-1 studies: systematic review and reanalysis. BMJ Open. 2018;8(5):e017641.
Starks MA, Sanders GD, Coeytaux RR, Riley IL, Jackson LR II, Brooks AM, et al. Assessing heterogeneity of treatment effect analyses in health-related cluster randomized trials: a systematic review. PLoS ONE. 2019;14(8):e0219894.
Jackson CH. flexsurv: a platform for parametric survival modeling in R. J Stat Softw. 2016;70:i08.
Gallacher D, Kimani P, Stallard N. Extrapolating parametric survival models in health technology assessment: a simulation study. Med Decis Mak. 2021;42(1):37–50.
Latimer N. NICE DSU technical support document 14: survival analysis for economic evaluations alongside clinical trials-extrapolation with patient-level data. Report by the Decision Support Unit. 2011.
Gallacher D, Auguste P, Connock M. How do pharmaceutical companies model survival of cancer patients? A review of NICE single technology appraisals in 2017. Int J Technol Assess Health Care. 2019;35(2):160–7.
Bell Gorrod H, Kearns B, Stevens J, Thokala P, Labeit A, Latimer N, et al. A review of survival analysis methods used in NICE technology appraisals of cancer treatments: consistency, limitations and areas for improvement. Med Decis Mak. 2019;39(8):899–909.
Gallacher D, Kimani P, Stallard N. Extrapolating parametric survival models in health technology assessment using model averaging: a simulation study. Med Decis Mak. 2021;41(4):476–84.
Cox DR. Regression models and life-tables. J Roy Stat Soc Ser B (Methodol). 1972;34(2):187–202.
Nagashima K, Noma H, Sato Y, Gosho M. Sample size calculations for single-arm survival studies using transformations of the Kaplan–Meier estimator. Pharm Stat. 2021;20:499–511.
Redwood S, Gill PS. Under-representation of minority ethnic groups in research—call for action. Br J Gen Pract. 2013;63(612):342–3.
Gallacher D, Stallard N, Kimani P, Gökalp E, Branke J. Development of a model to demonstrate the impact of NICE cost-effectiveness assessment on health utility for targeted medicines. Health Econ. (Under review).
Royston P, Parmar MK. Flexible parametric proportional-hazards and proportional-odds models for censored survival data, with application to prognostic modelling and estimation of treatment effects. Stat Med. 2002;21(15):2175–97.
Klijn SL, Fenwick E, Kroep S, Johannesen K, Malcolm B, Kurt M, et al. What did time tell us? A comparison and retrospective validation of different survival extrapolation methods for immuno-oncologic therapy in advanced or metastatic renal cell carcinoma. Pharmaco Economics. 2021;39:345–56.
Connock M, Armoiry X, Tsertsvadze A, Melendez-Torres GJ, Royle P, Andronis L, et al. Comparative survival benefit of currently licensed second or third line treatments for epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) negative advanced or metastatic non-small cell lung cancer: a systematic review and secondary analysis of trials. BMC Cancer. 2019;19(1):392.
Gallacher D, Auguste P, Royle P, Mistry H, Armoiry X. A systematic review of economic evaluations assessing the cost-effectiveness of licensed drugs used for previously treated epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) negative advanced/metastatic non-small cell lung cancer. Clin Drug Investig. 2019;39(12):1153–74.
Gray J, Sullivan T, Latimer NR, Salter A, Sorich MJ, Ward RL, et al. Extrapolation of survival curves using standard parametric models and flexible parametric spline models: comparisons in large registry cohorts with advanced cancer. Med Decis Mak. 2021;41(2):179–93.
Guyot P, Ades AE, Beasley M, Lueza B, Pignon J-P, Welton NJ. Extrapolation of survival curves from cancer trials using external information. Med Decis Mak. 2017;37(4):353–66.
Bullement A, Latimer NR, Bell GH. Survival extrapolation in cancer immunotherapy: a validation-based case study. Value Health. 2019;22(3):276–83.
Ouwens MJNM, Mukhopadhyay P, Zhang Y, Huang M, Latimer N, Briggs A. Estimating lifetime benefits associated with immuno-oncology therapies: challenges and approaches for overall survival extrapolations. Pharmacoeconomics. 2019;37(9):1129–38.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The open access fee for this article was paid by Warwick Evidence who are funded by NIHR award 14/25/05.
Conflict of interest
Daniel Gallacher, Peter Kimani, and Nigel Stallard have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
The code for this paper can be accessed online (https://github.com/daniel-g-92/heterogeneity).
Author contributions
DG generated the research idea, designed and conducted the simulation study, and drafted the manuscript. PK and NS helped develop the study design and contributed to the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gallacher, D., Kimani, P. & Stallard, N. Biased Survival Predictions When Appraising Health Technologies in Heterogeneous Populations. PharmacoEconomics 40, 109–120 (2022). https://doi.org/10.1007/s40273-021-01082-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-021-01082-x