
Abstract
Background and Objectives
Cefozopran is a parenteral cephalosporin with a broad spectrum of activity against Gram-positive and Gram-negative bacteria. The objective of this study was to evaluate the pharmacokinetics of cefozopran after single- and multiple-dose intravenous administration in healthy subjects, to provide clinical guidance in its application.
Methods
This was a single-center, open-label, randomized, two-phase study conducted in 12 subjects. In the single-dose phase, subjects were randomly assigned to receive single doses of 0.5, 1.0 and 2.0 g of injected cefozopran hydrochloride in a three-way crossover design with a 5-day washout period between administrations. In the multiple-dose phase, subjects received 2.0 g every 12 h for 4 days. Plasma and urine pharmacokinetic samples were assayed by a validated high-performance liquid chromatography–tandem mass spectrometry method. Pharmacokinetic parameters were calculated and analyzed statistically. Safety assessments were conducted throughout the study.
Results
Twelve healthy volunteers (six males and six females) were enrolled and completed the study. Following a single 1-h intravenous infusion of 0.5, 1.0 or 2.0 g cefozopran, maximum plasma concentration (C max) and area under the plasma concentration–time curve from time zero to the time of the last measurable concentration (AUClast) increased in a dose-proportional manner. The mean half-life in plasma (t ½) was in the range of 1.20–2.80 h. Cefozopran was mainly excreted in its unchanged form, with no tendency for accumulation, via the kidney, and varied from 65.99 to 73.33 %. No appreciable accumulation of either drug occurred with multiple intravenous doses of cefozopran, and pharmacokinetic parameters for cefozopran were similar on days 1 and 4. No serious adverse events were reported. Adverse events were generally mild.
Conclusion
Cefozopran was safe and well tolerated in the volunteers and displayed linear increases in the C max and AUClast values.
Similar content being viewed by others


Cefozopran was safe and well tolerated in healthy Chinese volunteers when administered as a single dose or as multiple doses. |
The dose-dependent increase in exposure between 0.5 and 2.0 g cefozopran can be considered dose proportional. |
Cefozopran exhibited no accumulation with repeated administration. |
1 Introduction
Infection remains an important cause of morbidity and mortality in patients with cancer and other underlying myelosuppressive diseases [1]. During the past decade, a number of cephalosporins with broad spectra of activity and resistance to hydrolysis by β-lactamases have been developed. However, the clinical usefulness of these antibiotics has been limited because of their rather weak activities. Cefozopran is a parenteral, fourth-generation cephalosporin that has broad spectrum activity against Gram-positive and Gram-negative organisms [2, 3]. Moreover, cefozopran has comparatively good activity against Enterococci and Pseudomonas aeruginosa, which are refractory to other cephalosporins [4]. Since the late 1990s, cefozopran has been clinically available in Japan for the treatment of various infections such as pneumonia, sepsis, urinary-tract infections, and intra-abdominal infections in adult patients [5, 6]. The reason why this drug exhibits strong anti-microbial activity is assumed to be that it is stable against β-lactamase and that its action of inhibiting cell wall peptidoglycan cross-bridge formation is strong because it has a powerful affinity for penicillin binding proteins 1 and 2 of Staphylococcus aureus as well as for penicillin binding protein 3 of Escherichia coli and P. aeruginosa [7, 8].
In addition to its clinically useful antibacterial activity, the pharmacokinetic characteristics of the drug have been studied in healthy Japanese volunteers [9, 10] and adult patients [11–13] previously. Its blood concentration half-life is observed to extend as renal function is deteriorated, and this tendency is especially obvious in patients who are of old age or who have renal dysfunction. Further, serious side effects such as shock, anaphylactoid symptoms and acute insufficiency have been reported [7]. Its development is now underway in China; however, data for the pharmacokinetics of cefozopran in Chinese are still limited. A previous study in healthy Chinese volunteers [14] showed dose-dependent increases in cefozopran plasma concentration following 0.5-h intravenous administration. However, the pharmacokinetics of cefozopran following 1-h infusion administration in healthy Chinese subjects was not assessed.
The present study was, therefore, conducted to evaluate the safety, tolerability and dose proportionality of the pharmacokinetics of cefozopran following 1-h infusion administration of single doses of injected cefozopran hydrochloride (0.5, 1.0 and 2.0 g) and multiple doses of 2.0 g every 12 h for 4 days in healthy Chinese subjects.
2 Methods
2.1 Study Population and Design
This study was a single-center, open-label, two-part (parts A and B) study in healthy adult subjects. Part A was a randomized three-way crossover, open-label, single-dose study in which 12 subjects received each of the following three treatments (1-h infusion of injected cefozopran hydrochloride) in a randomized sequence: 0.5–1.0–2.0 g, 1.0–2.0–0.5 g, and 2.0–0.5–1.0 g according to a computer-generated randomization schedule (SAS 9.1, SAS Institute Inc., Cary, NC, USA). Treatments were separated by 5-day washout periods. Following the single-dose phase, volunteers assigned to continue into the multiple-dose phase (part B), during which they were confined to the study center, received 1-h intravenous infusions of injected cefozopran hydrochloride at 2.0 g every 12 h (q12h) for 4 days. On day 4, subjects received only a single dose of their assigned treatment.
Healthy men and women, between 18 and 45 years of age, were eligible to participate in this study. No subjects with clinically significant abnormal findings on electrocardiogram (ECG), clinical laboratory evaluations, or physical examination were included in the study. The permitted body mass index (BMI) was 19–25 kg/m2 inclusive. Female subjects needed to have a negative urine pregnancy test and be using appropriate contraception. The subjects had not received any medications within 14 days prior to the current study and had not participated in any study within 3 months prior to the current study. Subjects who had a history of hypersensitivity, allergy, serious adverse drug reaction, especially to any β-lactam antibiotic, were excluded from the study. Each subject was informed about the nature of and risks associated with the study, and signed an informed consent form before participating.
The protocol and written informed consent in the study was approved by the Ethical Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (approval No.: 2010-EC-84). The study was conducted in accordance with the Guideline for Good Clinical Practice by the China Food and Drug Administration (CFDA) and conformed to the Declaration of Helsinki (World Medical Association 2008).
2.2 Study Drug
Jiangsu Zhengda Tianqing Pharmaceutical Co., Ltd. (Jiangsu, China) supplied 0.5-g injected cefozopran hydrochloride (lot 100701, expiration 2012-06-30). The required doses for each treatment were added to normal saline solution to obtain a final infusion volume of 100 mL.
2.3 Sample Collection
For the single-dose phase, blood samples (4 mL) were collected from an indwelling venous catheter into heparinized tubes immediately before (within 15 min of) the start of infusion, 30 min after the start and at completion of infusion, and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8 and 12 h after completion of infusion for each treatment. The blood samples were centrifuged at 3,000×g for 10 min at 4 °C, and the plasma samples were stored at −70 °C until analyzed. Urine samples were collected from 0 h (pre-dose) and from 0 to 2, 2 to 4, 4 to 7, 7 to 13, and 13 to 24 after the start of infusion for each treatment. At the end of the interval, the urine bottles were shaken and the overall urinary excretion was measured and noted with accuracy. Approximately 10 mL of urine from each volunteer was collected and stored at −70 °C until analysis.
For the multiple-dose phase, samples were collected on day 3 pre-dose, immediately before the start of infusion for each dose on that day. On day 4, blood samples were drawn before the start of infusion, 30 min after the start and at completion of infusion, and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8 and 12 h after the infusion. Other experimental conditions were the same as in the single-dose phase.
2.4 Safety Assessment
Tolerability was evaluated using subject interviews and adverse-event (AE) monitoring during the study period. Vital signs, physical examinations, clinical laboratory tests (e.g., routine hematology, blood biochemistry and urine analysis) and 12-lead ECG was performed for each subject at screening and at the completion of the study. AEs were evaluated at the time of each blood draw on the basis of direct observation, spontaneous reports, and nonspecific inquiry. The physicians were responsible for determining the clinical significance of the AEs. All information, including undesirable symptoms or medical conditions after dosing, were recorded on the case-report form by investigators regardless of the suspected relationship to the study drugs.
2.5 Bioanalytical Methods
Plasma and urine concentrations of cefozopran were quantified by a sensitive and specific liquid chromatography–tandem mass spectrometry (LC–MS/MS) method. The assays were validated according to US Food and Drug Administration guidelines on bioanalytical method validation [15]. Plasma samples (200 μL) were combined with 10 μL of 968.6 μg/mL niacin in methanol, which was used as the internal standard (IS). To this mixture, 600 μL methanol was added. This new mixture was vortexed for 30 s and then centrifuged at 10,500g for 5 min. The supernatant (2 μL) was injected into the LC–MS/MS system for analysis. Urine samples (500 μL) were combined with 10 μL of 11 mg/mL niacin in methanol. Then 10 μL of the mixture was transferred into 1.5 mL polypropylene tubes, to which 990 μL methanol was added, and vortexed for 30 s. This new mixture was centrifuged at 10,500g for 5 min. The supernatant (2 μL) was injected into the LC–MS/MS system for analysis.
Liquid chromatography was performed on a Shimadzu LC-20AD system equipped with an autosampler, a degasser, a thermostatted column, and a binary pump (Shimadzu, Tokyo, Japan). The system was coupled to the API 4000 via a TurboIonspray electrospray ionization (ESI) interface for mass analysis and detection. Data acquisition was performed with Analyst version 1.4.2 software (Applied Biosystems). A ZORBAX SB-Aq column (4.6 × 250 mm, 5 μm) was used for chromatographic separation. The mobile phase consisted of methanol (A)—water containing 5 mM ammonium format and 0.25 % formic acid (B) (90:10, v/v). The flow rate was 0.80 mL/min and the column temperature was set at 25 °C.
All detections were carried out with the mass spectrometer in positive ESI mode. The multiple reaction monitoring transitions were m/z 516.1/120.2 for cefozopran and m/z 124.2/80.1 for niacin. The optimized instrumental parameters for mass spectral acquisition were as follows: collision gas at 6 psi, curtain gas at 5 psi, ion source gas 1 at 50 psi, ion source gas 2 (nitrogen) at 45 psi, dwell time of 200 ms, ion spray voltage of 4,000 V, and temperature of 350 °C.
Under the analytical conditions mentioned above, cefozopran and IS were well separated and there was no interference from human plasma and urine. The assay for plasma samples was linear over a range of concentrations from 0.395 to 276.3 μg/mL (r 2 = 0.9989). Separate sets of analytical quality control samples (for concentrations of 0.790, 39.48 and 197.5 μg/mL) were used during plasma sample analysis to assess assay precision and accuracy. The intra- and inter-day precision (relative standard deviation, %RSD) were ≤3.23 and ≤8.36 %, respectively, with accuracy (relative error) in the range −6.27 to 5.88 % in plasma standards. The calibration curve range for urine samples was 37.20–7441 μg/mL, and the intra- and inter-batch relative standard deviations (SDs) were less than 3.47 and 9.44 % in urine standards, respectively. The stability results showed that plasma and urine samples were stable in 3 freeze/thaw cycles, short-term (4 h, room temperature) and long term storage (−70 °C, 120 days).
2.6 Pharmacokinetic Data Analysis
The pharmacokinetic parameters were calculated using WinNonlin version 6.3 (Pharsight Corporation, Mountain View, CA, USA). For the single-dose period, the plasma concentration–time data profiles were constructed, and the pharmacokinetic parameters of cefozopran were processed by a standard non-compartmental model: the peak plasma concentration (C max) and the time to reach C max (t max) were determined directly from experimental observations. The elimination rate constant (k e) was obtained as the linear regression of log-transformed concentration versus time data in the terminal portion of the curve. Drug elimination half-life (t ½) was then calculated as ln2/k e. The area under the plasma concentration–time curve (AUC) from time zero to last measurable time point (AUClast) was calculated using the linear trapezoidal rule and was extrapolated to infinity (AUC0–∞) according to the relationship AUC0–∞ = AUClast + C t/k e, where C t is the last measurable concentration of drug in plasma at time. The total body clearance (CL) was calculated as the ratio of dose to AUC0–∞. The apparent volume of distribution (V) was calculated as the ratio of clearance to k e. Cumulative urinary excretion of cefozopran was obtained from urine data.
In the multiple-dose period, the time to reach peak concentration at steady state (t max,ss) and maximum and minimum plasma concentration at stead state (C max,ss and C min,ss) were determined directly from experimental observations. The accumulation ratio was calculated as the ratio of AUC0–τ,ss to AUC0–τ (single dose of 2.0 g), and the fluctuation ratio was calculated as (C max,ss − C min,ss)/C avg, where C avg is the average steady-state drug concentration during multiple dosing, which is calculated as AUC0–τ,ss/τ, where τ is the dosing interval (12 h). The t ½, CL and volume of distribution at steady state (V SS) were calculated using the same method as that used for the single-dose period.
2.7 Statistical Analysis
All data is expressed as the mean and SD. Statistical analysis was performed using the SPSS (version 16.0, SPSS Inc., Chicago, USA). Log-transformed pharmacokinetic parameters AUC and C max were analyzed to determine dose proportionality using the power model, PK = A × (Dose)β, where PK is the pharmacokinetic parameter, A is the intercept and β is the dose-proportionality coefficient. A 95 % confidence interval (CI) for the dose-proportionality parameter β was then calculated. Note that a slope of 1 would correspond to perfect dose proportionality [16]. Pharmacokinetic parameters were compared among dose levels using analysis of variance (ANOVA). The sex-related differences in the main pharmacokinetic parameters were investigated using ANOVA. P < 0.05 was considered significant.
3 Results
3.1 Subjects
A total of 12 healthy subjects, six men and six women, were enrolled in and completed the study. The mean age (range) of subjects was 22.5 (19–26) years, mean body weight (range) was 57.4 (51.0–73.0) kg, mean height (range) was 1.64 (1.53–1.76) m and mean BMI (range) was 21.2 (19.6–23.6) kg/m2.
3.2 Safety and Tolerability Results
No serious AEs occurred during the study, and all subjects were in good compliance with the protocol. There were no clinically meaningful changes from baseline to final assessment values in results related to blood pressure, pulse rate, body temperature, physical examination findings, serum chemistry measures and hematology results.
Two AEs of proteinuria (2/12, 17 %) were reported during the trial. The aforementioned AEs were mild and considered not related to the study drug, and the subjects recovered without treatment.
3.3 Pharmacokinetics Results
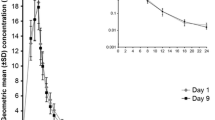
The mean plasma concentration–time profiles after single and multiple doses are shown in Fig. 1. Pharmacokinetic parameters after single- and multiple-dose administration are shown in Table 1. As shown in the slope β from the power model and the scatter diagrams (Table 2; Fig. 2, respectively), cefozopran C max and AUC were linear over the dose range. Plasma t ½ was independent of the dose and dosing duration, averaging 2.15 h (range 1.20–2.93 h) (Fig. 3). Negligible drug accumulation occurred with the multiple-dose regimens, as evidenced by the minimal change in AUC after 4 days of repeated dosing (Table 1). Clearance, which was primary renal and independent of dose and dose duration, averaged 3.53 L/h (±34.8 %) after a single intravenous dose and 4.27 L/h (±27.2 %) after the last of multiple doses. The pharmacokinetic parameters comparisons between the male and female subjects (Table 3) suggested there were no significant gender differences in the process of drug metabolism. The large majority of cefozopran (70.09 % following a single intravenous dose) was excreted in the urine as unchanged cefozopran.

Plots of the mean plasma concentration–time curves of intravenous cefozopran for the three dose groups (0.5, 1.0 and 2.0 g) after a single dose and on the fourth day after multiple doses of 2.0 g (a linear scale, b semi-log scale)

Scatter diagrams of the relationship between the dose and a the log-normal maximum plasma drug concentration (lnC max) and b the log-normal area under the plasma concentration–time curve (lnAUClast)

Plots of geometric mean (standard deviation) half-life of cefozopran after single or multiple intravenous doses
4 Discussion
A similar study has been conducted previously in healthy Chinese subjects. However, unlike the findings from the study in China by Guo, which used high-performance liquid chromatography as the analytical method, the present study employed a more robust method, LC–MS/MS, to quantify the plasma concentration of cefozopran. Moreover, their study had a parallel design rather than the cross-over design we used for our present study. The cross-over design can eliminate intersubject differences and decrease the number of subjects. Finally, we provided the first assessment of the pharmacokinetics of cefozopran in Chinese healthy subjects following 1-h intravenous infusion of 0.5, 1.0 and 2.0 g cefozopran and multiple 1-h intravenous infusions of 2.0 g cefozopran.
In this study, the pharmacokinetics, safety and tolerability of cefozopran were evaluated in healthy Chinese subjects. In the current study, cefozopran was generally safe and well tolerated at the dose regimens studied. The pharmacokinetic profile of cefozopran after single-dose administration was predictable and dose proportional within the dose range evaluated. The slope β of the regression lines was slightly less than 1 (Table 2). However, 95 % CIs were close to 1 for both parameters, with the 95 % CIs for C max and AUClast including 1, indicating dose proportionality. Therefore, it can be concluded that the increases in cefozopran exposure between 0.5 and 2.0 g were roughly dose proportional.
Following a single 1-h intravenous infusion of 0.5, 1.0 or 2.0 g injected cefozopran hydrochloride, the C max and AUC values in our study presented here (as show in Table 1) were higher than the values previously reported for Japanese [10] and Chinese [14] volunteers (in Japanese, C max mean values of 29.1, 68.3 and 118 μg/mL, and AUC0–∞ mean values of 72.4, 180 and 295 μg·h/mL, respectively; in Chinese, C max mean values of 48.27, 77.99 and 171.59 μg·h/mL, and AUC0–∞ mean values of 95.23, 157.27 and 350.28 μg·h/mL, respectively). This variation might be due to differences in infusion time, infusion volume, the manufacture of the study drugs and analytical method for measuring the plasma cefozopran concentration. It should be noted that in the previous study in Japanese, subjects with normal renal function were enrolled. However, the patients’ accompanying diseases might interfere with the results. In the previous study in Chinese, healthy subjects with a wider range in age (19–35 years) were selected, compared with the age range between 19 to 26 years in the present study. Thus, age and accompanying diseases might also be an important reason for this difference. Moreover, the drug was delivered by infusion pump in our study, which allowed for a greater level of control, accuracy and precision in drug delivery. CL, V and t ½ did not vary significantly with dosage. The elimination half-lives proved to be independent of the dose, with values of 1.20–2.80 h, which was close to the values reported in previous studies [9, 14] and less than the value of 4.8 h that was reported in renal dysfunction patients [17]. Cefozopran was eliminated almost entirely through urinary excretion; this observation suggests a need for dosage adjustment in patients with the most severe degrees of renal impairment.
Following concomitant administration of multiple doses of cefozopran as a 1-h intravenous infusion q12h for 4 days, no appreciable accumulation was detected for cefozopran (ratio of day 4 to day 1 area under the curve from time zero to 13 h [AUClast] 0.85–1.46). These observations are consistent with the relatively short half-life of cefozopran. The pharmacokinetic parameters for cefozopran were similar on days 1 and 4 in this study.
The favorable safety and predictable pharmacokinetic profile of cefozopran in this study and the excellent predicted probability of target attainment at the likely clinical dose regimen of 2.0 g every 12 h support further clinical application in humans for the treatment of bacterial infections.
5 Conclusion
Cefozopran is generally well tolerated in Chinese young, healthy male and female subjects. The dose-dependent increase in exposure between 0.5 and 2.0 g of cefozopran can be considered roughly dose proportional. Cefozopran exhibited no accumulation with repeated administration. Compared with a single dose, multiple doses showed similar pharmacokinetics and tissue distribution patterns. Sex did not appear to affect the pharmacokinetic properties of cefozopran.
References
Hughes WT, Armstrong D, Bodey GP, et al. 2002 guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clin Infect Dis. 2002;34:730–51.
Iwahi T, Okonogi K, Yamazaki T, et al. In vitro and in vivo activities of SCE-2787, a new parenteral cephalosporin with a broad antibacterial spectrum. Antimicrob Agents Chemother. 1992;36:1358–66.
Ikeda K, Morikawa N, Kuribayashi M, et al. Real-time therapeutic drug monitoring of cefozopran in plasma using high-performance liquid chromatography with ultraviolet detection. J Pharm Biomed Anal. 2007;45:811–6.
Sato T, Kobayashi R, Yasuda K, et al. A prospective, randomized study comparing cefozopran with piperacillin-tazobactam plus ceftazidime as empirical therapy for febrile neutropenia in children with hematological disorders. Pediatr Blood Cancer. 2008;51:774–7.
Ikawa K, Morikawa N, Matsuda S, et al. Peritoneal penetration and pharmacodynamic exposure of intravenous cefozopran in abdominal surgery patients. Int J Antimicrob Agents. 2007;30:352–5.
Ikawa K, Nomura K, Morikawa N, et al. Pharmacokinetic-pharmacodynamic target attainment analysis of cefozopran in Japanese adult patients. J Infect Chemother. 2008;14:130–6.
Kitahashi T, Furuta I. Development and validation of a MEKC method for the direct determination of cefozopran in human serum. J Pharm Biomed Anal. 2004;34:409–14.
Iizawa Y, Okonogi K, Hayashi R, et al. Therapeutic effect of cefozopran (SCE-2787), a new parenteral cephalosporin, against experimental infections in mice. Antimicrob Agents Chemother. 1993;37:100–5.
Paulfeuerborn W, Muller HJ, Borner K, et al. Comparative pharmacokinetics and serum bactericidal activities of SCE-2787 and ceftazidime. Antimicrob Agents Chemother. 1993;37:1835–41.
Yamamoto T, Adachi Y, Nagamine Y, et al. Clinical phase I study of cefozopran. Shinryou Shinyaku (Treat New Med). 1993;30:281–304.
Kobayashi H, Moritono S, Hara K. Significant role of new cephem antibiotics: focused on cefozopran. Jpn J Antibiot. 1997;50:807–20.
Ikeda K, Ikawa K, Morikawa N, et al. Determination of total cefozopran concentrations in human peritoneal fluid by HPLC with cefepime as an internal standard: comparative pharmacokinetics in the fluid and plasma. J Pharm Biomed Anal. 2009;49:1075–81.
Ikawa K, Mikamo H, Morikawa N, et al. Penetration into and exposure of cefozopran in pelvic retroperitoneal space exudate. J Infect Chemother. 2008;14:170–1.
Guo WW, Shen Q, Qin YP, et al. Pharmacokinetics of injected cefozopran hydrochloride in healthy volunteers. Sichuan Da Xue Xue Bao Yi Xue Ban. 2012;43:711–4.
US Food and Drug Administration. Guidance for industry bioanalytical method validation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf. Accessed 6 Mar 2014.
Limoges D, Dieterich HA, Yeh CM, et al. A study of dose-proportionality in the pharmacokinetics of the oral direct renin inhibitor aliskiren in healthy subjects. Int J Clin Pharmacol Ther. 2008;46:252–8.
Takenaka T, Watanabe T, Hayashi T, et al. Pharmacokinetics and dosage planning of cefozopran in patients with renal insufficiency. Jpn J Chemother. 1993;41:147–53.
Acknowledgments
The authors deeply appreciated the contributions of the study team during the study. This study was sponsored by Jiangsu Zhengda Tianqing Pharmaceutical Co., Ltd. This research was funded by the Key Technologies R&D program of 12th Five-year Plan (2012ZX09302-003-03), PR China. The authors have indicated that they have no conflicts of interest with regard to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wu, G.L., Shentu, J.Z., Zhou, H.L. et al. Pharmacokinetics of Cefozopran by Single and Multiple Intravenous Infusions in Healthy Chinese Volunteers. Drugs R D 15, 63–70 (2015). https://doi.org/10.1007/s40268-014-0075-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-014-0075-3