
Abstract
Fabry Disease (FD) is an X-linked lysosomal storage disease that emerges as a result of the mutations in the galactosidase A gene encoding alpha-galactosidase. The peripheral nervous system (PNS) involvement manifests itself as acroparesthetic complaints due to the small-fiber involvement. Our goal was to assess the PNS involvement of 14 patients with FD both clinically and electrophysiologically besides the other systemic features. 14 patients (11 female and 3 male) of the same family whose enzyme level and genetic mutation analysis confirmed the FD diagnosis were evaluated retrospectively in terms of systemic and neurological findings of the FD. Neurological examination and nerve conduction studies were performed to evaluate the PNS involvement. PNS involvement was more common in females. Eight of the patients had acroparesthesia. The neurological examinations of all patients were normal. Two patients presented sensory axonal polyneuropathy, one of whom had no acroparesthesia. Other patients with acroparesthesia had normal nerve conduction studies. There was no significant relationship between the presence of acroparesthesia and the results of conduction studies (p > 0.05). Acroparesthetic complaints in patients with normal results were attributed to small-fiber involvement. Since small-diameter nerve fibers cannot be evaluated by routine conduction studies, especially in the early stages of FD, these studies may be normal. Early diagnosis through the symptoms such as acroparesthesia may contribute to the survival of the patient by preventing and/or delaying the development of renal, cardiac, and cerebrovascular diseases, which are the main causes of morbidity and mortality.
Similar content being viewed by others

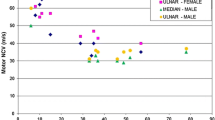
References
Desnick RJ, Ioannou YA, Eng CM (2014) α-Galactosidase a deficiency: Fabry disease. In: Vogelstein B (ed) The metabolic and molecula rbases of inherited disease, 8th edn. TheMcGraw-Hill Companies, NewYork, pp 3733–3774
Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, Sunder-Plassmann G, FOS Investigators (2009) Natural course of Fabry disease: changing pattern of causes of death in FOS—Fabry outcome survey. J Med Genet 46(8):548–552
Burlina AB, Polo G, Salviati L, Duro G, Zizzo C, Dardis A, Bembi B, Cazzorla C, Rubert L, Zordan R, Desnick RJ, Burlina AP (2018) Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inherit Metab Dis 41(2):209–219. https://doi.org/10.1007/s10545-017-0098-3(Epub 2017 Nov 15)
Winchester B, Young E (2006) Biochemical and genetic diagnosis of Fabry disease, chapter 18. In: Mehta A, Beck M, Sunder-Plassmann G (eds) Fabry disease: perspectives from 5 years of FOS. Oxford PharmaGenesis, Oxford
Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W (1973) Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, andleukocytes. J Lab Clin Med 81(2):157–171
Germain DP (2002) Fabry’s disease (alpha-galactosidase-A deficiency): physiopathology, clinical signs, and genetic aspects. J Soc Biol 196(2):161–173
Deegan PB, Bähner F, Barba M, Hughes DA, Beck M (2006) Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy, chapter 30. In: Mehta A, Beck M, Sunder-Plassmann G (eds) Fabry disease: perspectives from 5 years of FOS. Oxford PharmaGenesis, Oxford
MacDermot J, MacDermot KD (2001) Neuropathic pain in Anderson-Fabry disease: pathology and therapeutic options. Eur J Pharmacol 429(1–3):121–125
Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S (2007) Ocular manifestations of Fabry’s disease: data from the Fabry outcome survey. Br J Ophthalmol 91(2):210–214
Baig S, Edward NC, Kotecha D et al (2017) Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace. https://doi.org/10.1093/europace/eux261(Epub ahead of print)
Kayıkçıoğlu M, Şimşek E, Kalkan Uçar S, Bayraktaroğlu S, Onay H, Sözmen E, Çoker M (2017) Fabry disease: an overlooked diagnosis in adult cardiac patients. Turk Kardiyol Dern Ars 45(6):549–555
El-Abassi R, Singhal D, England JD (2014) Fabry’s disease. J Neurol Sci 344(1–2):5–19. https://doi.org/10.1016/j.jns.2014.06.029
Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M, European FOS Investigators (2006) Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 43(4):347–352
Arends M, Hollak CE, Biegstraaten M (2015) Quality of life in patients with Fabry disease: a systematic review of the literature. Orphanet J Rare Dis 10:77
Eng CM, Guffon N, Wilcox WR et al (2001) International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 345(1):9–16
Schiffmann R, Kopp JB, Austin HA 3rd et al (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285:2743–2749
Hughes DA, Elliott PM, Shah J, Zuckerman J, Coghlan G, Brookes J, Mehta AB (2008) Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 94(2):153–158
Schiffmann R, Hughes DA, Linthorst GE, Ortiz A, Svarstad E, Warnock DG, West ML, Wanner C (2017) Conference participants. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 91(2):284–293. https://doi.org/10.1016/j.kint.2016.10.004
Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP et al (2016) Treatment of Fabry disease with the pharmacological chaperone migalastat. N Engl J Med 375:545–555
Hughes DA, Nicholls K, Shankar SP, Sunder- Plassmann G, Koeller D, Nedd K, Vockley G, Hamazaki T et al (2018) Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 55(6):422–429
Senocak Tasci E, Bicik Z (2015) Safe and successful treatment with agalsidase beta during pregnancy in Fabry disease. Iran J Kidney Dis 9(5):406–408
Schiffmann R, Moore DF (2006) Neurological manifestations of Fabry disease, chapter 22. In: Mehta A, Beck M, Sunder-Plassmann G (eds) Fabry disease: perspectives from 5 years of FOS. Oxford PharmaGenesis, Oxford
Laaksonen SM, Röyttä M, Jääskeläinen SK, Kantola I, Penttinen M, Falck B (2008) Neuropathic symptoms and findings in women with Fabry disease. Clin Neurophysiol 119(6):1365–1372
Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, vanSchaik IN (2012) Small fiber neuropathy in Fabry disease. Mol Genet Metab 106:135–141
Pellissier JF, Van Hoof F, Bourdet-Bonerandi D, Monier-Faugere MC, Toga M (1981) Morphological and biochemical changes in muscle and peripheral nerve in Fabry’sdisease. Muscle Nerve 4:381–387
Liguori R, Incensi A, de Pasqua S et al (2017) Skin globotriaosylceramide 3 deposits are specific to Fabry disease with classical mutations and associated with small fibre neuropathy. PLoS ONE 12(7):e0180581
Lakomá J, Rimondini R, FerrerMontiel A, Donadio V, Liguori R, Caprini M (2016) Increased expression of Trpv1 in peripheral terminals mediates thermal nociception in Fabry disease mouse model. Mol Pain 12:1–16
Kummer KK, Kalpachidou T, Kress M, Langeslag M (2018) Signatures of altered gene expression in dorsal root ganglia of a Fabry disease mouse model. Front Mol Neurosci 25(10):449
Low M, Nicholls K, Tubridy N et al (2007) Neurology of Fabry disease. Intern Med J 37(7):436–447
Dütsch M, Marthol H, Stemper B, Brys M, Haendl T, Hilz MJ (2002) Small fiber dysfunction predominates in Fabry neuropathy. J Clin Neurophysiol 19(6):575–586
Lacomis D (2002) Small-fiber neuropathy. Muscle Nerve 26:173–188
Terkelsen AJ, Karlsson P, Lauria G, Freeman R, Finnerup NB, Jensen TS (2017) The diagnostic challenge of small fibre neuropathy: clinical presentations, evaluations, and causes. Lancet Neurol 16(12):954
Akpinar ÇK, Türker H, Bayrak O, Cengiz N (2015) Electroneuromyographic features in Fabry disease: a retrospective review. Noro Psikiyatr Ars 52(3):258–262
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
İnan, R., Meşe, M. & Bicik, Z. Multidisciplinary approach to Fabry disease: from the eye of a neurologist. Acta Neurol Belg 120, 1333–1339 (2020). https://doi.org/10.1007/s13760-019-01138-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-019-01138-y