
Abstract
Few data are available concerning the composition of biofilms found at the surface of filtration membranes, which, to some extent, explains the long-term failure of numerous strategies developed to control biofouling. This preliminary study intended to design a metagenomic tool targeting the 16S rRNA gene in order to unravel a general portrait of bacterial communities found on spiral-wound membranes used in the dairy industry. A total of seven spiral-wound membrane elements (ultrafiltration, nanofiltration, or reverse osmosis) at the end of their useful lifetimes were collected from different dairy plants. Targeted analysis of the 16S rRNA genes of the metagenome extracted from the membranes revealed their bacterial diversity via high-throughput sequencing technology (Miseq, Illumina). It was found that the nature of the filtered fluid (milk, whey, water) explained 58.6 % of the variance observed between communities found on membranes. Treatments applied on dairy fluids (milk pasteurization, whey bleaching or whey ultrafiltration) induced a selective pressure that affected the diversity of bacterial communities found on membranes and the proportions of spore-former bacteria among them. This work provides the first complete bacterial portrait of the biofilm composition of spiral-wound membranes used in the dairy industry. It suggests that the nature of the filtered fluid and potentially filtration operating parameters may be important elements to consider in order to design new cleaning strategies or preventive measures targeting biofouling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Membrane separation processes improve the efficiency of dairy processors by enabling the recovery of all milk constituents and coproducts (whey, milk ultrafiltration permeate, effluents) (Mistry and Maubois 2004). Membrane installations in dairy processing plants are designed to operate continuously for 18 to 24 h daily under processing conditions (transmembrane pressure, recirculation rate, temperature, etc.) that are specific to each filtered fluid (milk, whey, whey permeate) and the technology used (microfiltration, ultrafiltration, nanofiltration, or reverse osmosis) (Jirjis and Luque 2010). At the end of the filtration step, a cleaning-in-place (CIP) is performed on membranes with acid and alkaline solutions in order to ensure hygienic operations and recover membrane performance (Tang et al. 2010).
However, even with daily care, long-term fouling of the membranes remains a major issue affecting the performance of membrane systems and leads to expensive membrane replacements (Jirjis and Luque 2010; Anand et al. 2014). For most dairy fluids, short- and long-term fouling results from the accumulation of proteins (Bégoin et al. 2006) and minerals (Anand et al. 2014) on membrane surfaces. However, concerns around bacterial adhesion warrant further investigation in the dairy industry because of the great resilience of biofilms exposed to cleaning procedures (Tang et al. 2009; Hassan et al. 2010; Anand et al. 2012; Anand and Singh 2013; Anand et al. 2014) and the high bacterial counts observed in filtered products (Anand et al. 2012). Biofouling, through biofilm development, has been reported in desalination plants (Matin et al. 2011; Khan et al. 2013; Levi et al. 2016) and membrane bioreactors (Ivnitsky et al. 2007; Malaeb et al. 2013; Vanysacker et al. 2014b). Initially, it requires the formation of a conditioning film and the adhesion of specific bacteria (pioneer bacteria) equipped with numerous adhesion strategies (Ivnitsky et al. 2007). The pioneer bacteria grow and modify surface properties, producing extra polymeric substances (EPS) that affect membrane performance and permit recruitment of other bacteria as the biofilm matures (Lappin-Scott and Costerton 1989; Ivnitsky et al. 2007; Vanysacker et al. 2014a). In the dairy industry, biofouling may also result in serious quality issues for the filtered fluid and subsequent dairy products because of the presence of spore-forming or pathogenic bacteria on membranes (Burgess et al. 2010; Anand et al. 2014) and their concurrent microbial enzyme production (Raats et al. 2011; Teh et al. 2014). However, only a few studies described the microbial composition of biofilm detected on dairy processing membranes, and they have been limited to microscopy (Hassan et al. 2010) or culture-based observations (Tang et al. 2009; Tang et al. 2010; Anand et al. 2012; Anand and Singh 2013). Considering that the majority of bacteria in environmental samples are usually unculturable (Amann et al. 1995), the use of culture-independent approaches, such as metagenomics, is essential.
Consequently, the main objective of this preliminary study was to characterize the compositional diversity of biofilms found on industrial dairy processing membranes in order to understand these complex communities and the factors leading to biofouling.
2 Materials and methods
2.1 Sampling industrial membranes
A total of seven spiral-wound membranes (SWMs) were collected in four Canadian dairy processing plants named D1, D4, E2, and F3, at the end of their useful lifetime (between 119 and 920 days of use) (Table 1). These membranes were used to filter pasteurized milk (pM), bleached or fresh cheese whey (respectively bW and fW), ultrafiltration whey permeate (P), or a blend of condensed water and permeate from reverse osmosis of dairy fluids (the term “water blend” will be used to simplify the text, Wa) (Table 1). Ultrafiltration (UF), nanofiltration (NF), or reverse osmosis (RO) SWMs were collected (Table 1).
Before being sampled, SWMs were frozen at −20 °C in the different dairy plants and carried frozen to Université Laval (Québec city, QC, Canada) until analysis. Among the SWMs, the final treatment applied in the different dairy plants on membrane was a rinsing with tap water for D4RO-fW and D4RO-Wa while other SWMs were cleaned with the specific CIP procedure of each plant. The dairy manufacturers provided information on their membranes such as their age, the filtered fluid, the temperature of the feed during filtration, and their daily usage times (Table 1), while other proprietary operating parameters and details about cleaning conditions were kept confidential.
Samples of 150 cm2 were taken in duplicate with a sterile scalpel at four (4) locations on each unrolled SWM (total of eight samples, 1200 cm2): at the inlet and outlet sides, at the inner part of the membrane, close to the permeate tube, and at the outer part of the membrane. Samples from each section were cut into 1-cm2 pieces with sterile scissors to fit in a 15-mL sterile polypropylene tube for DNA extraction. No visual sign of the presence of biofilms was detected, except for the uncleaned D4RO-fW membrane.
2.2 DNA extraction method
DNA extraction was performed following the methodology of Ferrera et al. (2010), including an enzymatic pretreatment and a phenol-chloroform extraction but adapted according to other authors (Ercolini et al. 2003) as described below. Membrane samples were soaked in 3.8 mL of TES buffer (50 mmol∙L−1 Tris–HCl, 1 mmol∙L−1 EDTA, 8.7% w/v sucrose; pH 8.0). Lytic enzymes were added (200 μL of 40 mg∙mL−1 lysozyme, 50 μL of 5 U∙mL−1 mutanolysine, and 10 μL of 10 mg∙mL−1 RNAse A) to the tubes maintained at 37 °C and incubated for 60 min (Ercolini et al. 2003; Ferrera et al. 2010). Subsequently, 500 μL of 10 mg∙mL−1 proteinase K, 50 μL of 10 % w/w SDS, and 450 μL of TES buffer were added. The tubes were incubated at 55 °C for 60 min, then at 65 °C for 10 min. Next, 900 μL of 5 mol∙L−1 NaCl and 900 μL of TES were added and tubes were held on ice. The phenol-chloroform extraction was performed as specified by Ferrera et al. (2010) with the following modifications: no isoamyl alcohol was used and the centrifugation steps were conducted at 13,000×g at 4 °C for 10 min. The polymeric phase of the membrane samples dissolved completely in phenol. DNA in the aqueous supernatant was precipitated using 1/10 vol of 8 M LiCl and 1 vol of pure ice-cold isopropanol, the latter being selected to precipitate the low amounts of DNA extracted from SMWs even if it increased the salt concentration in the DNA samples. Tubes were chilled for 1 h at −20 °C. DNA was recovered after centrifugation (15,000×g for 30 min at 4 °C) and washed twice with ice-cold 70% v/v ethanol to remove residual salts. The DNA pellet was dried and resuspended in 50 μL of sterile deionized water. DNA yield and quality (purity) were assessed using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware, USA). DNA extractions were done in duplicate for each membrane section (eight extractions per SWM). For each membrane section, the sample having the highest DNA extraction yield between the two duplicates was selected for high-throughput sequencing (four samples per SWM). The absence of PCR inhibitor was assessed prior to sequencing by amplifying the selected samples by PCR (universal bacterial primers 27F and 1492R) and migrating amplicons on a 0.8% w/v agarose gel.
2.3 16S rRNA Gene sequencing
Targeted amplification of the 16S rRNA gene, equimolar pooling, and sequencing were performed at the Plate-forme d’Analyses Génomiques (IBIS, Université Laval, Québec City, Canada). Briefly, amplification of the V6-V8 region of the gene encoding 16S rRNA (Comeau et al. 2011) was performed using a two-step, dual-indexed PCR approach specifically designed for Illumina instruments (Illumina, San Diego, California, USA) (Table 2). In the first step, the gene-specific sequence was fused to the Illumina TruSeq sequencing primers and PCR was carried out in a total volume of 25 μL containing 1× Q5 buffer (New England Biolabs, Ipswich, MA, USA), 0.25 μmol∙L−1 of each primer, 200 μmol∙L−1 of each dNTP, 1 U of Q5 High-Fidelity DNA polymerase (NEB) and 1 μL of the template DNA. The PCR parameters were 30 s at 98 °C, followed by 35 cycles (10 s at 98 °C, 10 s at 55 °C, 30 s at 72 °C), and a final extension of 2 min at 72 °C. The PCR reaction was purified using the Axygen PCR Clean-up kit (Axygen Biosciences, Union City, CA, USA). The quality of the purified PCR product was determined on a 1% w/v agarose gel. The second PCR step, performed with a 50- to 100-fold dilution of the previously purified PCR product as a DNA template, allowed the addition of barcodes (dual-indexed) and the missing sequence required for Illumina sequencing. The PCR parameters were 30 s at 98 °C, followed by 12 cycles (10 s at 98 °C, 10 s at 55 °C, 30 s at 72 °C), and a final 2-min extension at 72 °C. PCR reactions were purified as described above, and quantification and quality assessment were performed using the Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware, USA) and a DNA7500 Bioanalyzer chip (Agilent Technologies, Santa Clara, CA, USA), respectively. Barcoded amplicons were then pooled at equimolar concentration for sequencing by the Illumina’s MiSeq sequencer.
2.4 Sequence accession number
The 1,369,080 raw reads have been deposited in the NCBI Sequence Read Archive under accession number SRP065116.
2.5 Metadata processing and statistical analysis
Raw sequences were processed with Mothur v1.34.4 following Schloss’s MiSeq SOP (available at http://www.mothur.org/wiki/MiSeq_SOP) (Schloss et al. 2009; Kozich et al. 2013). Computations were made on the supercomputer Colosse from Université Laval, managed by Calcul Québec and Compute Canada. After assembling forward and reverse amplicons (make.contigs), sequences containing ambiguous bases (maxambig = 0) and sequences that were too short (minlength = 400) or too long (maxlength = 440) were removed (screen.seqs). Remaining sequences were aligned (align.seqs) for the V6-V8 region (obtained with pcr.seqs) of a reference database (SILVA bacteria release 102). Chimera reads were removed using the UCHIME program (Edgar et al. 2011). A naive Bayesian classifier (classify.seqs) assigned a taxonomy file (RDP 16S rRNA reference v14) to the trimmed sequences with 1000 iterations (iters = 1000) (Mizrahi-Man et al. 2013), and a minimum bootstrap value of 80 % was set as recommended (Manes et al. 2011). Since bacterial primers were used, sequences belonging to Chloroplast, Mitochondria, Eukaryota, and Archae were discarded (remove.lineage). Sequences were clustered (cluster.split) using the average neighbor algorithm method to the order level (taxlevel = 4) with a minimum of 97% of similarity (cutoff = 0.03), which is commonly accepted for classifying sequences at the genus level (Comeau et al. 2011).
Within-sample diversity (Alpha diversity) was analyzed with Good’s coverage estimator (Good 1953) index, as described by Tully and Heidelberg (2013); the Chao-1 richness estimator (community richness), as described by Katra (Katra et al. 2014); and the inverse Simpson diversity index (community diversity) with the same number of sequences in each sample (sub.sample) (Schloss et al. 2009). Rarefaction curves were made using R software (v3.2.0; http://www.r-project.org/) with the data generated by Mothur (rarefaction.single). The ThetaYC calculator based on the relative abundance of each operational taxonomic unit (OTU) was used to generate a dendrogram showing similarities/dissimilarities between samples (beta diversity) (Tully and Heidelberg 2013). A web-based tool (iTol) was further used to add a stacked bar chart to the dendrogram figure (Letunic and Bork 2011). Jaccard and Bray-Curtis dissimilarity calculators were also tested, but, as per Tully and Heidelberg (2013), they were not discussed since their outputs were very similar to those of ThetaYC. Finally, the variance in bacterial communities was calculated by permutational analysis of variance (PERMANOVA test, Vegan package of R) based on the ThetaYC distance matrix (Anderson 2001). Considering the heterogeneity of the biofilms detected on filtration membranes (Hassan et al. 2010), the results of the four membrane sections were merged for the presentation of bacterial ratio and diversity indexes. However, the PERMANOVA test was done on unmerged samples to maximize the degrees of freedom.
3 Results
3.1 Ecological comparisons of the SWMs
Total genomic DNA amounts recovered from the SWM samples varied between 0.17 ng·cm−2 (E2UF-bW) and 1091.06 ng·cm−2 (D4RO-fW). DNA extraction on F3UF-bW resulted in the highest extraction yield on a cleaned membrane (9.65 ng·cm−2) (Table 1). The average A260/A280 and A260/A230 ratios of the DNA samples of each SWM varied from 1.28 to 1.93 and from 0.32 to 1.04, respectively (Table 1).
The number of OTUs identified on SWMs varied between 515 and 2452 (Table 1), with flattened rarefaction curves indicating a sufficient sequencing depth for most of the samples but only for partial depth for F3UF-bW (Supplementary material, Fig. S1). Good’s coverage index was greater than 99% for all samples and there were low ratios of unclassified OTUs at the phylum level (ranging from 0.01 to 2.10%) (Table 3). A higher number of OTUs (higher richness) was observed (Sobs) and estimated (Chao 1 index) on the oldest SWM sampled (F3UF-bW, collected after 930 days of operation), even if its rarefaction curve was not completely saturated. The lowest Sobs was found on the youngest SWM (E2UF-bW), collected after 113 days of operation (Table 1).
The membranes E2UF-pM1 and E2UF-pM2 were cleaned applying the same industrial procedure. Membrane E2UF-pM1 was older (458 compared to 322 days for membrane E2UF-pM2), but their theoretical richnesses were not statistically different (2648 and 3840 for E2UF-pM1 and E2UF-pM2, respectively, 95% confidence interval). Comparing these two membranes, the highest Sobs was observed on E2UF-pM2, which had a longer daily usage (18 h instead of 15 h for E2UF-pM1).
3.2 The composition of bacterial communities on dairy processing SWMs
Various diversity levels were observed among SWM, mostly depending on the nature of the filtered fluid (Table 1). The highest diversity (Inverse simpson diversity index) was found in communities formed on milk processing membranes (8.41 E2UF-pM1 and 14.10 for E2UF-pM2). The diversity decreased on bleached cheese whey processing membranes (5.76 for E2UF-bW and 4.63 for F3UF-bW). The lowest diversity was observed on UF whey permeate processing membrane (1.21 for D1NF-P). The level of diversity found on both membranes from dairy plant D4 (D4RO-fW and D4RO-Wa) was similar (1.49 and 1.51, respectively) but not comparable to other membranes since they were not subject to a complete cleaning cycle but were only rinsed (Table 1). The uncleaned membrane used for fresh cheese whey filtration (D4RO-fW) had a significantly lower diversity index than both cleaned membranes used to filter bleached cheese whey (E2UF-bW and F3UF-bW) (Table 1).
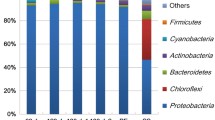
Proteobacteria, Firmicutes, and Actinobacteria were the three major phyla observed on the SWMs but the dominant phylum differed according to the filtered fluid. In the case of milk processing membranes, Proteobacteria were dominant with relative abundances of 63.62 and 62.68% for samples E2UF-pM1 and E2UF-pM2, respectively (Table 3). The highest proportion of Actinobacteria was also found in those samples. On both E2UF-pM1 and E2UF-pM2, Methylobacterium sp. was the most abundant genus and accounted for 23 and 14% of the global population, respectively. The dominant Bacilli genera identified were Streptococcus sp., Enterococcus sp., and Lactococcus sp. while Acinetobacter sp., Cronobacter sp., and Klebsiella sp. were the major γ-Proteobacteria found (relative abundance >5%) (Table 3). In the case of E2UF-pM1, less diversity was found and only Lactococcus sp., Psychrobacter sp., and Arthrobacter sp. had a relative abundance higher than 5 %.
Bacterial communities of SWMs used for cheese whey (raw and bleached), whey permeate, and water blend filtration were dominated by the Firmicutes phylum, essentially because of the presence of a high ratio of the Bacilli class. Except for both membranes from the plant D4, spore-forming bacilli dominated these bacterial communities (Table 3). For example, the E2UF-bW membrane showed a great abundance of Paenibacillus sp. (28.37 %) and Brevibacillus sp. (17.81 %). These genera were found at low ratio on milk processing membranes from the same plant, E2 (relative abundances of 0 to 0.02%). The α-Proteobacteria Methylobacterium sp. was the only one to be substantially present on both milk and bleached whey filtration membranes used in plant E2.
Spore-forming Alicyclobacillus sp. was the most abundant genus on the F3UF-bW membrane (49.36%; Table 3). The UF whey permeate filtration membranes (D1NF-P) had the greatest proportion of spore-forming bacteria with 93.18% of Anoxybacillus sp. (Table 3). No spore-forming bacteria were detected on the uncleaned membrane used to filter raw cheese whey (D4RO-fW). Genera possibly related to lactic acid bacteria (LAB) such as Streptococcus sp. (83.02%), Lactobacillus sp. (10.78%), and Lactococcus sp. (2.82%) were detected instead. The same phenomenon was observed with D4RO-Wa for which the bacterial community was mainly composed of Streptococcus sp. (81.56%) and Lactococcus sp. (4.12%).
Statistically, according to a PERMANOVA test, 58.6, 14.2, and 27.2% of the variations observed would be respectively explained by the nature of the filtered fluid, the temperature of the feed, and other parameters (Table 4). Among these other parameters, it is suggested that the plant environment where the SWMs were sampled may have had an effect, with divisions occurring between the SWMs of each plant on the ThetaYC similarity dendrogram (Fig. 1). For example, based on the relative proportions of their OTUs, the community identified on the whey processing membrane in the plant E2 (E2UF-bW) was closer to those found on milk processing membranes (E2UF-pM1 and pM2) sampled in the same plant as the one found on F3UF-bW, which filtered the same type of fluid in a different plant (Fig. 1). Specific OTUs had a higher proportion in specific dairy plants, for example, Anoxybacillus sp. and Meiothermus sp. (D1 plant), Streptococcus sp. (plant D4), Methylobacterium sp. (E2), or Alicyclobacillus sp. (F3).

Combined stack chart and dendrogram presenting the proportion (%) of bacterial classes found on SWMs sampled and the similarity between the samples (ThetaYC calculator). The red numbers represent branch lengths. Distances below 0.05 were not shown
4 Discussion
The high-throughput sequencing approach selected for this study allowed to obtain an overview of the bacterial communities established on SWMs used in dairy processing plants. Since most of the SWMs were cleaned prior sampling, low DNA concentrations were detected on their surfaces. The A260/A230 average ratios of the DNA samples were low, indicating a possible salt contamination in them. However, they had a sufficient quality for the preparation of adequate libraries prior to sequencing (absence of PCR inhibitor and good A260/A280 average ratios) (Simbolo et al. 2013).
There are controversial results regarding the effect of sample freezing on DNA extraction yields or the accuracy of high-throughput sequencing data. Indeed, the freezing processes of the membrane samples for this study could affect the DNA extraction yield and the proportions of some genera observed among bacterial communities observed on SWM (Metzler-Zebeli et al. 2016). However, it was demonstrated that freezing may only have minor impacts on the global portrait of a bacterial community (Fouhy et al. 2015), on which this study focused.
The short read lengths (400–440 bp) enabled a thorough taxonomic assignment for all samples at the genus level, with an overall low abundance of 1% of unclassified phyla. In comparison to many studies previously conducted on biofouling in water treatment plants (including desalination and waste water treatment plants), biofilm composition in the dairy industry is different, most notably because of the high proportion of Firmicutes, which are absent from water treatment plants (Chen et al. 2004; Ivnitsky et al. 2007; Huang et al. 2008; Matin et al. 2011; Khan et al. 2013; Levi et al. 2016).
As reported by Tang et al. (2009) and Anand et al. (2012), the results of this study confirm the resilience of biofilms on SWMs after the cleaning processes used in the dairy industry, possibly indicating inadequate cleaning strategies and/or high intrinsic resistance of the bacteria and biofilms themselves. Considering this, the richness (and the diversity) of the communities on the SWMs would be expected to continuously increase during the lifetimes of these membranes (Manes et al. 2011). Accordingly, the oldest SWM (F3UF-bW) had the highest Sobs and estimated number of OTUs (Chao1 estimator), while the youngest had the lowest (E2UF-bW).
4.1 The effect of operating conditions
This study shows the preponderant impact of the nature of the filtered fluid or the combination of treatments, like heating (milk pasteurization) (Burgess et al. 2010), bleaching (whey discoloration), or ultrafiltration of the feed (bacteria removal before filtration). The bacterial community found on pasteurized milk processing membranes had the closest similarity to natural raw milk microflora of the province of Quebec and to pasteurized cheese ecosystems, in comparison to the other fluids filtered (Rasolofo et al. 2010; Wolfe et al. 2014). Bacilli (Lactococcus sp. and Streptoccus sp.) and γ-Proteobacteria (Klebsiella sp., Acinetobacter sp., and Psychrobacter sp.) found on milk processing membranes could be associated with their natural presence in raw milk and their natural resistance to pasteurization since they were found in both milk and cheese rind ecosystems or in biofilms on post-pasteurization lines (Sharma and Anand 2002; Quigley et al. 2012; Bokulich and Mills 2013; Quigley et al. 2013). The presence of γ-Proteobacteria on milk processing membranes could be favored by the cold filtration environment (17 °C, Table 1), as observed in milk ecosystems during long cold storage at 4 °C, in contrast with fresh raw milk dominated by Gram-positive bacteria (such as Bacilli) at the farm (Raats et al. 2011; Quigley et al. 2013).
The prevalence of the Methylobacterium sp. in E2UF-pM1 and E2UF-pM2 bacterial communities (23 and 14%, respectively; Table 3) could be attributed to the water used during the milk processing chain, from the farm to the plant. Methylobacterium sp. is well adapted to survive in a filtration environment due to its resistance to cleaning agents, which makes it the principal bacterium identified in pink bathroom biofilms (Yano et al. 2013).
Cheese whey in plants E2 and F3 is submitted to a moderate heat treatment prior to its filtration, partly to reduce its microbial load but mainly to reduce chemical fouling (mineral equilibria) (Cheryan 1998). In addition, this whey is bleached by strong oxidizers to remove residual colorant-stabilizing cheese color (Kang et al. 2010). Compared to the milk filtering SWMs studied, a lower ratio of γ-Proteobacteria was observed on whey processing membranes. Therefore, it is hypothesized that the selective pressure induced by the presence of strong oxidizing agents may have influenced the growth of the most sensitive microorganisms found in the whey prior to filtration, enhancing the relative abundance of the Bacilli class found on E2UF-bW and F3UF-bW (Table 3). This led to the growth of more resistant spore-forming bacteria (Bacillus sp., Brevibacillus sp., Paenibacillus sp., and Alicyclobacillus sp.) (Burgess et al. 2010). Interestingly, the filtrations conducted in these two plants were performed at different operating temperatures (48 °C for E2UF-bW and <20 °C for F3UF-bW), which may have modulated, to some extent, the composition of these biofilms. Additional experiments are needed to address the exact impact of these differences even if the PERMANOVA test suggested a significant effect of the temperature of the feed.
The low diversity ecosystem found on UF whey permeate membrane D1NF-P was composed primarily (93.18%) of the thermophilic spore-forming bacteria Anoxybacillus sp. The UF treatment prior to NF certainly contributed to the low diversity observed on D1NF-P since virtually no bacteria should normally cross a UF membrane (Mistry and Maubois 2004). Anoxybacillus sp. was first identified in extreme environments such as hot springs and is often found as a single-species biofilm in milk powder processing lines (Burgess et al. 2010). Its presence on D1NF-P could be due to its intrinsic resistance to alkaline conditions similar to those found during industrial CIP cleaning.
Managers of dairy plant D4 expressed their long-term challenges in restoring the water flux of their membranes after cleaning. Biofouling was suspected for D4RO-fW, which filtered a fluid containing starter cultures and several nutrients. Conversely, filtration of an oligotrophic fluid by D4RO-Wa led to the formation of a similar community. Both membranes were dominated by Streptococcus sp. and were operated at 40 °C, a temperature known to be near optimal for the growth of streptococci such as S. thermophilus (Beal et al. 1989). The D4RO-fW and D4RO-Wa membranes were not cleaned prior to sampling, and consequently, comparisons with membranes from other plants were not possible. However, since these membranes were the only ones to be operated at 40 °C, it is suggested that changing the temperature of the feed in this system may impact the biofilm composition on D4 membranes and, therefore, prevent and control biofouling. This aspect is under investigation in our laboratory with the use of a greater number of membrane samples and replicates operated in a controlled environment.
5 Conclusions
This preliminary study is the first to present a targeted metagenomic analysis of the bacterial composition of biofilms formed on dairy processing membranes. The results suggested a possible effect on biofilm composition caused by the operational environment where the SWMs were used. Similarities were observed at the class level between communities on membranes that filtered the same type of fluid. In addition, similarities were observed between biofilms sampled in the same plant, according to ThetaYC calculator, suggesting that the environmental microflora could also impact biofilm composition. However, additional studies are needed with a larger number of membrane samples and true replicates generated under known cleaning conditions to address the potential impact of the natural microflora of each plant on the composition of their biofilms, similar to what has been observed for cheese microflora (Bokulich and Mills 2013).
This study intended to determine a general qualitative portrait of the biofilms found on SWMs in the dairy industry. Obviously, an analysis including a quantitative aspect would be of great interest in the future considering the major differences between the DNA concentrations observed in this study. However, because of the clear differences observed between the taxonomic composition of each SWMs, it appears that the impact of each operating parameter (fluid type, temperature of the feed, velocity, turbulence, membrane type, etc.) should also be studied in more detail to understand the factors leading to biofouling and to develop new preventive measures and cleaning strategies specifically designed for the dairy industry.
References
Amann R, Ludwig W, Schleifer K (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Mol Biol Rev 59:143–169
Anand S, Singh D (2013) Resistance of the constitutive microflora of biofilms formed on whey reverse-osmosis membranes to individual cleaning steps of a typical clean-in-place protocol. J Dairy Sci 96:6213–6222. doi:10.3168/jds.2013-7012
Anand S, Hassan A, Avadhanula M (2012) The effects of biofilms formed on whey reverse osmosis membranes on the microbial quality of the concentrated product. Int J Dairy Technol 65:451–455. doi:10.1111/j.1471-0307.2012.00848.x
Anand S, Singh D, Avadhanula M, Marka S (2014) Development and control of bacterial biofilms on dairy processing membranes. Compr Rev Food Sci Food Saf 13:18–33. doi:10.1111/1541-4337.12048
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x
Beal C, Louvet P, Corrieu G (1989) Influence of controlled pH and temperature on the growth and acidification of pure cultures of Streptococcus thermophilus 404 and Lactobacillus bulgaricus 398. Appl Microbiol Biotechnol 32:148–154. doi:10.1007/BF00165879
Bégoin L, Rabiller-Baudry M, Chaufer B, Hautbois M-C, Doneva T (2006) Ageing of PES industrial spiral-wound membranes in acid whey ultrafiltration. Desalination 192:25–39. doi:10.1016/j.desal.2005.10.009
Bokulich NA, Mills DA (2013) Facility-specific “house” microbiome drives microbial landscapes of artisan cheesemaking plants. Appl Environ Microbiol 79:5214–5223. doi:10.1128/AEM.00934-13
Burgess SA, Lindsay D, Flint SH (2010) Thermophilic bacilli and their importance in dairy processing. Int J Food Microbiol 144:215–225. doi:10.1016/j.ijfoodmicro.2010.09.027
Chen C-L, Liu W-T, Chong M-L, Wong M-T, Ong SL, Seah H, Ng WJ (2004) Community structure of microbial biofilms associated with membrane-based water purification processes as revealed using a polyphasic approach. Appl Microbiol Biotechnol 63:466–473. doi:10.1007/s00253-003-1286-7
Cheryan M (1998) Ultrafiltration and microfiltration handbook, Second edition. CRC press
Comeau AM, Li WKW, Tremblay JÉ, Carmack EC, Lovejoy C (2011) Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS One 6:e27492. doi:10.1371/journal.pone.0027492
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi:10.1093/bioinformatics/btr381
Ercolini D, Hill PJ, Dodd CER (2003) Bacterial community structure and location in stilton cheese. Appl Environ Microbiol 69:3540–3548. doi:10.1128/AEM.69.6.3540-3548.2003
Ferrera I, Massana R, Balagué V, Pedrós-Alió C, Sánchez O, Mas J (2010) Evaluation of DNA extraction methods from complex phototrophic biofilms. Biofouling 26:349–357. doi:10.1080/08927011003605870
Fouhy F, Deane J, Rea MC, O’Sullivan Ó, Ross RP, O’Callaghan G, Plant BJ, Stanton C (2015) The effects of freezing on faecal microbiota as determined using Miseq sequencing and culture-based investigations. PLoS One 10:1–12. doi:10.1371/journal.pone.0119355
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264. doi:10.1093/biomet/40.3-4.237
Hassan AN, Anand S, Avadhanula M (2010) Microscopic observation of multispecies biofilm of various structures on whey concentration membranes. J Dairy Sci 93:2321–2329. doi:10.3168/jds.2009-2800
Huang L-N, De Wever H, Diels L (2008) Diverse and distinct bacterial communities induced biofilm fouling in membrane bioreactors operated under different conditions. Environ Sci Technol 42:8360–8366. doi:10.1021/es801283q
Ivnitsky H, Katz I, Minz D, Volvovic G, Shimoni E, Kesselman E, Semiat R, Dosoretz CG (2007) Bacterial community composition and structure of biofilms developing on nanofiltration membranes applied to wastewater treatment. Water Res 41:3924–3935. doi:10.1016/j.watres.2007.05.021
Jirjis BF, Luque S (2010) Practical aspects of membrane system design in food and bioprocessing applications. In: Cui ZF, Muralidhara HS (eds). Membrane technology, a practical guide to membrane technology and applications in food and bioprocessing, First edn. Butterworth-Heinemann, Elsevier, pp 179–212
Kang EJ, Campbell RE, Bastian E, Drake MA (2010) Invited review: annatto usage and bleaching in dairy foods. J Dairy Sci 93:3891–3901. doi:10.3168/jds.2010-3190
Katra I, Arotsker L, Krasnov H, Zaritsky A, Kushmaro A, Ben-Dov E (2014) Richness and diversity in dust stormborne biomes at the southeast Mediterranean. Sci Rep 4:1–7. doi:10.1038/srep05265
Khan MT, de O Manes C-L, Aubry C, Gutierrez L, Croue JP (2013) Kinetic study of seawater reverse osmosis membrane fouling. Environ Sci Technol 47:10884–10894. doi:10.1021/es402138e
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the Miseq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi:10.1128/AEM.01043-13
Lappin-Scott HM, Costerton JW (1989) Bacterial biofilms and surface fouling. Biofouling 1:323–342. doi:10.1080/08927018909378120
Letunic I, Bork P (2011) Interactive tree of life v2 : online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi:10.1093/nar/gkr201
Levi A, Bar-Zeev E, Elifantz H, Berman T, Berman-Frank I (2016) Characterization of microbial communities in water and biofilms along a large scale SWRO desalination facility: site-specific prerequisite for biofouling treatments. Desalination 378:44–52. doi:10.1016/j.desal.2015.09.023
Malaeb L, Le-Clech P, Vrouwenvelder JS, Ayoub GM, Saikaly PE (2013) Do biological-based strategies hold promise to biofouling control in MBRs? Water Res 47:5447–5463. doi:10.1016/j.watres.2013.06.033
Manes C-LDO, West N, Rapenne S, Lebaron P (2011) Dynamic bacterial communities on reverse-osmosis membranes in a full-scale desalination plant. Biofouling 27:47–58. doi:10.1080/08927014.2010.536980
Matin A, Khan Z, Zaidi SMJ, Boyce MC (2011) Biofouling in reverse osmosis membranes for seawater desalination: phenomena and prevention. Desalination 281:1–16. doi:10.1016/j.desal.2011.06.063
Metzler-Zebeli BU, Lawlor PG, Magowan E, Zebeli Q (2016) Effect of freezing conditions on fecal bacterial composition in pigs. Animals 6:18. doi:10.3390/ani6030018
Mistry VV, Maubois J-L (2004) Application of membrane separation technology to cheese production. In: Fox PF, McSweeney PLH, Cogan TM, Guinee TP (eds) Cheese: Chemistry, Physics and Microbiology, Third edition, Volume 1: General Aspects. Elsevier, pp 261–285
Mizrahi-Man O, Davenport ER, Gilad Y (2013) Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PLoS One 8:e53608. doi:10.1371/journal.pone.0053608
Quigley L, O’Sullivan O, Beresford TP, Ross RP, Fitzgerald GF, Cotter PD (2012) High-throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl Environ Microbiol 78:5717–5723. doi:10.1128/AEM.00918-12
Quigley L, McCarthy R, O’Sullivan O, Beresford TP, Fitzgerald GF, Ross RP, Stanton C, Cotter PD (2013) The microbial content of raw and pasteurized cow milk as determined by molecular approaches. J Dairy Sci 96:4928–4937. doi:10.3168/jds.2013-6688
Raats D, Offek M, Minz D, Halpern M (2011) Molecular analysis of bacterial communities in raw cow milk and the impact of refrigeration on its structure and dynamics. Food Microbiol 28:465–471. doi:10.1016/j.fm.2010.10.009
Rasolofo EA, St-Gelais D, LaPointe G, Roy D (2010) Molecular analysis of bacterial population structure and dynamics during cold storage of untreated and treated milk. Int J Food Microbiol 138:108–118. doi:10.1016/j.ijfoodmicro.2010.01.008
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi:10.1128/AEM.01541-09
Sharma M, Anand SK (2002) Characterization of constitutive microflora of biofilms in dairy processing lines. Food Microbiol 19:627–636. doi:10.1006/fmic.2002.0472
Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A (2013) DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One 8:e62692. doi:10.1371/journal.pone.0062692
Tang X, Flint SH, Brooks JD, Bennett RJ (2009) Factors affecting the attachment of micro-organisms isolated from ultrafiltration and reverse osmosis membranes in dairy processing plants. J Appl Microbiol 107:443–451. doi:10.1111/j.1365-2672.2009.04214.x
Tang X, Flint SH, Bennett RJ, Brooks JD (2010) The efficacy of different cleaners and sanitisers in cleaning biofilms on UF membranes used in the dairy industry. J Memb Sci 352:71–75. doi:10.1016/j.memsci.2010.01.063
Teh KH, Flint S, Palmer J, Andrewes P, Bremer P, Lindsay D (2014) Biofilm—an unrecognised source of spoilage enzymes in dairy products? Int Dairy J 34:32–40. doi:10.1016/j.idairyj.2013.07.002
Tully BJ, Heidelberg JF (2013) Microbial communities associated with ferromanganese nodules and the surrounding sediments. Front Microbiol 4:1–10. doi:10.3389/fmicb.2013.00161
Vanysacker L, Boerjan B, Declerck P, Vankelecom IFJ (2014a) Biofouling ecology as a means to better understand membrane biofouling. Appl Microbiol Biotechnol 98:8047–8072. doi:10.1007/s00253-014-5921-2
Vanysacker L, Declerck P, Bilad MR, Vankelecom IFJ (2014b) Biofouling on microfiltration membranes in MBRs: role of membrane type and microbial community. J Memb Sci 453:394–401. doi:10.1016/j.memsci.2013.11.024
Wolfe BE, Button JE, Santarelli M, Dutton RJ (2014) Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158:422–433. doi:10.1016/j.cell.2014.05.041
Yano T, Kubota H, Hanai J, Hitomi J, Tokuda H (2013) Stress tolerance of Methylobacterium biofilms in bathrooms. Microbes Environ 28:87–95. doi:10.1264/jsme2.ME12146
Acknowledgments
The authors want to thank Agropur, Parmalat, and Saputo for providing the SWM elements for this project. Also, we are grateful to Barb Conway for the editing of this manuscript and to Marc-André Gardner and Félix-Antoine Fortin (Calcul Québec) for their support with the use of supercomputer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the NSERC-Novalait Industrial Research Chair on Process Efficiency in Dairy Technology (Grant IRCPJ 461630–12 to Yves Pouliot) and the Canadian Dairy Commission and Novalait through a M. Sc. scholarship to Julien Chamberland (Grant 187,210). The operation of the supercomputers of Calcul Québec is funded by the Canada Foundation for Innovation (CFI), Le ministère de l’Économie, de l’Innovation et des Exportations du Québec (MEIE), RMGA, and the Fonds de recherche du Québec—Nature et technologies (FRQ-NT).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Fig. S1
(PNG 231 kb)
About this article

{kind=link}
Cite this article
Chamberland, J., Lessard, MH., Doyen, A. et al. A sequencing approach targeting the 16S rRNA gene unravels the biofilm composition of spiral-wound membranes used in the dairy industry. Dairy Sci. & Technol. 96, 827–843 (2017). https://doi.org/10.1007/s13594-016-0305-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13594-016-0305-2