
Abstract
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS), characterized by demyelination, gliosis, and neurodegeneration. While the currently available disease-modifying therapies effectively suppress the immune attack on the CNS, there are no therapies to date that directly mitigate neurodegeneration. Glucagon-like peptide-1 (GLP-1) is a small peptide hormone that maintains glucose homeostasis. A novel GLP-1 receptor (GLP-1R) agonist, NLY01, was recently shown to have neuroprotective effects in the animal models of Parkinson’s disease and is now in a phase 2 clinical trial. In this study, we investigated the therapeutic potential of NLY01 in a mouse model of MS, experimental autoimmune encephalomyelitis (EAE). Our data show that NLY01 delays the onset and attenuates the severity of EAE in a prevention paradigm, when given before disease onset. NLY01 inhibits the activation of immune cells in the spleen and reduces their trafficking into the CNS. In addition, we show that NLY01 suppresses the production of chemokines that are involved in leukocyte recruitment to the site of inflammation. The anti-inflammatory effect of NLY01 at the early stage of EAE may block the expression of the genes associated with neurotoxic astrocytes in the optic nerves, thereby preventing retinal ganglion cell (RGC) loss in the progressive stage of EAE. In the therapeutic paradigm, NLY01 significantly decreases the clinical score and second attack in a model of relapsing–remitting EAE. GLP-1R agonists may have dual efficacy in MS by suppressing peripheral and CNS inflammation, thereby limiting neuronal loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by demyelination, gliosis, and neurodegeneration. It most often presents initially with a relapsing–remitting course, where the relapsing phases are defined by inflammatory attacks of peripheral immune cells directed against the myelin sheaths wrapped around neurons [1]. The progressive stage of MS usually evolves from the relapsing–remitting stage and involves worsening disability at the later stage. In the last few decades, several disease-modifying therapies have emerged that suppress new relapses and slow down the disease progression in people with MS [1, 2]. These therapies have broad immunomodulatory effects, mainly targeting lymphocyte activation and trafficking to the CNS. However, their therapeutic potential is mainly limited to the inflammatory phase of MS, while their benefits in the progressive phase of the disease are limited [3]. Neurodegeneration is the hallmark of progressive MS, characterized by continuous neurological deterioration and permanent disability. Several studies have recently demonstrated that the degenerative mechanisms underlying the progression of MS are distinct from the inflammatory mechanisms that cause the relapsing–remitting course. One of the potential mechanisms involved in the progression of MS is the chronic inflammation of glial cells, including microglia and astrocytes that contribute to neurodegeneration [4,5,6]. Activated microglia release C1q, TNFα, and IL-1α, polarizing astrocytes away from their supportive and neuroprotective phenotype towards an inflammatory profile that promotes neurodegeneration [4]. Therefore, targeting the immunopathological mechanisms that are involved in the chronic activation of glia could be a potential treatment to promote neuroprotection.
Glucagon-like peptide-1 (GLP-1) is a small peptide hormone that is mainly secreted from L cells in the intestine and increases insulin secretion following food intake. GLP-1 has a very short half-life (about 2 min) and acts through binding to its receptor (GLP-1R), which is expressed in pancreatic islets, gastrointestinal tract, heart, CNS, and lymphoid organs (thymus, bone marrow, lymph node, and spleen) [4, 7−9]. Several long-acting GLP-1R agonists have been developed that are currently approved for the treatment of type-2 diabetes including exenatide (exendin-4), liraglutide, lixisenatide, and dulaglutide [10]. Interestingly, GLP-1 is also an active neuropeptide, produced in the nucleus of the solitary tract in the hindbrain, where it acts as a key regulator of many autonomic and neuroendocrine functions [11]. Extensive studies have demonstrated that activation of the GLP-1R signaling axis leads to protection against neuroinflammation, oxidative stress, and neurotoxicity [12]. GLP-1 has recently gained significant attention as a novel drug candidate for the treatment of neurodegenerative diseases, due to its anti-inflammatory and neuroprotective actions [13,14,15]. We have recently developed a novel long-acting GLP-1R agonist, NLY01, which is a PEGylated form of exendin-4 [16]. NLY01 has an impressive half-life of 38 h in mice and 12.5 days when administered subcutaneously in humans. Recent studies show that NLY01 penetrates the blood–brain barrier, ameliorates clinical symptoms, and protects neurodegeneration in mouse models of Parkinson’s disease (PD) [17] and Alzheimer’s disease (AD) [18]. It has been shown that NLY01 inhibits inflammatory microglial activity and blocks the conversion of astrocytes towards the neurotoxic phenotype that causes neuronal death [17, 18]. Given the pathological role of peripheral immune cells and inflammatory glia in MS, we hypothesized that NLY01 may suppress neuroinflammation and prevent neurodegeneration in MS. In this study, we investigated the neuroprotective and anti-inflammatory effects of NLY01 in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS.
Materials and Methods
Mice
C57BL/6 J and SJL/J mice were purchased from The Jackson Laboratory. The animals were housed in the pathogen-free animal facility at the Johns Hopkins University School of Medicine. The mice were kept in 12 h/12 h light/dark cycles and fed with standard food and water. All experimental protocols were performed in accordance with the National Institutes of Health guidelines for the use of experimental animals and were approved by the Johns Hopkins Institutional Animal Care and Use Committee.
EAE Induction
Chronic EAE was induced in 9-week-old C57BL/6 J mice with myelin oligodendrocyte glycoprotein (MOG35–55) immunization as previously described [5]. Briefly, mice were immunized on day 0 subcutaneously at two sites over the lateral abdomen with an emulsion of MOG (200 μg) in complete Freund’s adjuvant (CFA) containing 400 μg of Mycobacterium tuberculosis H37RA (Difco Laboratories). Pertussis toxin (250 ng; List Biological Labs, Campbell, CA, USA) was intraperitoneally injected on days 0 and 2. To induce relapsing–remitting EAE, female SJL/J mice (8-week-old) were immunized subcutaneously with 100 µg of PLP139-151 peptide with CFA containing 200 µg Mycobacterium tuberculosis. All immunized mice were weighed and scored daily using the established standard scoring scale from 1 to 5 as follows: 0, no signs of disease; 1, loss of tail tonicity; 2, loss of tail tonicity and mild paralysis of hind limbs; 3, paralysis of hind limbs; 4, hind limb paralysis and mild paralysis of forelimbs; and 5, complete paralysis or death [19]. Mice were sacrificed at post-immunization days (PID) 6, 11, or 42, corresponding to the early or late phase of EAE.
NLY01 Treatment
NLY01 was obtained from Neuraly Inc. (Gaithersburg, MD). Mice were treated subcutaneously with NLY01 (10 mg/kg) or the vehicle (phosphate-buffered saline, PBS) twice weekly. In the prevention paradigm, we started the treatment NLY01 or vehicle at the time of EAE immunization or at PID 8. In the treatment paradigm, mice were treated with NLY01 or vehicle individually once a clinical score of 1 was reached.
Flow Cytometric Analysis of Peripheral Immune Cells
Mice were euthanized with isoflurane, and spleens were collected in a FACS buffer containing PBS buffer containing 2% fetal bovine serum (FBS) and 2 mM EDTA. Single-cell suspensions were generated by passing the spleen through a 70-μm nylon sieve and red blood cells (RBC) were lysed using RBC lysis buffer (BioLegend). Cells were incubated with anti-mouse CD16/32 antibody (Fc Block, BioLegend) for 15 min prior to staining with fixable live–dead stain for 20 min at room temperature. The cells were then stained with antibodies against surface markers (30 min at room temperature). All the antibodies were purchased from BioLegend. Flow cytometry was performed on a MACSQuant 10 (Miltenyi Biotec) or a 3-laser Cytek® Aurora spectral flow cytometer. The data were analyzed using the FlowJo 10.6.0 software.
Flow Cytometric Analysis of CNS-Infiltrating Mononuclear Cells
Mice were euthanized with isoflurane and then perfused transcardially with cold HBSS without cations. Spinal cords were flushed from the column with hydrostatic pressure. Tissue was mechanically dissociated by passing through a 70-μm nylon sieve and then enzymatically digested with collagenase IV (0.5 mg/ml, Worthington) and DNAse I (100 U/ml, Worthington) for 30 min at 37 °C with mixing after 15 min. After washing the enzymes out, cells were separated from myelin debris using debris removal solution (Miltenyi Biotec) and the cellular pellet was collected. The cells were stained with viability dye and antibodies against surface markers and run on a MACSQuant 10 flow cytometer (Miltenyi Biotec). The data were analyzed with the FlowJo software.
Flat Mount Retina and Retinal Ganglion Cell Counting
Mice were anesthetized with isoflurane gas and perfused transcardially with 30 ml PBS. After perfusion, the eyes were removed from the eye socket using curved forceps and immediately placed into 4% paraformaldehyde (PFA) for 4 h and then transferred to 30% sucrose in PBS until the tissue sank. Whole retinas were dissected from the eyes and then processed for retinal ganglion cell (RGC) staining as described previously [5]. Briefly, retinas were permeabilized with PBS containing 3% Triton X-100 and blocked with PBS containing 5% normal goat serum and 1% Triton X-100 for 1.5 h at room temperature with shaking. Sections were incubated with anti-Brn3a antibody (Synaptic Systems #411 003, 1:1000) for 72 h at 4 °C with shaking. After washing with PBS, the sections were then incubated with a secondary antibody, Alexa Fluor 488 (Invitrogen #A11001, 1:1000) for 1 h at room temperature followed by nucleus staining with Hoechst. The sections were mounted using the Aqua-Poly/Mount reagent (Polysciences, Warrington, PA, USA). The whole-mount retinas were imaged using a Zeiss Axio Observer Z1 epifluorescence microscope, and images were saved in TIF format. In each retina section, 12 regions (central, middle, and peripheral; 4 locations each) were selected and the number of RGCs was determined using a semi-automated MATLAB algorithm previously developed by our group [5].
Quantification of Gene Expression by qPCR
RNA was isolated from the hindbrain and optic nerves using the RNeasy Plus Mini Kit and RNeasy Plus Micro Kits (QIAGEN), respectively, and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad). The expression levels of the target genes were quantified using iQ SYBR Green Supermix (Bio-Rad) and primer sequences (Integrated DNA Technologies, IDT) that are listed in Supplemental Table S1. The expression levels of target genes were normalized to the expression of β-actin and calculated based on the comparative cycle threshold Ct method (2−ΔΔCt).
Western Blotting
Following dissection of the brain and spinal cord, tissues were lysed with ice-cold RIPA lysis buffer (Sigma-Aldrich) with protease and phosphatase inhibitors (Boston BioProducts, Inc.). After centrifuging the tissue lysate at 14,000 g for 10 min at 4 °C, the protein concentration of the supernatant was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were run on SDS polyacrylamide gels (10%) and transferred onto 0.45-µm nitrocellulose membranes with the Trans-Blot Turbo system (Bio-Rad Laboratories, Hercules, USA). Blots were blocked with 5% bovine serum albumin (Sigma-Aldrich) in Tris-buffered saline (TBS, Quality Biological) solution with 0.002% Tween 20 for 1 h at room temperature. Blots were then incubated in primary antibody diluted in 5% bovine serum albumin TBS solution with 0.002% Tween 20 overnight at 4 °C. Blots were then incubated with LiCOR secondary antibodies for 30 min at room temperature and were visualized using the LiCOR Odyssey CXL imaging system software (Lincoln, NE, USA). For each protein of interest, the density value was normalized to the related density of the loading control to obtain the integrated density values. The primary antibodies were anti-GLP-1R (Thermo Fisher Scientific #PA5-33,591, 1:1000) and anti-β-actin (Millipore-Sigma #A2228, 1:2000).
T Cell Activation and DifferentiationIn Vitro
Spleens and lymph nodes were removed from 3 naïve mice, and single-cell suspensions were generated by passing cells through a 70-μM cell strainer (BD Biosciences). T cells were isolated using negative selection with a CD4+ T-Cell Isolation Kit (MojoSort, BioLegend) following the manufacturer’s protocol and cultured in Roswell Park Memorial Institute media (RPMI) 1640 (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS (Gemini Bio-Products), 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific), and 0.5 μM 2-mercaptoethanol (Invitrogen). To measure proliferation via flow cytometry, CD4 + T cells previously labeled with cell proliferation dye eFluor 450 (Thermo Fisher) were cultured in 96-well flat-bottom plates previously coated with 1 µg/ml anti-CD3 in the presence of 2 μg/ml anti-CD28 antibody (BD Biosciences) ± NLY01 (0.5, 5, 50 μM) at a density of 150,000 cells/well. To specifically activate T cells by antigen, we isolated CD4 T cells from MOG-specific TCR transgenic mice (2D2 mice) and activated with MOG-pulsed splenocytes. Splenocytes were prepared from wild-type spleen and incubated with MOG (20 µg/ml) and the drug (NLY01 or vehicle) for 30 min at 37 °C. CD4+ T cells were then added to splenocytes (1:1 ratio) and incubated for 72 h at 37 °C.
To differentiate CD4+ T cells into T helper 1 (Th1), cells were treated with recombinant IL-2 (PeproTech, 5 ng/ml), IL-12 (PeproTech, 10 ng/ml), and anti-IL-4 antibody (BioLegend, 10 µg/ml) for 3 days. For polarizing into regulatory T cells (Treg), CD4+ T cells were treated with recombinant human TGFβ (Invitrogen, 10 ng/ml) and murine IL-2 (PeproTech, 10 ng/ml) for 5 days.
For flow cytometric analysis of surface markers, cells were stained with a viability dye (Live/Dead Aqua, Thermo Fisher) followed by fluorochrome-conjugated antibodies (BioLegend) and analyzed on a flow cytometer (MACSQuant Analyzer X, Miltenyi Biotec). For intracellular staining of IFNγ, cells were collected and stimulated with cell stimulation cocktail plus protein transport inhibitors (Thermo Fisher Scientific) for 5 h at 37 °C. Following stimulation, cells were washed and stained for the surface markers in FACS buffer for 30 min at room temperature. Cells were then fixed using Intracellular (IC) Fixation Buffer (BD Bioscience) at 4 °C for 30 min followed by permeabilization and staining with anti-IFNγ antibody (BioLegend) for 30 min at room temperature. After washing with permeabilization buffer and FACS buffer, cells were analyzed by flow cytometry. For staining nuclear markers (FoxP3, Tbet, and Ki67), cells were incubated with FoxP3 fixation/permeabilization buffer (Thermo Fisher Scientific) for 1 h at 4 °C. After washing with FoxP3 permeabilization buffer (Thermo Fisher Scientific), cells were stained with intracellular markers as described above. The samples were run on a MACSQuant flow cytometer (Miltenyi Biotec), and data were analyzed with the FlowJo 10 software.
Data Analyses
Statistical analyses were completed using GraphPad Prism 8.3.0. Comparisons of EAE scores between groups in the in vivo treatment studies by performing a Mann–Whitney U test on the cumulative scores. Other comparisons between groups were performed using Student’s t test, and P-values < 0.05 were considered statistically significant.
Results
NLY01 Delays the Onset and Attenuates the Severity of EAE in a Prevention Paradigm
To evaluate whether NLY01 treatment affects the onset of EAE, C57BL/6 J mice were immunized with MOG35-55/CFA and treated with NLY01 or vehicle. In the early prevention paradigm, we started NLY01 treatment at the time of MOG35-55/CFA immunization (PID 0) and recorded the daily behavioral scores and the weight of mice for 42 days. The results revealed that NLY01 significantly delayed the onset and attenuated the severity of EAE (Fig. 1a). However, starting the treatment at the later time point (PID 8) resulted in a modest effect of NLY01 on the clinical scores of EAE, which was not significant compared to the effect of the vehicle (Fig. 1b).

NLY01 delays the onset and attenuates the severity of EAE in an early prevention paradigm. C57BL/6 mice were immunized with MOG35–55/CFA emulsion and pertussis toxin to induce EAE. (a) The behavioral plot and cumulative scores of the mice treated with NLY01 or vehicle twice weekly starting at the day of EAE immunization (PID 0) and continued for 42 days. The behavior scores are presented as mean ± SEM of three independent experiments (vehicle, n = 26; NLY01, n = 25); ****P ≤ 0.0001 by Mann–Whitney U test. (b) The behavioral plot and cumulative scores of the mice treated with NLY01 or vehicle twice weekly starting at day 8 (PID 8) and continued for 42 days. The behavior scores are presented as mean ± SEM. Each dot represents one mouse
GLP-1R Is Expressed in the Periphery and the CNS During EAE
After finding that NLY01 affected disease onset and severity in the prevention paradigm, we next tested whether MOG immunization affects the expression of GLP-1R during the course of EAE. We first evaluated the expression of GLP-1R in the lymph node at the early stage of EAE when priming of the immune response occurs. We found no significant difference in the percentage of CD45+GLP-1R+ cells between naïve and EAE mice at PID 8 using flow cytometry (Fig. 2a, b). We also quantified the expression of Glp-1r by qPCR and found no difference in the mRNA levels of Glp-1r in the lymph nodes of naïve and EAE mice at PID 8 (Fig. 2c). In the spinal cord, we evaluated the expression of GLP-1R at PID 8 by western blot and found no change between the naïve and EAE mice. However, GLP-1R expression was significantly decreased in the spinal cord of sick mice (clinical scores, 1.5–3.25; average 2.37) at the late stage of EAE (PID 42) (Fig. 2d). The expression of GLP-1R remained unchanged in the brain at the early and late stages of EAE compared with that of naïve mice (Fig. 2e).

GLP-1R is expressed in the periphery and the CNS of EAE mice. (a) Representative flow cytometry plots demonstrating the percentage of CD45+GLP1R+ cells in the lymph node at PID 8. (b) The percentage of CD45+GLP-1R+ cells in the lymph nodes quantified by flow cytometry. (c) The mRNA levels of Glp1r in the lymph nodes of naïve and EAE mice quantified by qPCR. (d) The protein levels of GLP-1R in the spinal cord and (e) the brain of EAE mice at PID 8 and PID 42 compared with those of naïve mice. The intensity of GLP-1R band was normalized to the level of β-actin and presented as mean ± SEM. Each dot represents one mouse; *P ≤ 0.05 by Student’s t test
NLY01 Suppresses the Activation of Immune Cells in the Periphery
Next, we examined whether the effect of NLY01 on the early stage of EAE was associated with its anti-inflammatory activity in the periphery. We evaluated the activation of myeloid cells in the spleen of mice treated with NLY01 or the vehicle during the priming phase of EAE (PID 6) and found a significant reduction in the spleen weight and the absolute number of CD45+ splenocytes in mice treated with NLY01 compared with those of vehicle-treated mice (Fig. 3a, b). Among CD45+ cells, the percentage and absolute counts of CD11b+ cells, including neutrophils (CD11b+Ly6G+ cells), activated myeloid cells (CD11b+MHCII+), and dendritic cells (CD11b+CD11c+), were significantly diminished by NLY01 treatment (Fig. 3c). At the pre-onset stage, PID 11, flow cytometry revealed that NLY01 significantly reduced the percentage of activated myeloid cells (CD11b+MHCII+), mature dendritic cells (CD11c+MHCII+), and effector T cells (CD4+CD44+) in the spleen of NLY01-treated mice compared with that of the vehicle group (Fig. 4). We found no change in the percentage of CD4+CD25+FoxP3+ Treg in the NLY01 group compared to that in the vehicle group (Fig. S1). Our in vitro findings revealed that NLY01 had no direct effect on the activation, proliferation, and differentiation of T cells into Th1 or Treg (Fig. S2). At PID 42, the percentage of T cells and activated myeloid cells in the spleen were comparable between groups (Fig. S3a).

NLY01 reduces the activation of immune cells in the spleen at the very early stage of EAE. EAE mice were treated with NLY01 or vehicle twice weekly starting at the time of immunization. The mice were euthanized on day 6 after immunization (PID 6). (a) Representative image of spleen from EAE mice treated with NLY01 or vehicle and euthanized after 6 days. (b) The spleen weight and the absolute counts of live cells and CD45+ cells in the treatment groups at PID 6. (c) Representative flow cytometric analysis of CD11b+ myeloid cells in the spleen of EAE mice treated with NLY01 or vehicle at PID 6. All the data are presented as mean ± SEM. Each dot represents one mouse. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, determined by Student’s t test

NLY01 reduces the activation of immune cells in the spleen at the pre-onset stage of EAE. The mice were treated with NLY01 or vehicle twice weekly at the time of immunization. The mice were euthanized on day 11 after immunization (PID 11). Representative flow cytometric analysis of CD11b+ myeloid cells and CD4+ T cells in the spleen of EAE mice treated with NLY01 or vehicle. All the data are presented as mean ± SEM. Each dot represents one mouse. *P ≤ 0.05, **P ≤ 0.01, determined by Student’s t test
NLY01 Inhibits the Activation and Trafficking of Immune Cells into the CNS
Since we found evidence for anti-inflammatory activity of NLY01 in the spleen, we next investigated whether NLY01 would suppress the infiltration and trafficking of activated immune cells into the CNS. In the brain, we found a lower percentage of infiltrating leukocytes (CD45high) and monocytes (Clec12a+) in the NLY01-treated group compared with that in the vehicle group (Fig. 5a, b). While the percentage of activated myeloid cells (CD11b+MHCII+) in the brain did not change following NLY01 treatment, we found a lower percentage of effector/memory T cells (CD4+CD44+) in the brain of mice treated with NLY01 compared to that of mice treated with the vehicle (Fig. 5a, b). Similar results were observed in the spinal cord of NLY01-treated mice compared to the vehicle group at PID 11 (Fig. 5c). The percentage of effector/memory T cells and activated myeloid cells in the CNS of NLY01- or vehicle-treated mice were comparable at the late stage of EAE (PID 42) (Fig. S3). Since astrocyte-derived chemokines play important roles in the recruitment of immune cells into the CNS [20], we tested whether NLY01 treatment reduced astrogliosis and the expression of chemokines associated with recruiting leukocytes in the brain of EAE mice. We found that the expression of reactive astrocyte markers Lcn2 and Gfap was significantly downregulated in the hindbrain of EAE mice by NLY01 treatment (Fig. 5c). Moreover, we found a significant reduction in the expression of Ccl8 and Cxcl1 chemokines in the hindbrain of mice treated with NLY01 while the expression of Cxcl10 remained unchanged (Fig. 5c). We found, however, no significant difference in the expression of genes associated with the neurotoxic astrocyte profile and activated monocyte/microglia in the hindbrain of mice treated with NLY01 compared with that of the vehicle group at PID 11 (Fig. S4).

NLY01 reduces the activation and trafficking of immune cells into the CNS. EAE mice were treated with NLY01 or vehicle twice a week starting at the time of immunization. The mice were euthanized 11 days after immunization (PID 11). (a) Representative flow cytometric plots and the percentage of infiltrating immune cells into the brain of EAE mice treated with NLY01 or vehicle. (b) The percentage of infiltrating myeloid cells and activated T cells in the spinal cord of EAE mice treated with NLY01 or vehicle. (c) The expression of reactive astrocyte markers and chemokines in the hindbrain of EAE mice treated with NLY01 or vehicle. All the data are presented as mean ± SEM. Each dot represents one mouse. *P ≤ 0.05, determined by Student’s t test
NLY01 Attenuates RGC Loss at the Late Stage of EAE
We have recently found the neurotoxic astrocyte profile in the optic nerve of EAE mice, which was associated with a significant RGC loss in the late stage of EAE [5]. Previous studies showed that NLY01 blocks microglial activation and the formation of neurotoxic astrocytes in PD and AD experimental models [17, 18]. Accordingly, we examined whether NLY01 may prevent RGC loss associated with the neurodegenerative stage of EAE. We found that NLY01 significantly reduced RGC loss in the retina of EAE mice, compared with the vehicle. The neuroprotective effect of NLY01 was more prominent in the central and peripheral regions of the retina (Fig. 6a, b). We next investigated whether NLY01 affects the expression of genes associated with the neurotoxic astrocytes at the onset of EAE. We quantified the mRNA levels of inflammatory mediators and neurotoxic astrocyte genes in the optic nerves from NLY01- or vehicle-treated mice at PID 11. We found that a number of genes associated with neurotoxic astrocytes, including C3, Ligp1, PSMB8, H2-T23, and H2-D1, were significantly downregulated in optic nerve samples from the NLY01 group compared to those from the vehicle group (Fig. 6c). The expression of Tnfα was markedly decreased in the optic nerves of NLY01-treated mice compared to that of the vehicle group (Fig. 6c).

NLY01 attenuates retinal ganglion cell (RGC) loss at the late stage of EAE. EAE mice were treated with NLY01 or vehicle twice a week starting at the time of immunization. The mice were euthanized at PID 42. (a) Retinal flat mounts were prepared from one eye per animal per time point. RGCs (Brn3a +) were counted using a semi-automated quantitative algorithm in 12 specified regions in the center, middle, and periphery of the retina. The mean RGC count of all regions was calculated for each animal. (b) The number of RGCs in the retina dissected from EAE mice treated with NLY01 or vehicle at PID 42. The comparisons are presented as the average of RGC counts of the total 12 regions as well as in the different regions of the retina. The data are pooled from 2 independent experiments. Each dot represents one mouse. (c) The expression of genes associated with neurotoxic astrocytes and reactive microglia in the optic nerve of EAE mice, quantified by qPCR. All the data are presented as mean ± SEM. Each dot represents one mouse. *P ≤ 0.05, **P ≤ 0.01 by Student’s t test
NLY01 Ameliorates the Severity of Relapsing–Remitting EAE in a Therapeutic Paradigm
To determine whether NLY01 would reduce the severity of clinical scores after the induction of EAE, we investigated NLY01 efficacy in a therapeutic paradigm, in which mice were treated with NLY01 after EAE onset. We studied the therapeutic effect of NLY01 in two EAE models: chronic progressive EAE in C57BL/6 mice and relapsing–remitting EAE in SJL mice. We immunized C57BL/6 mice with MOG35–55/CFA emulsion and pertussis toxin and started NLY01 treatment either at the onset of EAE (clinical score of 1) (Fig. 7). We found a modest effect of NLY01 on the clinical scores of chronic progressive EAE. However, we found a significant reduction in the severity of EAE in the relapsing–remitting model, in which SJL mice were immunized with PLP139-151 and treated with NLY01 or vehicle treatment at the peak of the disease (days 11–15). Following NLY01 treatment, the severity of the clinical score was dramatically reduced in the NLY01 group while it remained unchanged in the vehicle group (Fig. 7).

NLY01 in therapeutic paradigm. (a) C57BL/6 mice were immunized with MOG35–55/CFA emulsion and pertussis toxin to induce EAE. Mice were treated individually at the onset of EAE (clinical score of 1). The clinical scores of the mice presented as line graph, NLY01 (n = 10) and vehicle (n = 9). The cumulative behavior scores are presented as mean ± SEM. Each dot represents one mouse. (b) NLY01 treatment in SJL relapsing remitting EAE model. SJL mice were immunized with PLP 139–151 and started NLY01 (10 mg/kg) or vehicle treatment at the peak of the disease (days 11–15). The cumulative scores were compared between phase 1 (P1; days 11–19) and phase 2 (P2; days 20–30) after treatment by NLY01 (n = 8) or vehicle (n = 7) using Wilcoxon matched-pairs signed rank test; **P ≤ 0.01
Discussion
GLP-1R agonists are already approved for the treatment of type 2 diabetes. Previous studies have demonstrated the potential neuroprotective effects of GLP-1R agonists in neurologic disorders such as AD and PD [14]. The current study identified the therapeutic benefit of a novel GLP-1R agonist, NLY01, in MS experimental models. We found that NLY01 inhibits peripheral immune response at the early stage and reduces neurodegeneration at the late phase of EAE.
We first examined the efficacy of NLY01 treatment in the MOG-induced EAE model, in which NLY01 was administered in an early prevention paradigm at the day of immunization. We found a significant delay in the onset and reduced severity of EAE in the NLY01 group compared with those of the vehicle group. However, no difference was detected in the clinical scores between NLY01 and vehicle groups when the treatments started later, at PID 8. This finding is consistent with a previous report showing that a GLP-1R agonist, dulaglutide, barely maintains its protective effect on the EAE clinical course when administrated at PID 9 [15]. This finding highlights the beneficial effect of NLY01 before the onset and at the early stage of EAE, when peripheral immune cells are activated in the periphery. A previous study reported a similar finding in MOG-immunized mice, showing that the treatment with dulaglutide (0.18 mg/kg, twice per week) delays EAE onset [15]. Similarly, in myelin basic protein-immunized Lewis rats, liraglutide (0.2 mg/kg, twice daily) was shown to be effective in delaying EAE onset [21]. Our results are consistent with previous studies and suggest that GLP-1R agonists suppress the early inflammatory pathways that initiate CNS pathology.
We next investigated whether the expression of GLP-1R changes during the course of EAE. It is shown that GLP-1R is highly expressed in microglia and relatively less in neurons and astrocytes [17]. In the spinal cord, we found no change in the level of GLP-1R at the early stage of EAE, while its expression was significantly decreased in the sick mice at the late stage of the disease. This result is consistent with a previous study, demonstrating that GLP-1R expression is downregulated in the damaged spinal cord upon EAE challenge [22]. Unlike that in the spinal cord, the GLP-1R expression remained unchanged in the brain during the course of EAE. We found no difference in the mRNA levels of Glp-1r in the lymph nodes of naïve and EAE mice at PID 8. These results suggest that the NLY01 receptor is highly expressed by peripheral immune cells and the CNS resident cells.
Since NLY01 effectively delayed the onset of EAE, we investigated whether its protective effect is mediated during the priming phase of the immune response in the periphery, where the immune cells are activated and begin to enter the CNS. We found a dramatic reduction in the spleen size and weight of the NLY01 treatment group compared with those of the vehicle group. More specifically, we found a significant reduction in the absolute numbers of CD45+ cells including neutrophils (CD11b+Ly6G+), activated myeloid cells (CD11b+MHCII+), and dendritic cells (CD11b+CD11c+) in the spleen of the NLY01 group compared with those of the vehicle group. This finding suggests that NLY01 suppresses the activation and expansion of innate immune cells in the myeloid lineage at the very early stage of EAE. It has been shown that immunization with MOG-CFA induces a significant expansion of circulating neutrophils and monocytes in the blood and spleen from preclinical time points through EAE onset [23]. This occurs in response to systemic upregulation of granulocyte colony-stimulating factor (G-CSF) and CXCL1 chemokine [23]. Our results are consistent with reports that GLP-1 attenuates neutrophil activation and decreases PMN accumulation in ischemic and reperfused myocardium [24]. These results extend the anti-inflammatory activities of GLP-1R agonists to the very early priming events including the activation of neutrophils, monocytes/macrophages, and dendritic cells, suggesting a broader mechanism of action.
The anti-inflammatory effect of NLY01 appears to be persistent in the spleen since a reduction in the percentage and absolute numbers of activated myeloid cells (CD11b+MHCII+) was still detectable at PID 11. Additionally, we observed a reduction in the percentage and absolute counts of effector/memory T cells (CD4+CD44+) in the spleen of mice treated with NLY01 compared with those treated with the vehicle. We found, however, no effect of NLY01 on the expansion of Treg cells in the spleen of EAE mice at PID 11. This finding is consistent with a previous study showing that a GLP-1R agonist, dulaglutide, did not change the percentage of Treg cells in the CNS of EAE mice [15]. Moreover, we found no direct effect of NLY01 on T cell activation and proliferation in vitro, suggesting that the observed effects in vivo may be mediated indirectly through other cell types. It is shown that dulaglutide reduces the number of macrophages (both M1 and M2) in the CNS [15]. These findings support the notion that GLP-1R agonists may prevent the activation of myeloid cells in the periphery at the early stage of EAE [15].
Next, we assessed whether NLY01 inhibits the infiltration of inflammatory cells into the CNS. A previous study showed dulaglutide treatment reduces lymphocyte infiltration into the CNS at PID 14 [15]. It was shown that the development of highly encephalitogenic Th1/Th17 cells in the CNS are significantly suppressed in dulaglutide-treated mice, suggesting a critical role in the modulation of T cell pathogenicity in the CNS [15]. We found a lower percentage of infiltrating leukocytes (CD45high cells) and effector/memory T cells (CD4+CD44+) in the CNS of NLY01-treated mice compared with that of vehicle-treated mice. These values are consistent with lower clinical scores in EAE mice receiving NLY01. We also found a decreased percentage of Clec12a+ cells in EAE mice treated with NLY01 compared with that of mice treated with the vehicle. Clec12a is particularly important for the migration of myeloid cells into the CNS tissues during inflammation [25]. We then tested whether NLY01 would affect the level of chemokines that recruit immune cells into the CNS. Our qPCR data showed a reduced expression of Ccl8 and Cxcl1 chemokines in the hindbrain of EAE mice treated with NLY01 compared to that of the vehicle group. Taken together, NLY01 treatment significantly reduces the infiltration of peripheral immune cells into the CNS. We also found that the expression of an astrocytic marker, Gfap, in the hindbrain of EAE mice treated with NLY01 was significantly reduced, suggesting the inhibitory effect of NLY01 on astrogliosis. Given the fact that astrocytes serve as a potent source of chemokines in the inflamed CNS, NLY01 might suppress leukocyte recruitment to the CNS via inhibiting astrocyte activation and inflammatory function. The gene expression analysis by qPCR warrants further analysis at the protein level.
We next investigated the potential therapeutic effect of NLY01 on neurodegeneration. Retinal degeneration and RGC loss are common features of many neurodegenerative disorders including MS [26]. Emerging evidence reveals that inflammatory microglia and astrocytes drive neuronal death in neurodegenerative diseases. Microglial inflammatory mediators such as C1q, TNFα, and IL-1α polarize astrocytes towards a neurotoxic phenotype that promotes neuronal death [4]. Neurotoxic astrocytes express a unique set of transcripts including the genes associated with the early complement cascade such as C3 [4], which leads to excess synaptic pruning during neurodegeneration [6]. Previous studies show that NLY01 blocks microglial activation and the formation of neurotoxic astrocytes in PD, AD, and glaucoma experimental models [17, 18, 27]. We hypothesized that NLY01 would interfere with the glial activation cascade, blocking the formation of neurotoxic astrocytes and preventing the RGC loss in EAE. We examined the level of RGC loss in NLY01- or vehicle-treated mice and found that NLY01 significantly reduced RGC loss at the late stage of EAE (PID 42), suggesting the neuroprotective effect of NLY01 following EAE progression. A recent study reported that several genes associated with the neurotoxic profile were highly expressed in astrocytes purified from the EAE optic nerve [28]. Consistent with this finding, we previously showed that the expression of genes associated with neurotoxic astrocytes is upregulated in the optic nerve at peak inflammation and are associated with RGC loss at the late stage of EAE (PID 42) [5]. To test whether NLY01 would block the formation of neurotoxic astrocytes, we measured the expression of neurotoxic astrocyte–associated transcripts in the optic nerve and found that NLY01 significantly decreased the expression of neurotoxic astrocyte genes including C3, Ligp1, Psmb8, and H2-T23 in the optic nerve at the early stage of EAE (PID 11). The role of NLY01 in blocking neurotoxic astrocytes and RGC death has been recently shown in the mouse model of glaucoma [27]. Our findings support the therapeutic potential of NLY01 in blocking neurotoxic astrocytes and preventing RGC loss in MS.
A recent study reported that intraperitoneal injection of exendin-4 (5 μg/kg, daily) into symptomatic mice (PID 29 to 42) significantly attenuated clinical score at the late stage of EAE [22]. To further ascertain the potential clinical relevance of NLY01 in MS, we explored its effects in a therapeutic paradigm using two distinct EAE models: MOG-induced chronic progressive EAE in C57BL/6 mice and PLP-induced relapsing–remitting EAE in SJL background. In MOG-immunized EAE, we found no difference in the clinical scores between NLY01 and vehicle groups when the treatments started at disease onset (clinical score 1). In relapsing–remitting EAE in SJL mice, treatment with NLY01 at onset of disease resulted in a significant decrease in the EAE clinical score and second relapse, suggesting that NLY01 has therapeutic potential in ongoing relapsing disease.
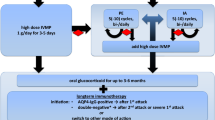
Taken together, these results support the hypothesis that NLY01 is protective in vivo through multiple mechanisms of action. We found no direct effect of NLY01 on T cell activation and proliferation in vitro, suggesting that the observed anti-inflammatory effect of NLY01 on T cells in EAE is likely due, in large part, to decreased activation of peripheral myeloid cells. It is possible that cell populations not studied here, such as myeloid-derived suppressor cells [29], contributed to the attenuation of EAE symptoms. In addition to its anti-inflammatory effect on peripheral immune cells, NLY01 reduces glial inflammation, both of which contribute to the observed therapeutic impact on EAE clinical score. Based on these observations, we suggest a dual efficacy of NLY01 in EAE: the suppression of both peripheral and CNS inflammation, thereby preventing neuronal loss (Fig. 8). At the early stage of the disease, NLY01 inhibits the activation and propagation of T cells and myeloid cells including neutrophils, dendritic cells, and monocyte/macrophages in the spleen and reduces their trafficking into the CNS. NLY01 suppresses the activation of astrocytes and the production of chemokines that are involved in leukocyte recruitment to the site of inflammation. At the late stage of the disease, NLY01 blocks the generation of neurotoxic astrocytes in the optic nerves, thereby preventing RGC loss. These findings warrant future studies focusing on these outcomes, along with ex vivo analyses of immune cell populations to provide additional mechanistic insight.

The putative anti-inflammatory and neuroprotective effects of GLP-1R agonists in MS. NLY01 may have dual efficacy in MS by suppressing both peripheral and CNS inflammation, thereby limiting neuronal loss
The data generated in our EAE animal model does not preclude the possibility that there might be unforeseen systemic side effects in people [30]. However, several PEGylated drugs are already FDA approved including PEGylated interferon β-1a for MS [31]. NLY01 contains a 50-kDa PEG molecule and was found to be well-tolerated in phase 1 clinical trial in healthy volunteers (NCT03672604). NLY01 is currently in phase 2 clinical trial in individuals with early untreated PD (NCT04154072).
The present study highlights the therapeutic potential of the GLP-1R agonists for preventing both neuroinflammation and neurodegeneration in MS. Optic neuritis is a common early symptom of MS and may lead to RGC damage and permanent visual loss. GLP-1R agonists may have therapeutic potential for optic neuritis when initiated prior to axonal injury to preserve neuronal function. The molecular mechanisms underlying the anti-inflammatory effects of NLY01 in the periphery and the CNS require further exploration. Since GLP-1R agonists are currently used for the treatment of type 2 diabetes, it could be rapidly translated for testing in MS.
References
Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. New England Journal of Medicine. 2018;378(2):169-80. https://doi.org/10.1056/NEJMra1401483.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron. 2018;97(4):742-68. https://doi.org/10.1016/j.neuron.2018.01.021.
Ciotti JR, Cross AH. Disease-Modifying Treatment in Progressive Multiple Sclerosis. Current treatment options in neurology. 2018;20(5):12. https://doi.org/10.1007/s11940-018-0496-3.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-7. doi:https://doi.org/10.1038/nature21029.
Jin J, Smith MD, Kersbergen CJ, Kam T-I, Viswanathan M, Martin K et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathologica Communications. 2019;7(1):125. doi:https://doi.org/10.1186/s40478-019-0767-6.
Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity. 2020;52(1):167-82.e7. https://doi.org/10.1016/j.immuni.2019.12.004.
Donnelly D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. British journal of pharmacology. 2012;166(1):27-41. https://doi.org/10.1111/j.1476-5381.2011.01687.x.
Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224-33. https://doi.org/10.2337/db13-1440.
Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia. 2010;53(4):730-40. https://doi.org/10.1007/s00125-009-1643-x.
Trujillo JM, Nuffer W, Ellis SL. GLP-1 receptor agonists: a review of head-to-head clinical studies. Ther Adv Endocrinol Metab. 2015;6(1):19-28. doi:https://doi.org/10.1177/2042018814559725.
Trapp S, Richards JE. The gut hormone glucagon-like peptide-1 produced in brain: is this physiologically relevant? Current opinion in pharmacology. 2013;13(6):964-9. doi:https://doi.org/10.1016/j.coph.2013.09.006.
Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. British journal of pharmacology. 2012;166(5):1586-99. https://doi.org/10.1111/j.1476-5381.2012.01971.x.
Grieco M, Giorgi A, Gentile MC, d'Erme M, Morano S, Maras B et al. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front Neurosci. 2019;13:1112-. https://doi.org/10.3389/fnins.2019.01112.
Batista AF, Bodart-Santos V, De Felice FG, Ferreira ST. Neuroprotective actions of glucagon-like peptide-1 (GLP-1) analogues in Alzheimer’s and Parkinson’s diseases. CNS drugs. 2019;33(3):209-23.
Chiou H-YC, Lin M-W, Hsiao P-J, Chen C-L, Chiao S, Lin T-Y et al. Dulaglutide Modulates the Development of Tissue-Infiltrating Th1/Th17 Cells and the Pathogenicity of Encephalitogenic Th1 Cells in the Central Nervous System. International Journal of Molecular Sciences. 2019;20(7):1584.
Kim TH, Jiang HH, Lim SM, Youn YS, Choi KY, Lee S et al. Site-Specific PEGylated Exendin-4 Modified with a High Molecular Weight Trimeric PEG Reduces Steric Hindrance and Increases Type 2 Antidiabetic Therapeutic Effects. Bioconjugate Chemistry. 2012;23(11):2214-20. https://doi.org/10.1021/bc300265n.
Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nature Med. 2018;24(7):931-8. https://doi.org/10.1038/s41591-018-0051-5.
Park J-S, Kam T-I, Lee S, Park H, Oh Y, Kwon S-H et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathologica Communications. 2021;9(1):78. https://doi.org/10.1186/s40478-021-01180-z.
Miller SD, Karpus WJ, Davidson TS. Experimental autoimmune encephalomyelitis in the mouse. Current protocols in immunology. 2010;Chapter 15:Unit 15.1. https://doi.org/10.1002/0471142735.im1501s88.
Rothhammer V, Quintana FJ. Control of autoimmune CNS inflammation by astrocytes. Seminars in immunopathology. 2015;37(6):625-38. https://doi.org/10.1007/s00281-015-0515-3.
DellaValle B, Brix GS, Brock B, Gejl M, Landau AM, Møller A et al. Glucagon-Like Peptide-1 Analog, Liraglutide, Delays Onset of Experimental Autoimmune Encephalitis in Lewis Rats. Front Pharmacol. 2016;7:433-. https://doi.org/10.3389/fphar.2016.00433.
Lee C-H, Jeon SJ, Cho KS, Moon E, Sapkota A, Jun HS et al. Activation of Glucagon-Like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by Reducing Neuroinflammatory Responses. Molecular neurobiology. 2018;55(4):3007-20. https://doi.org/10.1007/s12035-017-0550-2.
Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. 2015;212(1):23-35. https://doi.org/10.1084/jem.20141015.
Dokken BB, La Bonte LR, Davis-Gorman G, Teachey MK, Seaver N, McDonagh PF. Glucagon-like peptide-1 (GLP-1), immediately prior to reperfusion, decreases neutrophil activation and reduces myocardial infarct size in rodents. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2011;43(5):300–5. https://doi.org/10.1055/s-0031-1271777.
Sagar D, Singh NP, Ginwala R, Huang X, Philip R, Nagarkatti M et al. Antibody blockade of CLEC12A delays EAE onset and attenuates disease severity by impairing myeloid cell CNS infiltration and restoring positive immunity. Scientific reports. 2017;7(1):2707. https://doi.org/10.1038/s41598-017-03027-x.
Galetta KM, Calabresi PA, Frohman EM, Balcer LJ. Optical Coherence Tomography (OCT): Imaging the Visual Pathway as a Model for Neurodegeneration. Neurotherapeutics. 2011;8(1):117-32. https://doi.org/10.1007/s13311-010-0005-1.
Sterling JK, Adetunji MO, Guttha S, Bargoud AR, Uyhazi KE, Ross AG et al. GLP-1 Receptor Agonist NLY01 Reduces Retinal Inflammation and Neuron Death Secondary to Ocular Hypertension. Cell Reports. 2020;33(5):108271. https://doi.org/10.1016/j.celrep.2020.108271.
Tassoni A, Farkhondeh V, Itoh Y, Itoh N, Sofroniew MV, Voskuhl RR. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Scientific reports. 2019;9(1):10010. https://doi.org/10.1038/s41598-019-46232-6.
Melero-Jerez C, Suardíaz M, Lebrón-Galán R, Marín-Bañasco C, Oliver-Martos B, Machín-Díaz I et al. The presence and suppressive activity of myeloid-derived suppressor cells are potentiated after interferon-β treatment in a murine model of multiple sclerosis. Neurobiology of disease. 2019;127:13-31. https://doi.org/10.1016/j.nbd.2019.02.014.
Liu G, Li Y, Yang L, Wei Y, Wang X, Wang Z et al. Cytotoxicity study of polyethylene glycol derivatives. RSC Advances. 2017;7(30):18252-9. https://doi.org/10.1039/C7RA00861A.
Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. The Lancet Neurology. 2014;13(7):657-65. https://doi.org/10.1016/s1474-4422(14)70068-7.
Acknowledgements
We thank Neuraly for providing NLY01.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This study was funded by the US Department of Defense (DOD) #W81XWH-19–1-0622 grant to P.A.C. and The Fonds de recherche du Québec – Santé (FRQS) #270746 to M.G. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gharagozloo, M., Smith, M.D., Sotirchos, E.S. et al. Therapeutic Potential of a Novel Glucagon-like Peptide-1 Receptor Agonist, NLY01, in Experimental Autoimmune Encephalomyelitis. Neurotherapeutics 18, 1834–1848 (2021). https://doi.org/10.1007/s13311-021-01088-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01088-5