
Abstract
Two monoclonal antibodies (mAbs), aducanumab and lecanemab, have received accelerated approval from the US FDA for initiation of treatment in early Alzheimer's disease patients who have proven β-amyloid pathology (Aβ). One of these, lecanemab, has subsequently received full approval and other monoclonal antibodies are poised for positive review and approval. Anti-amyloid mAbs share the feature of producing a marked reduction in total brain Aβ revealed by amyloid positron emission tomography. Trials associated with slowing of cognitive decline have achieved a reduction in measurable plaque Aβ in the range of 15–25 centiloids; trials of agents that did not reach this threshold were not associated with cognitive benefit. mAbs have differences in terms of titration schedules, MRI monitoring schedules for amyloid-related imaging abnormalities (ARIA), and continuing versus interrupted therapy. The approximate 30% slowing of decline observed with mAbs is clinically meaningful in terms of extended cognitive integrity and delay of onset of the more severe dementia phases of Alzheimer’s disease. Approval of these agents initiates a new era in Alzheimer’s disease therapeutics with disease-modifying properties. Further advances are needed, i.e. greater efficacy, improved safety, enhanced convenience, and better understanding of ill-understood observations such as brain volume loss.
Similar content being viewed by others
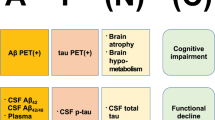

Anti-amyloid monoclonal antibodies produced marked lowering of brain β-amyloid plaque and slowing of clinical decline as measured by the Clinical Dementia Rating–Sum of Boxes and other clinical and functional measures. |
Anti-amyloid monoclonal antibodies produce amyloid-related imaging abnormalities (ARIA) that are usually asymptomatic but may be severe and require anticipatory management. |
Slowing of clinical decline has been observed when the β-amyloid lowering reaches 15–25 centiloids, a common measure of β-amyloid abundance in the brain. |
1 Introduction
Alzheimer’s disease (AD) is a complex, progressive, age-related, neurodegenerative disease that is becoming increasingly common in the United States and globally as the world’s population ages [1]. Progress is being made in the treatment of AD: the US FDA has approved five cognitive-enhancing drugs, one agent for reduction of agitation in AD, and two disease-modifying therapies (DMTs).
All currently approved DMTs for the treatment of AD are anti-amyloid monoclonal antibodies (mAbs). The two approved agents are aducanumab (Aduhelm®; Biogen, Cambridge, MA, USA), which received accelerated approval based on marked lowering of β-amyloid (Aβ) plaques as seen on amyloid positron emission tomography (PET) considered reasonably likely to predict clinical benefit [2]; and lecanemab (Leqembi®; Eisai Inc. and Biogen, Cambridge, MA, USA), which received accelerated approval based on a phase II study followed by standard approval based on clinical and biomarker data from a phase III study [3, 4]. Donanemab (Eli Lilly, Indianapolis, IN, USA), is currently under review for standard approval based on data derived from phase II and III clinical trials [5].
Anti-amyloid mAbs have some shared features; other characteristics distinguish among them. From a mechanism of action (MoA) point of view, all of them target high molecular weight fibrillar Aβ aggregates, produce marked Aβ lowering demonstrated on amyloid PET, and are associated with amyloid-related imaging abnormalities (ARIA) [6]. The mAbs differ in the type and range of amyloid species targeted and on pharmacokinetic (PK) parameters such as half-life, infusion frequency, and titration schedule [7, 8]. They overlap in having included early AD (mild cognitive impairment [MCI] due to AD and mild AD dementia) as the trial population, although the exact definition of early AD in terms of allowable severity differs among the mAb trials, and these differences may influence patient selection for treatment with a specific agent.
In this review, we describe the MoA of the mAbs; summarize the preclinical data for each of the agents; review the key features of the phase I, II, and III clinical trials; and note how similarities and differences among the mAbs may translate into the use of these agents in clinical practice (Table 1). We describe the approved agents and one drug (remternetug) in a current phase III trial, and describe gantenerumab, whose development program was terminated following negative phase III trials, where the trial data are informative regarding the clinical and biomarker aspects of mAbs. The efficacy and safety of drugs can be compared directly only if the agents are randomized to equivalent arms in the same clinical trial. Differences in entry criteria, sites and investigators participating in the trials, and participants recruited to the trials may affect the efficacy and safety observed. The summary information provided (Tables 2 and 3) should be interpreted with this caveat in mind.
2 Method
This is a narrative review based on interrogation of the literature addressing the five anti-amyloid mAbs included. Preclinical studies were identified through searches for ‘transgenic’ and related words that align with the non-clinical assessment of these agents. Data for the phase I, II, and III clinical trials were derived from ClinicalTrials.gov, with follow-up review of the primary publications presenting the main clinical and biomarker outcomes of the trials when these were available. Secondary literature was identified and used to augment the primary publications when they presented additional new information (e.g., were not re-analyses, reviews, follow-up analyses, or interpretations of the primary data).
3 Mechanism of Action of Anti-Amyloid Monoclonal Antibodies
For all mAbs, the mechanism of Aβ plaque reduction is hypothesized to be activation of microglia with phagocytosis of fibrillar Aβ and degradation through the endosomal/lysosomal system (Fig. 1). Each of the approved mAbs targets a different constellation of Aβ species. Aducanumab addresses a broad range of Aβ species with a greater affinity for high molecular weight species; lecanemab targets protofibrils with a 10:1 preference for protofibrils over plaque Aβ and a 100:1 higher affinity for protofibrils compared with Aβ monomers; and donanemab and remternetug target pyroglutamate Aβ present only in plaques [9, 10]. It is uncertain to what extent microglia remove only the species labeled by the mAb or if activated microglia may phagocytose both labeled and unlabeled protein aggregates including Aβ and tau. There is evidence that mAbs that address oligomeric Aβ (e.g., aducanumab) may interfere with Aβ aggregation and this mechanism may contribute to the therapeutic effect [9, 11].

© J Cummings; M de la Flor, PhD, illustrator)
Activation of resting microglia by anti-amyloid monoclonal antibodies (
Plaque Aβ, the only type of amyloid visualized by amyloid PET, is markedly reduced by all approved mAbs. The measure of Aβ in plasma and cerebrospinal fluid (CSF) reflects the presence of monomeric Aβ. There is no widely accepted measure of protofibrils or oligomers. The contribution of effects on these non-plaque Aβ species to the treatment benefit has not been determined.
4 History of Immunotherapy
In 1999, Schenk et al. made the seminal observation that active vaccination against full-length amyloid in amyloid-bearing PDAPP (platelet-derived growth factor [PDGF]-f3 promoter driving a human amyloid precursor protein [APP] minigene) mice led to a marked reduction in plaque Aβ [12]. This remarkable result led to additional verifying experiments in rodents [13] and an emerging consensus on the possibility of human application of immunotherapy. The first human trial (initiated in 2001) with the AN1792 vaccine was terminated when 6% of the participants developed meningoencephalitis ascribed to T-cell activation [14, 15]. Autopsy studies demonstrated reduction of Aβ plaques and decreased tau in neuronal processes [16]. No clinical benefit was shown [17]. The difficulties encountered with active vaccination resulted in developing passive immunotherapies that focus on specific Aβ epitopes. The first human trial, involving the mAb bapineuzumab, was begun in 2006. The trial showed no clinical benefit but was interpreted as indicating a response in apolipoprotein E4 (APOE4) non-carriers [18]. ARIA were observed for the first time in this trial. In response to the observations in the phase II trial, a pair of trials hosting E4 carriers and E4 non-carriers, respectively, were conducted without establishing benefit in either trial [19, 20]. Plaque reduction was demonstrated in a small trial using amyloid PET as an outcome [21].
Creating a ‘peripheral sink’ by engaging Aβ peripherally and creating flow from brain to plasma was hypothesized as a plausible approach to anti-amyloid mAb therapy and formed the basis of a pair of trials with solanezumab, a monomer-directed mAb in patients with mild-to-moderate AD [22, 23]. The trials were negative but suggested benefit in mildly impaired patients; a trial in this population also failed to demonstrate a treatment effect [24]. Trials of crenezumab, an mAb directed at monomers and oligomers, produced no drug-placebo difference in phase II trials involving mild-moderate AD [25, 26]. Similarly, phase III trials showed no benefit in participants with early AD [27]. Analyses of these trials, as well as data from the gantenerumab mAb development program [28], suggested that higher doses of mAbs might be needed. The next generation of mAb trials (described below) used doses four- to fivefold higher than those used in the initial studies.
The combination of higher doses, targeting high molecular weight Aβ species, and use of amyloid PET and CSF Aβ studies to verify the presence of the biological target in trial participants resulted in greater success in the mAb studies and FDA approval of two agents.
5 Aducanumab
5.1 Introduction
Aducanumab (BIIB037; Aduhelm™) is a human immunoglobulin (Ig) G1 monoclonal auto-antibody (IgG1-mAb) that binds to an N-terminal epitope formed by amino acids 3–7 of the Aβ42 (Aβ42) peptide, with a higher affinity for fibrillar aggregates compared with monomers [9]. Aducanumab received accelerated approval from the FDA in June 2021, making it the first approved Aβ-targeting mAb and the first approved DMT for AD [29]; it was approved in the United Arab Emirates (UAE) shortly after. The drug is indicated in early AD patients with MCI or mild dementia who have evidence of brain Aβ from amyloid PET or CSF studies [30]. Data from the clinical trials suggest that aducanumab at 10 mg/kg is the target dose [2, 30] delivered via intravenous infusions every 4 weeks.
5.2 Preclinical Studies
Preclinical studies in Tg2576 mice showed that an analog of aducanumab (BIIB037) crossed the blood–brain barrier (BBB), engaged its target, and cleared Aβ from the cortex and hippocampus of plaque-bearing mice [31]. BIIB037 selectively targets fibrillar Aβ with high selectivity and has sub-nanomolar affinities for aggregated forms of Aβ, including soluble oligomers and insoluble fibrils.
5.3 Phase I
The phase I clinical trial of aducanumab (NCT01397539) [32, 33] was a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and PK of a range of aducanumab doses (0.03, 1, 3, 10, 20, 30, and 60 mg/kg) in participants with mild to moderate AD. Three participants who received the 60 mg/kg dose developed ARIA, but a dose of ≤ 30 mg/kg was generally well tolerated without serious adverse events (SAEs).
A multiple-dose phase Ib clinical trial of aducanumab (PRIME, NCT01677572) [30, 34] randomized participants to receive a placebo or aducanumab (1, 3, 6, or 10 mg/kg) every 4 weeks via intravenous infusion for 1 year. This trial showed a dose- and time-dependent reduction in brain Aβ plaques, as measured by PET accompanied by a dose-dependent slowing of decline on the Clinical Dementia Rating–Sum of Boxes (CDR-SB) and Mini-Mental State Examination (MMSE). No downstream biomarkers were investigated in this study. No significant changes were observed on the Neuropsychological Test Battery (NTB) or the Free and Cued Selective Reminding Test (FCSRT) free recall [30, 34]. The main safety and tolerability findings involved ARIA, which usually resolved within 4–12 weeks.
5.4 Phase II
A phase II trial (EVOLVE, NCT03639987) with 52 participants aimed to assess the safety impact of continuing aducanumab dosing and ARIA in patients with MCI due to AD or with mild AD dementia [35]. Two groups (n = 26 each) had the aducanumab dose titrated up to 10 mg/kg via intravenous infusion and were followed up to week 54 with different ARIA management rules. The trial was discontinued in July 2019 based on a futility analysis conducted on phase III trials (NCT02477800 and NCT02484547) [36].
5.5 Phase III
Based on the findings of the PRIME trial, two identically designed 18-month randomized, double-blind, placebo-controlled, parallel-group studies were initiated: ENGAGE (NCT02477800) [37] and EMERGE (NCT02484547) [38]. The two trials enrolled 1653 and 1638 participants with MCI due to AD (80%) or mild AD (20%), respectively, at 348 sites in 20 countries. Specific inclusion criteria were [1] a baseline MMSE score of 24–30; (2) a CDR-SB global score of 0.5; and (3) positive amyloid PET. The participants were randomized into three groups in a ratio of 1:1:1 to receive low-dose aducanumab (3 mg/kg for APOE ε4 carriers, 6 mg/kg for non-carriers), high-dose aducanumab (10 mg/kg for APOE ε4 non-carriers, 6 mg for carriers), or placebo every 4 weeks for 76 weeks. Drug-placebo difference on the CDR-SB score at 78 weeks was the primary outcome measure. During the trials, the allowable dose for APOE ε4 carriers was increased to 10 mg/kg after safety of the lower doses was established.
Based on a futility analysis of data pooled from the first 50% of enrolled participants, the ENGAGE and EMERGE trials were halted [37, 38]. On subsequent analysis of a larger dataset, following a prespecified statistical analysis, the EMERGE trial met its primary endpoint in the high-dose aducanumab arm for the CDR-SB (22% decrease, p = 0.012) at week 78 [2]. This trial also met its secondary endpoints (MMSE, Alzheimer’s Disease Assessment Scale–13-item Cognitive Subscale [ADAS-Cog 13], and Alzheimer’s Disease Cooperative Study–Activities of Daily Living for Mild Cognitive Impairment [ADCS-ADL-MCI]) in the high-dose arm. The ENGAGE trial failed to meet its primary and secondary endpoints. Substudies from both trials demonstrated a dose- and time-dependent decrease in Aβ, as seen on amyloid PET with aducanumab treatment [2]. Effects on downstream biomarkers specific to AD (tau PET, CSF p-tau, and plasma p-tau 181) were observed in both studies [2]. Dose-related decreases were observed in CSF p-tau levels (e.g., high dose led to a reduction of 22.44 pg/mL and 13.19 pg/mL, as compared with placebo, in the EMERGE and ENGAGE studies, respectively) and plasma p-tau (13% and 16% decrease from baseline in the EMERGE and ENGAGE studies, respectively) [2]. Additionally, pooled results from a small sample of participants from both trials demonstrated dose-dependent reductions of tau PET standardized uptake value ratio (SUVR) in specific brain regions. These findings demonstrate that aducanumab directly affects both an upstream biomarker of AD (Aβ plaque) as well as downstream biomarkers of AD (CSF and plasma p-tau; tau PET). Reductions in amyloid PET SUVR were correlated with a reduction in plasma p-tau 181 levels. Together, these results support the hypothesis that Aβ accumulation triggers downstream tau pathology and subsequent clinical decline, and that removing aggregated Aβ in the brain via aducanumab treatment results in clinical benefit. The most common adverse effects in the high-dose arm included ARIA related to cerebral edema (ARIA-E) [35%] and intracerebral hemorrhage (ARIA-H) [19.1%]. The long-term extension of the PRIME study demonstrated that after 48 months of treatment with aducanumab, Aβ plaques decreased in a dose- and time-dependent manner, as measured by amyloid PET; results from the CDR-SB and MMSE also suggested clinical benefits [39, 40].
6 Lecanemab
6.1 Introduction
Lecanemab (BAN2401; Leqembi®) is a humanized IgG1 antibody based on the mouse mAb158 [41] that specifically binds to Aβ protofibrils. Lecanemab was granted FDA accelerated approval in January 2023, followed by full approval in July 2023, to be initiated in patients with MCI or mild AD dementia, shown to be Aβ positive by amyloid PET or CSF findings consistent with AD [29]. Lecanemab is administered to patients intravenously without titration in a weight-adjusted dose of 10 mg/kg biweekly [42]. The agent has been approved in Japan and is undergoing regulatory review in the European Union (EU), United Kingdom (UK), South Korea, and Canada.
6.2 Preclinical
Aβ protofibrils were clinically recognized as a pathogenic mechanism for AD in 2001 by Nilsberth et al. [43], who observed a novel APP mutation (dubbed ‘Arctic’) that accelerated Aβ protofibril formation and led to early-onset AD symptoms in mutation carriers. An in vivo study in transgenic mice expressing the Arctic and Swedish APP mutations (‘ArcSwe’) found that mAb158, originally developed to detect Aβ protofibrils, reduced both soluble Aβ protofibrils and insoluble Aβ plaques if administered early in disease progression [44]. Study of human post-mortem AD brains showed that mAb158 bound to similar soluble Aβ protofibrils (approximately 80–500 kDa) in the human samples, leading to development of the humanized BAN2401 [45].
6.3 Phase I
The phase I clinical trial of lecanemab (NCT01230853) enrolled 80 participants and sought to assess its safety, PK, and effect on plasma and CSF biomarkers [46]. Eligible participants had mild to moderate AD based on National Institute of Neurological and Communicative Diseases and Stroke–Alzheimer's Disease and Related Dementias Association (NINCDS-ADRDA) criteria and MMSE scores of 16–28. Participants were randomized into single and multiple ascending dose arms (six lecanemab and two placebo per cohort). The SAD study included 0.1, 0.3, 1, 3, 10, and 15 mg/kg, and the MAD study included 0.3, 1, and 3 mg/kg administered every 4 weeks, and 10 mg/kg biweekly. SAD and MAD were performed with staggered parallel cohorts; MAD was initiated once the dose was established as well tolerated in the SAD cohort.
Lecanemab was well tolerated in both dosage arms. PK analyses indicated lecanemab had a dose-proportional response and a 7-day serum half-life with doses ≥ 10 mg/kg. There was no observed ARIA-E; ARIA-H was observed in two cases in the SAD cohorts (one symptomatic), and six cases (all without symptoms) in the MAD cohorts (with one being placebo). Biomarker changes observed were limited to a mild increase in plasma Aβ1–40 [46].
6.4 Phase II
The phase IIb trial of lecanemab (NCT01767311) was an 18-month study that enrolled 856 subjects to determine the dose and efficacy of the treatment [3]. Participant eligibility required Aβ pathology confirmed by PET or CSF Aβ1-42 measurement, an MMSE ≥ 22 (22–28 in participating EU nations), and objective memory impairment (Weschler Memory Scale IV–Logical Memory II [WMS-IV LMII]) criteria. A unique Bayesian adaptive dose-finding trial design was used for the first 12 months to assess the primary endpoint, the change from baseline on the AD Composite Score (ADCOMS) [47]. Success at the 12-month endpoint required a dosage arm to have an 80% probability of slowing decline on the ADCOMS by 25% more than placebo. A total of 854 subjects were randomized in the study. Initially, 196 subjects were randomized into the placebo (n = 56) and five different dosage arms (2.5, 5, and 10 mg/kg biweekly, and 5 and 10 mg/kg monthly, n = 28 in each cohort). Thereafter, every 50 subjects were randomized into the dosage arms based on the ADCOMS performance of each cohort.
At 12 months, the trial failed to meet its primary endpoint, with the 10 mg/kg biweekly dose achieving a 64% probability of slowing ADCOMS decline by 25% more than placebo. The double-blind portion of the study continued until month 18. Frequentist analyses revealed statistically significant differences in favor of lecanemab on the ADCOMS and ADAS-Cog. Amyloid PET demonstrated Aβ plaque removal below the threshold for detection in 81% of participants. In an optional substudy of CSF biomarkers, the combined biweekly and monthly 10 mg/kg lecanemab arm showed higher Aβ1-42 and lower p-tau at 18 months compared with placebo. A notable amendment to the trial required the removal of APOE ε4 carriers from the 10 mg/kg biweekly arm as instructed by a regulatory agency, since these participants have the greatest risk for ARIA. This adjustment resulted in fewer APOE ε4 allele carriers in the high-dose arm of the trial. Aside from ARIA, the most common adverse events (AEs) were infusion reactions, which tended to be mild or moderate and responded to treatment or prophylaxis [3].
6.5 Phase III
‘CLARITY AD’ (NCT03887455) enrolled 1795 participants who were randomized 1:1 into a 10 mg/kg biweekly lecanemab arm (n = 898) or placebo arm (n = 897) [4]. Eligibility was determined by age (50–90 years), an MCI or mild AD diagnosis (National Institute on Aging–Alzheimer's Association [NIA-AA] criteria), a 1 standard deviation (SD) decrease in objective episodic memory below the age-adjusted mean (WMS-IV LMII), and Aβ positivity by PET or CSF Aβ1–42 measurement. CLARITY AD’s primary endpoint was the change from baseline on the CDR-SB at 18 months.
Lecanemab slowed decline on the CDR-SB by 0.45 points (+ 1.21 point change) compared with placebo (+ 1.66 point change). Other cognitive measures in the lecanemab arm (ADAS-Cog, ADCOMS, ADCS-ADL-MCI) had significantly slower decline than placebo at 18 months. Amyloid PET plaque levels were reduced on lecanemab (− 55.48 centiloid change) versus placebo (+ 3.64 centiloid change). All CSF and plasma biomarkers favored lecanemab over placebo except for neurofilament light (NfL), which showed no drug-placebo difference. Infusion-related reactions (26.4%), ARIA-H (17.3%), and ARIA-E (12.6%) were the most common AEs in the lecanemab dosage arm. Non-carriers of the APOE ε4 allele in the lecanemab arm had the lowest incidence of ARIA-H (11.9%) and ARIA-E (5.4%); ε4 heterozygotes had a higher incidence of both (ARIA-H: 14%; ARIA-E: 10.9%). APOE ε4 homozygotes had an incidence of ARIA-H and ARIA-E in 39% and 32.6%, respectively.
The FDA granted lecanemab accelerated approval based on its Aβ lowering in the phase IIb trial, which was considered reasonably likely to predict clinical benefit. Lecanemab was granted standard approval by the FDA based on the results of CLARITY AD.
7 Gantenerumab
7.1 Introduction
Gantenerumab (RO4909832; R1450) is the first fully human monoclonal IgG1 anti-Aβ antibody that underwent clinical development. It recognizes both the N-terminal portions and central amino acids of the Aβ peptide and initiates cell-mediated clearance via recruitment of microglia.
7.2 Preclinical Studies
A synthetic human combinatorial antibody library (HuCAL®; MorphoSys, Martinsried/Planegg, Germany) generated antibody fragments that were then screened for anti-Aβ effects [48]. Equilibrium binding studies of gantenerumab showed strong affinities for aggregated cerebral Aβ plaques and an ability to cross the BBB [49]. Preclinical studies suggested that gantenerumab could neutralize oligomer toxicity in rat brain. When APP751(Swedish)xPS2(N141I) transgenic mice received chronic treatment, gantenerumab reduced Aβ plaques via cell-mediated clearance and prevented new plaque formation [50].
7.3 Phase I
A phase I, randomized, double-blind, placebo-controlled SAD trial (NCT02711423) enrolled 18 participants at a single center to evaluate the safety, tolerability, and PK of subcutaneous gantenerumab. Healthy male volunteers between 18 and 45 years of age with a body mass index (BMI) of 20.0–32.0 kg/m2 were recruited for participation. The primary outcome measure was percentage of participants with AEs up to 12 weeks from baseline, while secondary outcome measures included maximum observed plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of gantenerumab [51]. The results from this trial are not available.
A phase I, randomized, double-blind, placebo-controlled MAD trial (NCT00531804) enrolled 60 participants with diagnoses of probable AD to receive 2 to 7 intravenous infusions of a placebo or gantenerumab (escalating doses of 60 or 200 mg) every 4 weeks. Primary outcome measures included AEs, laboratory parameters, vital signs, and PK parameters of the drug in plasma, while secondary outcome measures included CSF biomarkers and clinical efficacy measures [52]. This trial showed a dose-dependent reduction in Aβ levels as seen by Pittsburgh Compound B amyloid PET. Two participants in the 200 mg group were carriers of APOE ε4/ε4 and were observed to experience ARIA-E. Imaging findings were largely transient; ARIA was not well understood and subsequent clinical efficacy studies selected conservative doses, and participants were uptitrated in the absence of ARIA [53].
7.4 Phase II/III
SCarlet RoAD (NCT01224106) was a phase II/III, randomized, double-blind, placebo-controlled, parallel-group, 2-year trial with a 2-year open-label extension (OLE) that utilized this conservative dosing approach. Participants with prodromal AD, as exhibited by gradual decline in memory and an MMSE score of ≥ 24, were recruited to receive 105 mg or 225 mg subcutaneously every 4 weeks. Primary outcome measures were average change from baseline to week 104 on the CDR-SB and number of participants with AEs or SAEs, while secondary outcome measures included analyses of physical and cognitive function and neuropsychiatric symptoms [54]. ARIA-E was minimally observed in placebo groups (0.8%), with increased prevalence in the 105 mg cohort (6.6%) and the 205 mg cohort (13.5%). No differences between placebo and experimental groups were observed for primary or secondary clinical endpoints, and the study was discontinued early for futility. Analyses and modeling of the results of this study suggested that higher doses might be required to achieve efficacy, with long titration schedules to mitigate the risk of ARIA-E [28].
A phase II/III randomized, double-blind, placebo-controlled trial (DIAN-TU-001, NCT01760005) is currently investigating the effects of gantenerumab in participants with dominantly inherited Alzheimer disease (DIAD). This trial includes 52 participants receiving gantenerumab subcutaneously uptitrated to 1200 mg (n = 52) every 4 weeks for 4 years (comparative cohorts, solanezumab intravenously, n = 50; placebo, n = 40). Primary outcome measures include assessment of cognitive efficacy, measured by the change from baseline on the DIAN-Multivariate Cognitive Endpoint (DIAN-MCE) [55]. ARIA-E was observed in 10/52 participants receiving the active treatment. This OLE study of gantenerumab is continuing due to the unique application of the drug effects in individuals with DIAD as opposed to previously studied sporadic AD [56].
7.5 Phase III
The phase III studies GRADUATE I and II (NCT03444870 and NCT03443973, respectively) were randomized, double-blind, placebo-controlled, parallel-group studies of gantenerumab in prodromal to mild AD. There was a combined total of 1966 participants in these studies, with confirmed AD pathology as evidenced by CSF tau or Aβ42, or amyloid PET scan and abnormal memory function. Primary outcome measures included change from baseline to week 116 in the CDR-SB, while secondary outcome measures included changes in various cognitive and functional assessments [57]. Participants received a three-step titration over 9 months (120, 225, and 510 mg monthly) towards the target dosage of 1020 mg monthly; this titration structure was adopted to mitigate the risk of ARIA while maximizing participant exposure to the therapeutic. The titration scheduled was standardized for all participants, regardless of APOE ε4/ε4 carrier status. The trials failed to meet their primary endpoints, clinical trials were discontinued, and the development program was terminated [58].
Despite the negative outcomes of GRADUATE I and II, much was learned about the relationship between anti-amyloid mAbs and the treatment of AD. The mAb dose used in the gantenerumab phase III trials failed to reduce Aβ to the degree expected based on modeling of data from earlier trials. Reduction of Aβ below a threshold appears to be necessary to observe clinical slowing and therapeutic benefit associated with mAbs (discussed below).
8 Donanemab
8.1 Introduction
Donanemab (LY3002813; N3pG), a humanized IgG1 monoclonal antibody, developed from mouse mE8-IgG2a, recognizes N-terminal pyroglutamate Aβ, binding to deposited Aβ plaques and initiating microglial-mediated clearance. Currently, the donanemab phase III program for the treatment of early AD is complete and the agent is undergoing FDA review of clinical data supportive of standard approval; donanemab is also under review in the EU.
8.2 Preclinical Studies
Preclinical investigations of donanemab target engagement utilized a PDAPP mouse line genetically modified to develop Aβ plaques [59]. Following intraperitoneal injection of mE8-IgG2a, significant reduction in Aβ plaque was reported in a dose-dependent manner; treatment was not associated with microhemorrhages in these mice [59]. Post mortem brain tissue from AD and Down syndrome patients exhibited donanemab labeling of approximately one-third of plaques [60].
8.3 Phase I
A double-blind, randomized, placebo-controlled, phase Ia clinical trial investigated the safety, tolerability, and efficacy of donanemab in 100 patients with MCI or mild to moderate AD dementia (NCT01837641) [61]. The trial was designed as a seven-arm seamless SAD study transitioning into a MAD study. Cohort 1 received a sentinel dose of 0.1 mg/kg via intravenous infusion. After assessment of safety, the trial evaluated six doses, 0.3–10 mg/kg, administered by intravenous infusion, with one cohort receiving donanemab by subcutaneous injection. The cohort that received the 10 mg/kg dose of donanemab by intravenous infusion once per month demonstrated a 40–50% reduction in Aβ plaques. Two participants experienced ARIA-H and there were no reports of ARIA-E. The study demonstrated donanemab to have a shorter than expected half-life of about 10 days in the highest dose.
Phase Ib SAD and MAD studies enrolled 61 participants with MCI due to AD or mild to moderate AD dementia in a six-arm trial with doses ranging from 10 to 40 mg/kg administrated through intravenous infusion as a single or multiple dose regimen (NCT02624778) [62]. All cohorts demonstrated reductions in cerebral Aβ, with a sustained response at 72 weeks. ARIA-E was the most common AE, reported in 26% of participants receiving donanemab.
8.4 Phase II
A randomized, double-blind, phase II clinical trial enrolled 272 participants with early symptomatic AD (TRAILBLAZER-ALZ; NCT03367403) [5]. Inclusion criteria consisted of an MMSE score of 20–28 and demonstration of Aβ pathology by PET. Participants also had tau PET, and those with low or high levels of tau were excluded from the trial (high tau threshold was SUVR > 1.46). Donanemab was administered by intravenous infusion at 700 mg for the first three doses and 1400 mg thereafter, every 4 weeks for 72 weeks. The trial was originally designed to investigate donanemab independently and in combination with a BACE inhibitor, LY3202626; however, the arm evaluating LY3202626 was terminated due to the low probably of identifying statistical significance in slowing of cognitive decline. Following treatment with donanemab, participants showed a 25% slowing of cognitive decline on the primary measure of Integrated Alzheimer’s Disease Rating Scale (iADRS) score. Secondary outcome measures were change in cognitive and functional test scores and changes in Aβ levels. Results demonstrated an 85.06 centiloid reduction in Aβ plaques at 76 weeks, with 67.8% of participants having negative Aβ status (< 24.10 centiloids). Participants with sufficient lowering of Aβ levels (< 25 centiloids) were switched to placebo infusions. Post hoc analysis determined baseline Aβ levels to be directly associated with donanemab Aβ reduction and probability of clearance [63]. In participants who received donanemab, 6.1% reported symptomatic ARIA-E, significantly higher than the placebo group. Evaluation of plasma biomarkers demonstrated a significant decrease in p-tau 217 by 23% with donanemab treatment, whereas the placebo group had a 6% increase. Glial fibrillary acidic protein (GFAP) was significantly decreased by 12% with donanemab treatment, while the placebo group had a 15% increase [64]. Both changes in p-tau 217 and GFAP were positively correlated with Aβ plaque changes measured by PET [64]. There were no significant changes in plasma NfL or Aβ40/42 between the treated and placebo groups [64]. There is currently an active extension of this trial with 90 enrolled participants (TRAILBLAZER-EXT; NCT04640077).
8.5 Phase III
TRAILBLAZER-ALZ 2 (NCT04437511) is a phase III clinical trial of donanemab with an enrollment of 1800 participants. The inclusion criteria are similar to the phase II trial previously described; inclusion criteria were broadened to include individuals with high levels of tau to evaluate the effects of donanemab in this population.
The phase III trial is complete. In participants with intermediate tau levels (n = 1182) and clinical symptoms of AD, donanemab slowed the rate of cognitive decline by 35% in CDR-SB and led to 40% less decline in activities of daily living measured by iADRS [65]. ARIA-E appeared in 24% of participants who were treated with donanemab, with 6.1% experiencing symptomatic ARIA-E. ARIA-H occurred in 31% of participants receiving the drug and 13% receiving placebo. Based on these results, standard approval of donanemab by the FDA is anticipated [65, 66].
A phase III prevention study with donanemab is recruiting 3300 participants at risk for cognitive and functional decline due to AD (TRAILBLAZER-ALZ 3; NCT05026866). Inclusion criteria include intact cognitive functioning and tau PET consistent with the presence of Aβ and early-tau pathology. The primary outcome measure is Clinical Dementia Rating-Global Score (CDR-GS), with secondary outcome measures including several cognitive assessments.
Additionally, a phase III, open-label, two-arm comparison study evaluated the effects of donanemab compared with aducanumab (TRAILBLAZER-ALZ 4; NCT05108922). Two hundred participants with early symptomatic AD are enrolled, exhibiting gradual and progressive changes in memory, CDR-GS of 0.5 or 1, an MMSE score of 20–30, and abnormal Aβ levels on amyloid PET. The primary outcome measures include the percentage of participants who reach complete Aβ plaque clearance on PET, and secondary measures compare Aβ plaque levels between the drugs at various timepoints.
Another randomized, double-blind, placebo-controlled, phase III study is recruiting 1500 participants with early symptomatic AD to investigate the safety and efficacy of donanemab at 148 study sites globally (TRAILBLAZER-ALZ 5; NCT05508789). Inclusion criteria are the same as the TRAILBLAZER-ALZ 4 study. The primary outcome measure is changes in the iADRS score, and the secondary outcome measures include cognitive, functional, and neuropsychiatric assessments, as well as Aβ plaque removal and PK measures.
The most recent phase III clinical trial for donanemab is recruiting 800 participants to assess multiple dosing regimens and the effects of the drug on the frequency and severity of ARIA-E (TRAILBLAZER-ALZ 6; NCT05738486). Participants included in the study have gradual and progressive memory change, MMSE score of 20–28, and presence of Aβ by PET. The primary outcome of the study is the percentage of participants who experience ARIA-E in the first 24 weeks, while secondary outcome measures include frequency and severity of ARIA-E and ARIA-H, Aβ plaque removal, and PK measures.
After evaluation of the phase II clinical trial data, the FDA rejected donanemab for accelerated approval based on insufficient safety data; the study had fewer than 100 participants staying on the drug for 1 year [67]. With positive results emerging from the phase III TRAILBLAZER-ALZ 2 clinical trial, Eli Lilly has submitted an application for traditional approval of donanemab in the US and EU [65, 66].
9 Remternetug
9.1 Introduction
Remternetug (LY3372993) is an N3pG-AB monoclonal antibody, implying that it recognizes pyroglutamate Aβ, targeting Aβ plaques [68].
9.2 Phase I
A randomized, double-blind, placebo-controlled, phase I clinical trial has been completed with 36 healthy participants to evaluate the safety and tolerability of remternetug (NCT03720548). The trial was designed as a single- and multiple-dose, dose escalation study. Participants received remternetug monthly, with doses of 250, 700, 1400, or 2800 mg by intravenous infusion and compared with placebo; a titration group from 700 to 1400 mg was also included [69]. The study consisted of monthly doses for 6 months, followed by a 1-year extension. The primary outcome measure was number of participants who had one or more SAEs, while secondary outcome measures were PK and pharmacodynamic (PD) evaluations, including changes in Aβ burden by PET. Results showed a dose-dependent decrease in Aβ plaque by as much as 100 centiloids, with all participants receiving the 2800 mg dose of remternetug dropping below 24 centiloids within 3 months [69]. Safety data related to ARIA-E and ARIA-H are still blinded so comparisons between remternetug and placebo cannot be assessed [69].
A second phase I clinical trial is recruiting 224 individuals to investigate the safety and tolerability of remternetug in two parts (NCT04451408). Part A will include non-Japanese participants with AD, with inclusion criteria of gradual and progressive memory changes and MMSE score of ≥ 16. Part B will include healthy participants of first-generation Japanese origin with a BMI of 18–32 kg/m2. Remternetug will be administered as single or multiple doses, by intravenous infusion or subcutaneously, for about 61 weeks. The primary outcome measure is number of participants who had one or more SAEs in either part of the study. The secondary outcome measure for part A is changes in Aβ by PET, and secondary outcome measures for part B include assessment of PK/PD.
9.3 Phase III
A randomized, double-blind, placebo-controlled, phase III study is recruiting 600 participants with early symptomatic AD (NCT05463731). Inclusion criteria are gradual and progressive change in cognitive function, MMSE score of 20–28, plasma p-tau consistent with the presence of Aβ, and positive amyloid PET scan. One or two doses of remternetug or placebo will be administered by intravenous infusion or subcutaneously for 52 weeks. The primary outcome measure is the percentage of participants who reach Aβ plaque clearance on PET, and the secondary outcome measures are change of Aβ plaque on PET from baseline, time to reach Aβ plaque clearance, PK measure of trough serum concentration, and number of participants with treatment emergent antidrug antibodies. An extension period of an additional 52 weeks will be offered, where participants who have received placebo will receive remternetug and the participants who have received the drug will receive placebo. An open-label addendum safety cohort will enroll an additional 640 participants with early AD to receive remternetug either by intravenous infusion or subcutaneously.
10 Novel Agents in the Alzheimer’s Drug Development Pipeline
The agents described above are approved (aducanumab, lecanemab), under review (donanemab), terminated (gantenerumab), or in phase III (remternetug). Development of new anti-amyloid mAbs is an active area of the AD drug development pipeline. These agents aim to improve PK aspects, efficacy, safety, or convenience of mAbs. Subcutaneous (SC) administration is being explored. An SC formulation would eliminate the need for infusion, make home administration more feasible, and enable reaching a greater number of patients who may not reside near infusion centers.
Trontinemab and ABBV916 are in phase II clinical trials. Trontinemab combines gantenerumab with a ‘brain shuttle’ using the transferrin transporter to enhance BBB penetration and increase brain levels of the mAb [70]. This approach could improve efficacy while decreasing the amount of antibody (and the corresponding cost of production) required for treatment.
Phase I agents currently being assessed in single and multiple ascending dose trials include ACU193, SHR-1707, PMN310, and PRX012 [71].
11 Discussion
mAbs are the first DMTs for AD and among the first DMTs for any neurodegenerative disease. They demonstrate that changing the underlying biology of AD is possible and that reducing the pathological burden of the disease, in this case Aβ plaques, results in slowing of disease progression. mAbs are an unprecedented type of treatment for AD that redefine the paradigm of care from temporary relief of symptoms without an impact on the underlying biology of the disease to a combination approach with slowing of disease progression (applicable to early AD) based on Aβ reduction in conjunction with symptomatic benefits afforded by cholinesterase inhibitors and memantine. mAbs make unprecedented demands on healthcare systems for recognition of early AD to allow treatment of appropriate patients, PET or lumbar puncture to assess Aβ pathology before initiating therapy, infusion centers to administer mAbs, and multiple MRI scans in the initiation period of treatment to monitor for ARIA [72]. mAbs are the product of many years of investment in understanding AD and the role of Aβ in the pathogenesis of the disorder.
mAbs have important similarities and differences. They share the mechanistic target of fibrillar Aβ and the plaque reduction observed on PET. They differ in the specifics of the amyloid target—oligomers for aducanumab, protofibrils for lecanemab, and pyroglutamate Aβ for donanemab and remternetug. Remternetug and lecanemab development programs are exploring subcutaneous administration; all mAbs currently available or under review are administered by infusion. Lecanemab requires no titration and is administered at the same dose throughout treatment; donanemab features a two-step titration, and aducanumab requires a four-step titration [7, 8, 73]. Lecanemab is administered every other week after treatment initiation until the clinician and family terminate therapy; aducanumab is administered monthly until circumstances dictate stopping treatment; donanemab is infused monthly until amyloid plaque levels are undetectable on amyloid PET. ARIA occurs most commonly early in the treatment period and during titration, and the risk period is longest for aducanumab. ARIA has been observed in all trials of mAbs that lower plaque Aβ and that have been observed to reduce the rate of clinical decline. Among the completed trials, ARIA was reported most often with aducanumab treatment and least frequently with lecanemab. Infusion reactions occur with all the agents and were observed more commonly in the phase III trial of lecanemab than in other mAb trials. Some of the observed differences may be attributable to contrasts in trial design or participants and may not reflect drug-related distinctions.
While data from clinical trials are still preliminary, the available evidence indicates that end-of-trial Aβ levels above 25 centiloids predict the absence of slowing of disease progression regardless of the total amount of Aβ reduction achieved (e.g., patients starting at high Aβ levels may have substantial reductions but show no benefit if the end level is above 25 centiloids). This level was reached in positive lecanemab (phase II and III) and donanemab studies [3,4,5] but was not reached in negative gantenerumab studies [58]. Furthermore, this level was reached in the positive EMERGE study of aducanumab but not in the negative ENGAGE study of aducanumab [2]. Reaching the threshold of Aβ reduction required for slowing of disease progression is dose-related; trials with higher doses were more likely to attain the required decrease in Aβ burden [74]. Weight-adjusted dosing may be important to achieving the required exposure levels across populations of varying weights. These observations may help guide patient selection and treatment goals for mAbs.
The data available indicate that a threshold must be reached to change the biology of AD sufficiently to be associated with clinical benefit. The critical range of 15–25 centiloids has also been identified as important in natural history studies of AD [75, 76]. Patients with negative amyloid PET at baseline had future pathologic Aβ accumulation and decline on the Preclinical Alzheimer's Cognitive Composite if they reached an inflection point between 15 and 18.5 centiloids [77]. Aβ accumulation increases during the early phases of aggregation and plaque formation, and plateaus later in AD. Twenty-five centiloids coincides with the peak rate of Aβ accumulation [76]. This threshold relates to tau biology as well as Aβ biology. Twenty-five centiloids is the Aβ level associated with rising p-tau 231 and p-tau 217 levels to 2 SD above normal. Plasma GFAP, p-tau 181, and NfL do not rise to abnormal levels until later [78]. There is sharp rise in tau PET SUVR that begins when Aβ levels reach the vicinity of 25 centiloids [79]. Many older individuals with normal cognition have Aβ levels in excess of 25 centiloids, and dementia follows the rising amyloid levels at an interval of approximately 15–20 years, suggesting that secondary pathologies, such as tau aggregation, emerge in this interval and are associated with cognitive decline [80, 81]. Research indicates that exceeding an Aβ level of 25 centiloids coincides with cognitive impairment, cognitive benefit is observed between 15 and 25 centiloids, and a relationship to tau biology occurs at this level as a trigger for abnormal tau biomarkers.
In the clinical trials of donanemab, treatment was stopped when participants no longer had detectable levels of brain Aβ as determined by PET [5]. Aducanumab phase II and lecanemab phase II and III studies have continued treatment regardless of the magnitude of lowering demonstrated on amyloid PET. The donanemab target, pyroglutamate Aβ, is found only in plaques, whereas aducanumab and lecanemab engage oligomers or protofibrils, respectively, in addition to plaque Aβ. These target differences may warrant contrasting therapeutic approaches. Additional trials are needed to better define the optimal temporal regimen for each agent.
Aducanumab and lecanemab were approved using the accelerated approval pathway that accepts a biomarker change (e.g., Aβ lowering on amyloid PET) considered reasonably likely to predict clinical benefit. This pathway is used to make promising therapies available to patients with serious diseases and few therapeutic options while more compelling clinical data are generated [82]. Lecanemab received standard approval after a successful phase III study with positive primary and secondary clinical outcomes [4]. The relationship between Aβ lowering and slowing clinical decline has now been demonstrated in multiple trials with different MABs. Aβ lowering might be considered as an acceptable endpoint for secondary prevention trials of patients with preclinical AD, where clinical improvement cannot be shown in the absence of clinical deficits at baseline and delay of clinical decline may take too long to demonstrate in a trial of feasible duration. Changes in other biomarkers, including plasma biomarkers, might be considered as the basis for accelerated approval if multiple trials of multiple agents establish a predictive relationship with clinical benefit.
The meaningfulness of the degree of slowing of clinical decline observed in anti-amyloid MAB trials has been a subject of controversy [83]. The drug-placebo difference observed on clinical and functional measures is approximately 30% (Table 2). With data from the donanemab phase II trial, the drug-placebo difference was shown to translate into approximately 5 months delay of decline (‘time saved’) in an 18-month trial [84]. In the lecanemab phase III trial, the decline from baseline on the CDR-SB was 1.21 in the treatment group and 1.66 in the placebo group [4]. This 0.45 difference on the CDR-SB represents a 27% slowing in clinical decline in the treatment group compared with the placebo group. Given the slow progression of AD, a 27% additional slowing is difficult or impossible for clinicians, patients, and care partners to perceive. What can be appreciated in most circumstances is the time saved and longer residence in more mild stages of impairment [84, 85]. Modeling of the lecanemab outcomes with projection beyond an 18-month trial exposure period suggests delayed time to decline of 2.5 years to mild AD dementia [86]. Early treatment in the disease progression would provide the most benefit to the patient, demonstrating the importance of better screening and diagnostic tools for AD. It is anticipated that DMTs will produce widening treatment/no treatment differences over time, exhibiting greater cumulative benefit with longer treatment periods [87].
The risk versus benefit of treatment with mAbs must be considered by both the clinician and the patient and care partner. On average, the benefit of treatment is a 25–40% slowing of cognitive decline depending on the measure included in the clinical trials (as reviewed here). The major risk associated with mAb therapy is the occurrence of ARIA, which occurs in approximately 20–30% of individuals (higher in those with the APOE4 genotype, particularly homozygotes), with 5% of those receiving treatment experiencing symptoms of ARIA. Cerebral amyloid angiopathy (CAA), referring to the accumulation of Aβ in cerebral vasculature, could mediate the risk of ARIA from treatment with mAbs [88]. Additional adverse effects of mAbs could include headaches, seizures, confusion, and, in extreme cases, death [2, 4, 89]. The clinician must be aware of the risk profile of the patient, including the treatment candidate’s genotype, and the patient and caregiver must be fully informed of both the benefit and risk of treatment. The 25–40% slowing of decline is weighed against the unmodified inevitable descent into dementia, and the risk of ARIA is contextualized by the rare occurrence of severe reactions with fatal outcomes. The beneficial impact on patients’ lives is sufficient to warrant discussion of therapy in those who can be adherent to the required treatment regimens. Best practices for providing mAb treatment are available in appropriate use recommendations [8, 73].
Brain volume loss is observed in clinical trials of mAbs and other types of amyloid-related therapies. In mAb trials, the volume reduction is associated with the past occurrence of ARIA [89]. Volume loss observed in trials of β-site APP cleaving enzyme (BACE) inhibitors was non-progressive and greatest in amyloid-rich regions, suggesting a relationship to Aβ removal [90]. Brain atrophy from neuronal loss is less likely in the face of slowing of cognitive decline, but has not been excluded by brain autopsy studies. The etiology and biology of the volume loss requires careful study.
mAbs require sophisticated medical infrastructure for optimal and safe management. Diagnostic and treatment expertise, amyloid PET or CSF Aβ measures, MRI resources and interpretation, infusion centers, and intensive care units for the rare patient with life-threatening ARIA are required. Such resources are unavailable in many rural areas and in most low- and middle-income countries. Progress in blood tests that will allow a confident diagnosis of AD, and availability of subcutaneous formulations of mAbs, along with drugs with better efficacy and lower ARIA rates, are required to achieve global equity in AD care with DMTs.
Treatments with greater efficacy, greater safety, and more convenience are needed. Small molecule development programs are critical to meeting these goals. Amyloid has been validated as a therapeutic target by the mAbs, and small molecules with amyloid targets may succeed. There are many agents in the pipeline addressing brain inflammation and loss of synaptic integrity that have promise as therapeutics [71]. APOE4 plays a major role in the pathophysiology of AD, and drugs impacting the function of APOE4 may have therapeutic benefit. Anti-amyloid vaccines, DMT oral medications, and drugs for patients with more advanced forms of AD are all desirable outcomes of development programs to facilitate effective treatment of a broader range of patients with AD.
12 Conclusion
Anti-amyloid mAbs are the first DMTs for AD. There are two approved treatments, including aducanumab and lecanemab, whereas donanemab is undergoing regulatory review. mAbs have resulted in decreases of Aβ plaques and slowing of cognitive decline. Clinical trials investigating mAbs provide insight into the Aβ target species, drug administration and regimen, AEs, and relationship between Aβ plaques and cognitive decline. ARIA has been observed in all mAb clinical trials that resulted in Aβ plaque lowering. With 30% slowing of decline, the benefit for the patient may outweigh the associated risk with the treatment, although the clinician must be aware of the risk profile of the patient, and the patient and caregiver must be fully informed of both the benefit and the risk of treatment. Current mAbs represent a first step in the development of DMTs for AD and provide the foundation for development of safer, more efficacious, and more convenient approaches to disease modification.
References
Feigin VL, Nichols E, Alam T, Bannick MS, Beghi E, Blake N, et al. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–80.
Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis. 2022;9(2):197–210.
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res Ther. 2021;13(1):80.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21.
Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384(18):1691–704.
Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dementia. 2011;7(4):367–85.
Cummings J, Rabinovici GD, Atri A, Aisen P, Apostolova LG, Hendrix S, et al. Aducanumab: appropriate use recommendations update. J Prev Alzheimers Dis. 2022;9(2):221–30.
Cummings J, Apostolova L, Rabinovici GD, Atri A, Aisen P, Greenberg S, et al. Lecanemab: appropriate use recommendations. J Prev Alzheimers Dis. 2023;10(3):362–77. https://doi.org/10.14283/jpad.2023.30.
Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018;8(1):6412.
Söderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. Lecanemab, aducanumab, and gantenerumab—binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics. 2023;20(1):195–206.
Haddad HW, Malone GW, Comardelle NJ, Degueure AE, Kaye AM, Kaye AD. Aducanumab, a novel anti-amyloid monoclonal antibody, for the treatment of Alzheimer’s disease: a comprehensive review. Health Psychol Res. 2022;10(1):31925.
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173.
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–5.
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–62.
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54.
Nicoll JAR, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65(11):1040–8.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–23.
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–70.
Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8(1):18.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–33.
Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–72.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–21.
Zhang Y, Lee DHS. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist. 2011;17(2):163–73.
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378(4):321–30.
Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, et al. ABBY: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90(21):e1889–97.
Salloway S, Honigberg LA, Cho W, Ward M, Friesenhahn M, Brunstein F, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res Ther. 2018;10(1):96.
Ostrowitzki S, Bittner T, Sink KM, Mackey H, Rabe C, Honig LS, et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early Alzheimer disease: two phase 3 randomized placebo-controlled trials. JAMA Neurol. 2022;79(11):1113–21.
Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9(1):95.
FDA Office of the Commissioner. FDA. FDA; 2023 [cited 31 May 2023]. FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–6.
Bussiere T, Weinreb PH, Dunstan RW, Qian F, Arastu MF, Li M, et al. Differential in vitro and in vivo binding profiles of BIIB037 and other anti-abeta clinical antibody candidates. Neurodegener Dis. 2013. https://misc.karger.com/websites/NDD_2013_011_s_1/AbstractCD/pdf/787.pdf.
Biogen. A randomized, blinded, placebo-controlled single ascending dose study of the safety, tolerability, and pharmacokinetics of biib037 in subjects with mild to moderate Alzheimer’s disease. ClinicalTrials.gov; 2015 Mar [cited 23 Mar 2023]. Report No. NCT01397539. Available at: https://clinicaltrials.gov/ct2/show/NCT01397539.
Ferrero J, Williams L, Stella H, Leitermann K, Mikulskis A, O’Gorman J, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimer’s Dementia. 2016;2(3):169–76.
Biogen. A randomized, double-blinded, placebo-controlled multiple dose study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of BIIB037 in subjects with prodromal or mild Alzheimer’s disease. ClinicalTrials.gov; 2020 Jul [cited 23 Mar 2023]. Report No. NCT01677572. https://clinicaltrials.gov/ct2/show/NCT01677572.
Biogen. A phase 2, multicenter, randomized, parallel-group, double-blind, controlled study of aducanumab (BIIB037) in subjects with mild cognitive impairment due to Alzheimer’s disease or with mild Alzheimer’s disease dementia to evaluate the safety of continued dosing in subjects with asymptomatic amyloid-related imaging abnormalities. ClinicalTrials.gov; 2021 Aug [cited 30 Mar 2023]. Report No. NCT03639987. https://clinicaltrials.gov/ct2/show/NCT03639987.
Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79(1):13–21.
Biogen. A phase 3 multicenter, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of aducanumab (BIIB037) in subjects with early Alzheimer’s disease. ClinicalTrials.gov; 2021 Aug [cited 30 Mar 2023]. Report No. NCT02477800. https://clinicaltrials.gov/ct2/show/NCT02477800.
Biogen. A phase 3 multicenter, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of aducanumab (BIIB037) in subjects with early Alzheimer’s disease. ClinicalTrials.gov; 2021 Aug [cited 22 Mar 2023]. Report No. NCT02484547. https://clinicaltrials.gov/ct2/show/NCT02484547.
Herring WL, Gould IG, Fillit H, Lindgren P, Forrestal F, Thompson R, et al. Predicted lifetime health outcomes for aducanumab in patients with early Alzheimer’s disease. Neurol Ther. 2021;10(2):919–40.
von Rosenstiel P, Haeberlein SB, Castrillo-Viguera C, Chen T, O’Gorman J, Rajagovindan R, et al. Aducanumab 48-month analyses from PRIME, a phase 1b study in patients with early Alzheimer’s disease. 11th Clinical Trials on Alzheimer’s Disease Congress 2018; 2018 [cited 23 Aug 2023]; Barcelona, Spain. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8571451/.
Englund H, Sehlin D, Johansson AS, Nilsson LNG, Gellerfors P, Paulie S, et al. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J Neurochem. 2007;103(1):334–45.
FDA Office of the Commissioner. Eisai Newsroom. 2023. FDA Approves LEQEMBI™ (lecanemab-irmb) Under the Accelerated Approval Pathway for the Treatment of Alzheimer’s Disease [cited 31 May 2023]. https://media-us.eisai.com/2023-01-06-FDA-Approves-LEQEMBI-TM-lecanemab-irmb-Under-the-Accelerated-Approval-Pathway-for-the-Treatment-of-Alzheimers-Disease.
Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;4(9):887–93.
Lord A, Gumucio A, Englund H, Sehlin D, Sundquist VS, Söderberg L, et al. An amyloid-β protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;36(3):425–34.
Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE. 2012;7(2): e32014.
Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. Safety and tolerability of BAN2401—a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer’s Res Ther. 2016;8(1):14.
Satlin A, Wang J, Logovinsky V, Berry S, Swanson C, Dhadda S, et al. Design of a Bayesian adaptive phase 2 proof-of-concept trial for BAN2401, a putative disease-modifying monoclonal antibody for the treatment of Alzheimer’s disease. Alzheimer’s Dementia. 2016;2(1):1–12.
Rauchenberger R, Borges E, Thomassen-Wolf E, Rom E, Adar R, Yaniv Y, et al. Human combinatorial Fab library yielding specific and functional antibodies against the human fibroblast growth factor receptor 3. J Biol Chem. 2003;278(40):38194–205.
Novakovic D, Feligioni M, Scaccianoce S, Caruso A, Piccinin S, Schepisi C, et al. Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des Dev Ther. 2013;7:1359–64.
Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis. 2012;28(1):49–69.
Hoffmann-La Roche. A single-dose study to investigate the safety, tolerability, and pharmacokinetics (PK) of gantenerumab following subcutaneous (SC) administration in healthy volunteers. ClinicalTrials.gov; 2019 Apr [cited 30 May 2023]. Report No. NCT02711423. https://clinicaltrials.gov/ct2/show/NCT02711423.
Hoffmann-La Roche. A multiple ascending dose study of R1450 in patients with Alzheimer disease. ClinicalTrials.gov; 2016 Nov [cited 30 May 2023]. Report No. NCT00531804. https://clinicaltrials.gov/ct2/show/NCT00531804.
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207.
Hoffmann-La Roche. A study of gantenerumab in participants with prodromal Alzheimer’s disease (Scarlet Road). ClinicalTrials.gov; 2021 Dec [cited 30 May 2023]. Report No. NCT01224106. https://clinicaltrials.gov/ct2/show/NCT01224106.
Washington University School of Medicine. Dominantly inherited Alzheimer network trial: an opportunity to prevent dementia. a study of potential disease modifying treatments in individuals at risk for or with a type of early onset Alzheimer’s disease caused by a genetic mutation. Master Protocol DIAN-TU-001 (DIAN-TU). ClinicalTrials.gov; 2023 May [cited 30 May 2023]. Report No. NCT01760005. https://clinicaltrials.gov/ct2/show/NCT01760005.
Joseph-Mathurin N, Llibre-Guerra JJ, Li Y, McCullough AA, Hofmann C, Wojtowicz J, et al. Amyloid-related imaging abnormalities in the DIAN-TU-001 trial of gantenerumab and solanezumab: lessons from a trial in dominantly inherited Alzheimer disease. Ann Neurol. 2022;92(5):729–44.
Hoffmann-La Roche. Efficacy and safety study of gantenerumab in participants with early Alzheimer’s disease (AD). ClinicalTrials.gov; 2023 Apr [cited 30 May 2023]. Report No. NCT03444870. https://clinicaltrials.gov/ct2/show/NCT03444870.
Boess F, Sakaoka S, Abi-Saab D, Scelsi MA, Delmar P, Hofmann C, et al. Graduation study design: evaluation of once-weekly subcutaneous administration of gantenerumab on brain amyloid load. Alzheimer’s Dementia. 2021;17(S9): e052060.
DeMattos RB, Lu J, Tang Y, Racke MM, DeLong CA, Tzaferis JA, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;76(5):908–20.
Bouter Y, Liekefeld H, Pichlo S, Westhoff AC, Fenn L, Bakrania P, et al. Donanemab detects a minor fraction of amyloid-β plaques in post-mortem brain tissue of patients with Alzheimer’s disease and Down syndrome. Acta Neuropathol. 2022;143(5):601–3.
Lowe SL, Willis BA, Hawdon A, Natanegara F, Chua L, Foster J, et al. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimers Dement (N Y). 2021;7(1): e12112.
Lowe SL, Duggan Evans C, Shcherbinin S, Cheng YJ, Willis BA, Gueorguieva I, et al. Donanemab (LY3002813) phase 1b study in Alzheimer’s disease: rapid and sustained reduction of brain amyloid measured by florbetapir F18 imaging. J Prev Alzheimers Dis. 2021;8(4):414–24.
Shcherbinin S, Evans CD, Lu M, Andersen SW, Pontecorvo MJ, Willis BA, et al. Association of amyloid reduction after donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022;79(10):1015–24.
Pontecorvo MJ, Lu M, Burnham SC, Schade AE, Dage JL, Shcherbinin S, et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic alzheimer disease: a secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022;79(12):1250–9.
Eli Lilly and Company. Lilly—News Release. 2023. Lilly’s Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease. https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-significantly-slowed-cognitive-and-functional.
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512–27.
Eli Lilly and Company. Lilly—News Release. 2023. U.S. Food and Drug Administration Issues Complete Response Letter for Accelerated Approval of Donanemab. https://investor.lilly.com/news-releases/news-release-details/us-food-and-drug-administration-issues-complete-response-0.
Alzforum. 2023. Remternetug [cited 26 Apr 2023]. https://www.alzforum.org/therapeutics/remternetug.
Alzforum. 2023. International conference on Alzheimer’s and Parkinson’s diseases 2023: next goals for immunotherapy: make it safer, less of a hassle. https://www.alzforum.org/news/conference-coverage/next-goals-immunotherapy-make-it-safer-less-hassle.
Pardridge WM. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv. 2015;12(2):207–22.
Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement (N Y). 2023;9(2): e12385.
Cummings J. Anti-amyloid monoclonal antibodies are transformative treatments that redefine Alzheimer’s disease therapeutics. Drugs. 2023;83(7):569–76.
Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398–410.
Perneczky R, Jessen F, Grimmer T, Levin J, Flöel A, Peters O, et al. Anti-amyloid antibody therapies in Alzheimer’s disease. Brain. 2023;146(3):842–9.
Salvadó G, Molinuevo JL, Brugulat-Serrat A, Falcon C, Grau-Rivera O, Suárez-Calvet M, et al. Centiloid cut-off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimer’s Res Ther. 2019;11(1):27.
Jagust WJ, Landau SM. Temporal dynamics of β-amyloid accumulation in aging and Alzheimer disease. Neurology. 2021;96(9):e1347–57.
Farrell ME, Jiang S, Schultz AP, Properzi MJ, Price JC, Becker JA, et al. Defining the lowest threshold for amyloid-PET to predict future cognitive decline and amyloid accumulation. Neurology. 2021;96(4):e619–31.
Milà-Alomà M, Ashton NJ, Shekari M, Salvadó G, Ortiz-Romero P, Montoliu-Gaya L, et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med. 2022;28(9):1797–801.
Doré V, Krishnadas N, Bourgeat P, Huang K, Li S, Burnham S, et al. Relationship between amyloid and tau levels and its impact on tau spreading. Eur J Nucl Med Mol Imaging. 2021;48(7):2225–32.
Ossenkoppele R, Pichet Binette A, Groot C, Smith R, Strandberg O, Palmqvist S, et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med. 2022;28(11):2381–7.
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67.
Dunn B, Stein P, Temple R, Cavazzoni P. An appropriate use of accelerated approval—aducanumab for Alzheimer’s disease. N Engl J Med. 2021;385(9):856–7.
Cummings J, Aisen P, Lemere C, Atri A, Sabbagh M, Salloway S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res Ther. 2021;13(1):98.
Dickson SP, Wessels AM, Dowsett SA, Mallinckrodt C, Sparks JD, Chatterjee S, et al. ‘Time saved’ as a demonstration of clinical meaningfulness and illustrated using the donanemab TRAILBLAZER-ALZ study findings. J Prev Alzheimers Dis. 2023;10(3):595–9. https://doi.org/10.14283/jpad.2023.50.
Petersen RC, Aisen PS, Andrews JS, Atri A, Matthews BR, Rentz DM, et al. Expectations and clinical meaningfulness of randomized controlled trials. Alzheimer’s & Dementia. 2023;19(6):2730–6.
Tahami Monfared AA, Ye W, Sardesai A, Folse H, Chavan A, Aruffo E, et al. A path to improved Alzheimer’s care: simulating long-term health outcomes of lecanemab in early Alzheimer’s disease from the CLARITY AD trial. Neurol Ther. 2023;12(3):863–81.
Assunção SS, Sperling RA, Ritchie C, Kerwin DR, Aisen PS, Lansdall C, et al. Meaningful benefits: a framework to assess disease-modifying therapies in preclinical and early Alzheimer’s disease. Alzheimer’s Res Ther. 2022;14(1):54.
Sveikata L, Charidimou A, Viswanathan A. Vessels sing their ARIAs: the role of vascular amyloid in the age of aducanumab. Stroke. 2022;53(1):298–302.
Alves F, Kalinowski P, Ayton S. Accelerated brain volume loss caused by anti-β-amyloid drugs: a systematic review and meta-analysis. Neurology. 2023;100(20):e2114–24.
Sur C, Kost J, Scott D, Adamczuk K, Fox NC, Cummings JL, et al. BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer’s disease brain. Brain. 2020;143(12):3816–26.
Acknowledgements
Jeffrey Cummings is supported by NIGMS grant P20GM109025; NINDS grant U01NS093334; NIA grant R01AG053798; NIA grant P20AG068053; NIA grant P30AG072959; NIA grant R35AG71476; Alzheimer’s Disease Drug Discovery Foundation (ADDF); Ted and Maria Quirk Endowment; and the Joy Chambers-Grundy Endowment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jeffrey Cummings has provided consultation to Acadia, Actinogen, Acumen, AlphaCognition, Aprinoia, AriBio, Artery, Biogen, BioVie, Cassava, Cerecin, Diadem, EIP Pharma, Eisai, GemVax, Genentech, GAP Innovations, Janssen, Jocasta, Karuna, Lilly, Lundbeck, LSP, Merck, NervGen, Novo Nordisk, Oligomerix, Optoceutics, Ono, Otsuka, PRODEO, Prothena, ReMYND, Roche, Sage Therapeutics, Signant Health, Simcere, Suven, SynapseBio, TrueBinding, Vaxxinity, and Wren pharmaceutical, assessment, and investment companies. Davis Cammann, Jayde Powell, Amanda Leisgang Osse, and Jingchun Chen have no disclosures to declare in relation to this work.
Funding
No funding was received specifically for the development of this manuscript. Production of the manuscript is consistent with the lead author’s R35 award from the National Institute on Aging (NIA grant R35AG71476).
Ethics approval
No patient data are included in this article and all information is publicly available. No ethics approval is required.
Patient consent to participate/publish
No patients were included this this article. All information is available on ClinicalTrials.gov or derived from PubMed sources. No patient consent is required.
Availability of data and material
No database was developed for this article. All material cited is available on PubMed and cited in the references or derived from the publicly available registry ClinicalTrials.gov.
Code availability
No database was developed for this article. All material cited is available on PubMed and cited in the references or derived from the publicly available registry ClinicalTrials.gov.
Author contributions
All authors contributed to this article and approved the final manuscript. Each of the authors developed the section on at least one monoclonal antibody. Jeffrey Cummings conceived the article and produced the original outline. The outline was approved by all authors. Amanda Leisgang Osse managed the references and Mike de la Flor created the illustration.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cummings, J., Osse, A.M.L., Cammann, D. et al. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 38, 5–22 (2024). https://doi.org/10.1007/s40259-023-00633-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00633-2