
Abstract
Septic-associated encephalopathy (SAE) is a key manifestation of sepsis, ranging from delirium to coma and occurring in up to 70% of patients admitted to the ICU. SAE is associated with higher ICU and hospital mortality, and also with poorer long-term outcomes, including cognitive and functional outcomes. The pathophysiology of SAE is complex, and it may involve neurotransmitter dysfunction, inflammatory and ischemic lesions to the brain, microglial activation, and blood–brain barrier dysfunction. Delirium (which is included in the SAE spectrum) is mostly diagnosed with validated scales in the ICU population. There is no established treatment for SAE; benzodiazepines should generally be avoided in this setting. Nonpharmacological prevention and management is key for treating SAE; it includes avoiding oversedation (mainly with benzodiazepines), early mobilization, and sleep promotion.
Similar content being viewed by others

Introduction
Since ancient times, physicians have recognized that the central nervous system (CNS) is one of the first organs affected in sepsis, and its clinical manifestation is the so-called sepsis-associated encephalopathy (SAE) [1]. Sepsis, defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection [2], is a leading cause of ICU admission and death worldwide [3]. The incidence of SAE is about 50%. It varies from 8% to more than 70% of septic patients, according to the sepsis severity, patients’ profile, and SAE diagnostic criteria [4,5,6]. It can be the revealing manifestation of sepsis, and alteration in mental status has been identified as 1 of the 3 screening items for detecting sepsis [7]. Its pathophysiology comprises neuroinflammation, vascular changes, and metabolic failure leading to tissue lesions seen in animal models and observed in humans [1]. These mechanisms are not homogeneous in the brain and may lead to region-specific lesions. The centers involved in autonomic controls, arousal, awareness, and behavior are particularly affected, accounting for the clinical features of SAE varying from sickness behavior to consciousness impairment (i.e., ranging from delirium to coma) [1, 8]. SAE is characterized by changes in the electroencephalogram (EEG), with a nonconvulsive status epilepticus that can be detected in up to 20% of cases [9]. The radiologic lesions are uncommon, but white matter hypersignal and ischemic stroke can be observed in the subgroup of patients with septic shock ischemic lesions in about 30% of patients [10,11,12]. SAE is associated with increased mortality [4, 13] as well as long-term cognitive impairment—mainly affecting memory, attention, and verbal fluency [14, 15]—and psychological disorders, including depression, anxiety, and posttraumatic stress disorder (PTSD) [16, 17]. Its management relies mainly on general ICU good practices as specific treatment is still lacking.
The brain is an immune-privileged organ as it is less subject to the immune response, with consequences to make it tolerant to tumors and graft. Now the current thinking is that the CNS can be profoundly affected by severe systemic infections. Therefore, in this chapter, we will review the main characteristics of SAE and its pathophysiology, as well as its therapeutics and prognosis.
Epidemiology
Because of the absence of consensual definition of SAE, there is a great variation in its incidence across studies. Thus, 20 to 40% of septic patients admitted to the ICU will develop an encephalopathy. It has been reported that delirium is observed in up to 70% of elderly mechanically ventilated patients [14, 18]. The most consistent risk factors are age, previous cognitive impairment, kidney and liver failure, and sepsis severity [4, 14]. Similarly, bacteremia is accompanied by changes in neurological status, ranging from lethargy to coma, in about 70% of cases [4]. Finally, only 19% of the patients admitted to the ICU showed a normal EEG with alpha rhythm and 80% present EEG background or epileptic anomalies [19, 20].
It has been clearly shown that occurrence of encephalopathy increases the risk of death in septic patients, although the mechanisms underlying the relationships between SAE and mortality are not elucidated yet. Mortality rate is mainly associated with the clinical and electrophysiological severity of SAE.
Septic patients are at risk also of developing long-term cognitive impairment and psychological disorders. Thus, hospitalization for sepsis is associated with a 10% increase in the prevalence of cognitive impairment during 8 years [14, 21]. Attention, verbal fluency, executive function, verbal memory, and quick mental processing are the main cognitive functions impaired, whereas visual memory and visuoconstructive ability are usually spared [15, 22]. The psychological disorders include anxiety, depression, and PTSD [17, 23]. Sepsis even increases the risk for suicide within 2 years after its recovery [24]. These psychological and cognitive disorders dramatically impact quality of life and functional status [15]. At 1 year, up to 51% of septic patients have not returned to full-time employment [25]. It is considered that the cognitive dysfunction results from early sepsis-related insults of the hippocampus and frontal lobe [21, 26] and the psychological disorders from those involving the limbic system [1].
Pathophysiology
Systemic inflammation leads to deleterious effects on the brain parenchyma resulting in SAE. SAE can be evidenced clinically, electrophysiologically, or radiologically and can be modeled in animal studies.
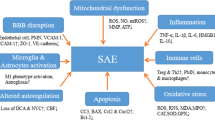
Specific pathways mediate this signal to the brain and triggers 3 processes which are neuroinflammation, ischemia, and cellular metabolic stress [27]. It is likely that complement system activation contributes to the BBB dysfunction [28]. Macro- and microcirculatory dysfunctions induce ischemia; the dysfunction of the vascular complex (which includes the endothelial cells, astrocytes, and blood–brain barrier) and the activation of microglia lead to neuroinflammation. These 2 processes add to other systemic factors (i.e., drug neurotoxicity, hypoxia, dysglycemia, renal or liver failure) and induce metabolic stress (Fig. 1). Oxidative stress closely linked to dysfunction of the mitochondria alters neuronal function and vitality [26, 29]. Specific brain regions are particularly sensitive to these processes, those involved in autonomic control, arousal, defining behavioral response to stress, and more complex cognitive functions such as memory and attention [30]. Strong neuronal activation is consistently evidenced in the hippocampus, the amygdala, the nucleus tractus solitarii, and the locus coeruleus [31]. An increased neuronal apoptosis associated with microglial activation has been specifically evidenced in these areas by the neuropathological study of the brain from patients who died from sepsis [32]. These findings provide the anatomic substrate of acute and long-term consequences of SAE.

Schematic view of the different pathophysiological processes observed or supposed during sepsis-associated encephalopathy. Vascular changes include blood–brain barrier dysfunction, neurovascular uncoupling, and strokes. Neuroinflammation includes microglial and astrocytic activation enhancing excitotoxicity and metabolic imbalance inducing neuronal cell death
Brain Signaling
The inflammatory signal in the brain involves the humoral and neural pathways. For example, the intraperitoneal inflammation signal is conveyed by vagal afferents to the medullary autonomic nuclei and the vagus nerve is able to modulate local and systemic inflammation (by its peritoneal and splenic innervations respectively). The neural pathways could also comprise other nerves and nuclei such as the trigeminal nerve and nucleus. The medullary autonomic nuclei can modulate more widely the response to the sepsis by their connections to other autonomic neuroendocrine and behavioral centers [32]. Areas deprived of BBB form the humoral pathway involving circumventricular organs and the area postrema. Inflammatory mediators are then allowed to traffic towards the local neuroendocrine and autonomic centers [33]. These 2 pathways orchestrate the inflammatory stress response clinically observable as the sickness behavior.
Blood–brain barrier (BBB) dysfunction is also observed during sepsis. It controls the blood–brain water, molecule, and ion balance and restrains immune cell, toxin, and pathogen crossing [34, 35]. Astrocytes and pericytes maintain its integrity. In septic shock, the neuropathological, electron micrograph examination and MRI studies confirm the BBB impairment, responsible for vasogenic edema in the white matter [11, 36]. Posterior reversible encephalopathy syndromes are evidenced in septic patients, corroborating these findings [37]. BBB impairment is also experimentally modeled, reproducing endothelial activation, complement activation, and the decrease in tight junction protein expression, namely occludin, ZO-1, ZO-2, claudin-3, and claudin-5 [38]. Also, an upregulation of aquaporine 4 has been evidenced, probably favoring brain edema in patients [36].
Neuroinflammation and Microglial Activation
In the brain, microglial cells are the main macrophage cells, representing most of the brain immune system. They also prune unused synapses and secrete neurotrophic factors contributing to synaptic plasticity. They also present phagocytic, migration, proliferation, and various mediator release capacities. Most of their various surface receptors interact with the peripheral immune system notably through cytokine ligation but can also sense damage- and pathogen-associated molecular patterns (DAMPs and PAMPs) [39].
Upon stimulation through circulating endotoxins or toll-like receptor pathways, microglial cells activate and present morphological, immunological, and metabolic changes. Morphological changes are mostly characterized by a shortening of their processes called deramification, up to an amoeboid form to the extreme. Various immunological phenotypes of microglia activation are observed in response to such activation, ranging from proinflammatory (M1) releasing proinflammatory cytokines (such as gamma interferon or tumor necrosis factor alpha) to anti-inflammatory (M2) ones releasing immune modulatory cytokines (such as IL4 or IL10). The neurotoxic consequences are usually considered secondary to proinflammatory phenotypes whereas anti-inflammatory phenotypes could be neuroprotective [39]. The neuronal activation and dysfunction is in part secondary to microglia activation; thus, the neuroinflammatory process contributes to long-term consequences of SAE. Cytokines, nitric oxide, excitatory gliotransmitters, and neurotoxic metabolites (such as reactive oxygen species) mediate the microglia-mediated neurotoxicity through an increase in neuronal excitability leading to hyperactivation and excitotoxicity [39, 40].
Animal [41, 42] and human [43, 44] studies during sepsis consistently show microglia activation without any known cerebral infection. This concept has recently been challenged as bacterial genomic material and living bacteria have been found in animals and humans without them leading to infectious encephalitis [45]. Whereas brain microbiota is considered absent in healthy subjects, this might be challenged in the critically ill [46]. Thus, targeting microbiota in the ICU could be a relevant therapy in the future [47, 48].
As microglia activation induces most probably brain damage during sepsis, its modulation seems a relevant approach for treating SAE [49, 50]. Promising experimental results have been published using various interventions including minocycline [51], cholinergic inhibition [52, 53], and vagal nerve stimulation [54]. Rivastigmine administration has been an original and convincing lead in the sepsis setting. It has been shown to reduce microglia activation by restoring the cholinergic inhibition. However, a randomized clinical trial evaluating rivastigmine has been prematurely interrupted for increasing mortality showing that manipulating the microglia may be hazardous [55]. The absence of a microglial phenotype biomarker limits clinical and experimental research allowing to classify easily such a dynamic phenomenon as microglial activation. Microglial activation has been characterized in Alzheimer disease using PET-CT, but its feasibility limits its transposition to septic patients until further improvements are made [56].
One dementia pathophysiology hypothesis is a neuroinflammatory cascade as microglial activation is consistently activated in such diseases. In these pathologies, microglial cells are considered as “primed” and overactivate after being submitted to a second hit. Sepsis can be considered the primer or the second hit [57]. This hypothesis can therefore account for the relationship between the rate of sepsis occurrence and intensity of the cognitive decline but also to its deleterious impact on patients with pre-existing neurodegenerative disease.
Astrocytes and Blood–Brain Barrier Dysfunction
Astrocytes represent the brain’s most numerous cells. They play a major role in brain homeostasis, and astrocyte dysfunction has been identified as a potential mechanism of SAE. BBB permeability, brain water balance, and microcirculatory cerebral brain flow (via the release of NO, prostaglandins, and arachidonic) are controlled by astrocytes [58]. They also participate in synaptic plasticity in the “tri-partite” synapse, releasing and reuptaking glutamate, GABA, and glycine. Astrocytes form extensive networks through gap junctions of connexins (Cx), channels permitting the bidirectional exchange of “gliotransmitters,” ions, or small molecules between astrocytes and neurons or in the extracellular milieu (hemichannels). In a neuroinflammatory context induced by LPS, microglial cell activation inhibits gap junction channels and open hemichannels [59]. Whether this occurs in SAE has to be evidenced.
In animal models of SAE, astrogliosis has been consistently evidenced [50]; however, these findings have never been reported in humans. Human studies rely solely on neuropathological findings and morphological analysis of glial fibrillary acid protein (GFAP) staining, which are insufficient to analyze the functionality of neuron–glia interaction and astrocytic network.
Ischemic Processes
Ischemic processes are separated in macrocirculatory dysfunction including hypotension, decreased cerebral flow, and impaired autoregulation—and microcirculatory impairment, characterized by neurovascular uncoupling impairment, disruption of the blood–brain barrier, and coagulation cascade activation. Septic shock induces ischemic damages observed in all cases of patients dead of septic shock which are associated in ~ 20% of cases with microhemorrhages associated to disseminated intravascular coagulopathy [60].
The cerebral blood flow (CBF) is maintained constant by autoregulation as mean systemic arterial pressure ranges between 60 and 150 mmHg. Below 60 mmHg, CBF decreases and induces olighemia whereas when MAP is above 150 mmHg, hyperhemia is observed. A decrease in the CBF has been consistently evidenced in septic shock, and an impairment of its autoregulation has been evidenced to be associated with delirium [29, 61].
Brain microcirculation is finely regulated through complex mechanisms. These depend on the gliovascular unit composed of endothelial cells, astrocytes, and pericytes [62]. When neuronal activity increases and consumes oxygen, the gliovascular unit couples the cerebral flow by vasodilation increasing locally the energy supply [62]. In experimental models of sepsis, the microcirculatory dysfunction has been evidenced by neurovascular uncoupling impairing cerebral vascular supply and leading to a deleterious metabolic crisis [26, 63].
In specific areas during sepsis, the metabolism is increased. For a normal mean blood pressure between 65 and 70 mmHg, the autoregulating mechanisms of the CBF allow the blood flow to match the energy demand. Sepsis can also impair cerebral autoregulation, compromising a major protection of the brain from ischemia. Thus, a decrease in mean arterial blood pressure below 70 mmHg can result in ischemic damage. Such disturbance could induce a mismatch between cerebral blood flow and metabolic demand [32].
Finally, such ischemic process could be involved in long-term cognitive decline as a similar mechanism to vascular dementia.
Mitochondrial Dysfunction
The metabolic and bioenergetics demands may result in oxidative stress and mitochondrial dysfunction during sepsis [26]. Indeed, early mitochondrial dysfunction has been shown in various brain regions of septic animals, resulting in reduced ATP generation and the production of oxygen/nitrogen reactive species [64]. This metabolic process is proapoptotic and involves the glial cells and neurons.
Neurotransmitter Dysfunction
Aside from mitochondrial dysfunction and oxidative stress, neurons are liable to excitotoxicity [65]. Neurotransmitter concentration in the synaptic cleft is the basis for excitotoxic processes occurring when neurotransmitter release is increased whereas its reuptake is diminished or insufficient [66]. The excitotoxic process is a pathological process occurring in neuroinflammation and ischemia but is also secondary to systemic perturbations such as hypoxemia, electrolyte disorders, dysglycemia, drug toxicity, and excess circulating neurotoxic amino acids (ammonium, tyrosine, tryptophan, and phenylalanine) [67, 68]. Excitotoxicity through glutamate receptors (N-methyl D aspartate receptor) induces functional impairment but also cell death, which could account for the clinical and electrophysiological features of SAE. Various synapses seem to be involved in the SAE, notably dopaminergic, β-adrenergic [69], the GABA [70], and the cholinergic ones [52, 71]. During delirium, a dopamine/choline imbalance is suspected [72] but its modulation with cholinergic drugs such as rivastigmine [55] or antidopaminergic drugs, like haloperidol [73], has not shown benefits in patients. On the other side, the risk of delirium is increased by benzodiazepines (GABA agonists) [74], but not reduced by noradrenergic drugs (i.e., dexmedetomidine) in septic patients [75].
Iatrogenic Factors
There are various drugs commonly administered in septic patients that can be neurotoxic, including antibiotics and sedative agents. Thus, antibiotics overdose has been shown to be associated with delirium. It has to be noted that antibiotic neurotoxicity is not always related to an overdose. Benzodiazepines have been clearly established to induce delirium. All pharmacological factors have to be systematically checked. ICU environment and sleep deprivation are also involved in the occurrence of delirium. The use of the ABCDEF bundle can be applied for controlling potential factors of delirium in septic patients [76].
Functional Neuroanatomy of SAE
A neuroanatomical approach to SAE helps in understanding not only SAE clinical features but also its associated increased mortality and long-term psychocognitive disorders (Fig. 2).

Early signs observed during sepsis (upper row), relying on specific structures (middle row) and associated with worst outcome (right row). Sickness behavior is considered as a physiological response to systemic inflammation; delirium and consciousness disorders are clinical signs of SAE. SAE = sepsis-associated encephalopathy, PTSD = posttraumatic stress disorder
Neuroanatomy of Response to Stress
During sepsis, the peripheral inflammation is transmitted to structures controlling autonomic and neuroendocrine systems interconnected to each other and to behavior and cognition centers, which in turn regulates the immune response through the “inflammatory reflex” [77,78,79,80]. The intensity of such response may be qualified in regard to the severity of sepsis as adapted, maladapted, or pathological (overactivated or blunted) [81, 82]. Such a distinction implies that recovery or not of sepsis might be influenced by the CNS response. However, there is no clinicobiological criteria to establish such a categorization. Through the neural pathway, the vagus nerve activates early the nucleus tractus solitarii and the locus coeruleus [31, 83, 84], which activate compensatory mechanisms in sepsis such as the control of blood pressure, heart rate, and arousal. These neural centers act as entry points to the CNS and stimulate the other autonomic nuclei and the behavioral and neuroendocrine centers. The precocity of the CNS activation by the neural route might explain that sickness behavior is one of the earliest features of sepsis (Fig. 3) [85].

Scheme for the response to stress network mobilization during sepsis. The peripheral inflammation signal is transmitted to the CNS through 3 main pathways (dark blue arrows) activating specific structures (light blue arrows), the circumventricular organs (CVO), or the vagus nerve nuclei. Behavioral, neuroendocrine, and autonomic structures are interconnected and stimulated, leading to the functional response (dark green arrows and rectangles). CNS = central nervous system, CVO = circumventricular organs, BBB = blood–brain barrier
Sickness behavior is a physiological response to a systemic inflammation that is clinically characterized by social withdrawal, impaired cognition (psychomotor retardation, impaired attention), altered alertness (anxiety, hypersomnia, fatigue, sleepiness), and eating disorders (anorexia, weight loss, thirst). Thus, a severe sickness behavior can mimic a hypoactive delirium [86]. Use of a specific scale for these entities is therefore clinically useful [87]. Interestingly, in brains of patients who died from septic shock, the neuropathological examination of response to stress structures consistently showed an increased number of apoptotic cells. This finding underlines their particular sensitivity to the excitotoxic process [32, 60, 85].
Brainstem and Mortality and Delirium
Four broad functions are controlled by the brainstem: brainstem reflexes, sleep–wake cycle, control of vital functions, and modulation of the immune response [88]. Brainstem dysfunction might then result in swallowing disorders, impaired arousal, cardiovascular sympathetic activity and respiratory drive dysfunction, and both cholinergic and adrenergic immunomodulation. These changes might account for the increased mortality, development of organ failure, and fluctuating wakefulness. We have found in deeply sedated critically ill patients that a specific pattern of heterogeneous brainstem reflex abolition was associated with higher mortality and delirium to the absence of oculocephalogyre reflex [89]. Also, a dysfunction of the reticular activating ascending substance has been suggested by the absence of EEG reactivity, and is predictive of death in septic patients [90]. In addition, critical illness increases brainstem latencies of auditory evoked potential suggesting common mechanisms of sepsis with other pathologies [91].
Finally, in septic patients, the decrease in heart rate variability is associated with bad outcome and reflects the sympathovagal imbalance [81]. Sepsis is also associated with an impairment in the baroreflex, controlled by medullar nuclei. Circulating inflammatory mediators can diffuse at the level of the area postrema, inducing an intense neuroinflammatory process leading to brainstem dysfunction. These mechanisms are supposed to account for the multifocal necrotizing leukoencephalopathy associated with marked apoptosis within the brainstem autonomic nuclei reported in a patient who died from septic shock [92].
Amygdala and Psychological Disorders
Postsepsis psychological disorders, also gathered under the name “postsepsis syndrome,” involve fear and anxiety circuits, notably the amygdala, the bed nucleus stria terminalis, the hypothalamus, or other brainstem nuclei [93]. Amygdala nuclei are involved in fear behavior expression (generalization, freezing) and traumatic memory formation as well as anxiety and depression symptoms. Also, within the response to stress network, amygdala mediates the response to stress network behavioral changes and the sickness behavior features (anorexia, anxiety, avoidance). In a model of LPS injections, the amygdala microcircuitry has been shown to define anorexic behavior [94]. Within the amygdala, microglia seems to be particularly involved [84], maybe explaining the efficiency of corticosteroids in the prevention of PTSD in septic patients [95]. The beneficial effects of corticoids might be explained on gene transcription but also through epigenetics pathways involved in aversive memory formation.
Hippocampus, Frontal Cortex, White Matter, and Cognitive Decline
Memory formation requires an intact hippocampus and frontal cortex. In septic animals, hippocampal dysfunction has been modeled and excitotoxicity secondary to long-term potentiation, gliosis, neuroinflammation, and increased production of reactive oxygen species (ROS) hypoxemia and ischemia lead to late neuronal death [51]. As these processes might be prolonged after sepsis control, it is not surprising that hippocampal dysfunction can progress toward atrophy [21]. Sepsis alters not only memorization but also other frontal cortex-specific cognitive functions such as attention, verbal fluency, and executive functions [14]. Micromacrovascular and neuroinflammation within the frontal cortex can contribute widely to cognitive impairment.
Finally, MRI studies in patients with septic shock showed white matter damages as expected according to the pathological findings [11]. This sepsis-induced axonopathy is largely unknown but might be similar to those of critical illness polyneuropathy. It is highly conceivable that it contributes to the long-term cognitive impairment.
Diagnosis
Clinical Features
SAE is defined by the combination of extracranial infection with clinical signs of neurological dysfunction. SAE clinical manifestation encompasses impairment of awareness, which ranges from delirium (50%) to coma (46%) [6]. Sepsis-related delirium is rather hypoactive than hyperactive. SAE be associated with focal deficits (18%) [11], seizures (10%) [20, 96], nonepileptic myoclonus, tremor, or asterixis [96]. CAM-ICU and ICSDC can be used for diagnosing delirium and GCS and FOUR for monitoring coma. In deeply sedated patients, the BRASS score can be helpful for detecting brainstem dysfunction [18].
It has to be underlined that sickness behavior is the first manifestation of sepsis, which can be distinguished from hypoactive delirium with the help of an appropriate scale. Second, the ICU physician must look for sepsis in any patient who develops change in mood, behavior, or consciousness. The suspicion of a brain infection must prompt the ICU physician to perform brain imaging and lumbar puncture. Finally, changes in the mental status of a septic patient can be related to other causes besides infection (i.e., electrolyte disturbances, drug overdose or withdrawal, vitamin deficiency, etc.).
Neurophysiological Features
There are 2 prospective electrophysiological studies in septic patients applying the ACNS guidelines [20, 96, 97]. A recent systematic review indicates that EEG and evoked potentials are sensitive, but not specific, to SAE [9].
Electroencephalogram
The electrophysiological classification developed by Young et al. for the classification of SAE severity takes mainly into account the background activity and includes 4 grades (grade 0, normal EEG; grade 1, predominant theta activity; grade 2, predominant delta activity; grade 3, predominant triphasic waves; and grade 4, suppression) [19]. In a prospective study based on 20-min standard EEG done within the first 3 days after ICU admission, Azabou et al. detected delta and theta predominant rhythms in 33% and 48%, triphasic waves in 6%, and suppression in less than 3% among 110 septic patients [20]. Low voltage and absence of reactivity were seen in 65% and 25% of cases, respectively. The predictors of mortality and occurrence of delirium were absence of EEG reactivity, a delta-predominant background, periodic discharges (PDs), Synek grade ≥ 3, and Young grade > 1 at day 1 to 3 following admission. In a study evaluating continuous EEG in 98 severely septic patients, Gilmore et al. detected nonconvulsive status (NCS), PD, and lack of reactivity in 11%, 25%, and 28% of cases, respectively [96]. Only the lack of reactivity was associated with mortality at 1 year.
The incidences of pseudoepileptic discharges (PED) and epileptic seizures vary between 19 and 48% and 9 to 50% [98], respectively. The discrepancies between studies in terms of sepsis severity, time, duration and type of EEG recording, and EEG criteria account for these wide ranges. It has to be noted that electrographic seizures are always associated with PED, which are predictive, when generalized and frequent, of subsequent electrographic seizure. Continuous EEG monitoring has evidenced that 75–95% of epileptic events are detected in the first 48 h [10].
There is no strong correlation between clinical manifestation and EEG findings. Thus, clinical seizures are exceptionally reported in septic patients, indicating that electrographic seizures are mostly nonconvulsive [96]. If electrographic seizures are more frequent in patients with delirium at time of EEG, EEG does not show epileptic activity in most comatose and delirious septic patients. Moreover, EEG can be abnormal even without changes in neurological status [10].
These findings suggest that EEG changes are rather markers of severity than pathogenic. Indeed, the prognosis value of EEG patterns has been clearly evidenced. Mortality increased from 0% in the case of normal EEG to 50 to 67% in the case of triphasic waves or suppression. Generalized PED and absence of reactivity are predictive of mortality but also subsequent occurrence of delirium after adjustment to severity of sepsis and sedation [20]. Finally, it has been reported that sepsis slightly increases the risk of long-term epilepsy. There is no evidence for recommending a preventive antiepileptic therapy as well as no clear recommendation on how to treat electrographic seizure in septic patients. However, we think continuous EEG should be used, whenever it is available, in septic patients in order to detect nonconvulsive seizures, especially in case of renal or hepatic insufficiency. All potential epileptogenic factors must be assessed in septic patients, including antibiotic neurotoxicity, drug overdose or withdrawal, electrolyte disturbances, etc. Finally, we recommend brain imaging in case of electrographic seizure or malignant EEG pattern.
Evoked Potentials
Subcortical (i.e., N20–N23 interlatency) and cortical (N20–N70 interlatency) pathways of somatosensory evoked potential (SSEP) are impaired early in 34% and 84% of 68 septic patients [99]. Evoked potentials are useful for assessing SAE in sedated critically ill patients, as, in contrast to EEG, their latencies are slightly altered by sedatives. Thus, increased P14–N20 SSEP interlatency was associated with mortality in this population [91].
Brain Imaging
There is retrospective or prospective imaging studies on a homogeneous and large cohort of septic patients. The only prospective MRI study was performed on 72 septic shock patients who developed an acute brain dysfunction defined by focal neurological sign (18%), seizure (10%), coma (46%), and delirium (49%). The MRI was normal in half of cases in this selected cohort. New white matter hyperdensities (suggestive of vasogenic edema) and ischemic stroke were observed in half of the remaining cases [11]. Sepsis can be complicated by posterior reversible encephalopathy syndrome (PRESS) [37]. Because of the risk of transporting septic patients, brain imaging should be indicated upon relevant clinical deterioration, including focal neurological deficit, seizure, and unexplained impaired consciousness. MRI is more specific and sensitive than brain CT scan, which conversely is easier and may complete a body scan. Transcranial Doppler imaging could also predict the occurrence of SAE with good diagnostic performance; however, these results are not found consistent, because of discrepancy in population, severity of sepsis, time of the test, and TCD criteria.
Biomarkers
Biomarkers might be helpful for detecting and monitoring SAE by targeting different structures involved in the pathophysiological processes. Some markers of systemic inflammation can be associated with the occurrence of SAE, such as C reactive protein or procalcitonin. There are also various markers of brain damage. N-Terminal propeptide of CNP (NT-proCNP), protein S100b, and neuron-specific enolase (NSE) or neurofilament, are respectively biomarkers of endothelial dysfunction, microglial activation, and brain injury with axonal damage [100,101,102]. They can be measured in the plasma or in the CSF. Although their increase can correlate with the severity of SAE, their sensibility and specificity remain low, limiting their clinical relevancy. One may argue that their combined assessment might improve their clinical and prognosis value.
Management
The treatment of SAE is based both on the management of sepsis and on the correction of potential neurotoxic factors and management of seizure, delirium, and coma. There is no specific treatment of SAE. Recent randomized clinical trials have not evidenced any benefit of statins and dexmedetomidine for preventing delirium in septic patients [103, 104], despite their respective anti-neuroinflammatory and noradrenergic/anti-apoptotic properties. If rivastigmine was theoretically interesting because of its potential microglial effect, it has been shown to increase mortality without reducing delirium in ICU patients, including septic ones. The SAILS and HARP2 trials failed to further demonstrate such effects in an interventional trial [103, 105].
There is no specific recommendation on pharmacological management of sepsis-associated delirium and seizure. It has to be reminded that fever can be due to neuroleptic malignant syndrome and choice of the anti-epileptic drug depends on renal and liver organ failure. To date, no specific sedative agent is particularly recommended, except that sedation has to be avoided or discontinued whenever possible [106]. The effect of sevoflurane on mortality and inflammatory parameters is currently assessed (NCT03643367). Ketamine is potentially neuroprotective in SAE but has not yet been tested in interventional trials. Enhanced sensitivity towards benzodiazepines is present in systemic inflammatory processes. Dexmedetomidine, an alpha agonist agent, has been shown to increase delirium-free days and reduce 28-day mortality rate in septic shock [104]. However, a recent large study showed that early goal-directed sedation based on dexmedetomidine did not show any benefit in critically ill patients [75].
While SAE could result from a mismatch between energy demand and cerebral blood flow, a personalized target of mean arterial pressure defined upon Doppler criteria could be evaluated in further work. However, such an approach has never been evaluated.
The nonpharmacological interventions recommended for preventing or treating delirium should be applied in septic patients, including early mobilization [107], discontinuation of sedation [106], rehydration, management of physical and psychological discomfort [108], and avoidance of prodelirious drugs.
Conclusion
SAE is a major complication of sepsis. It is characterized by changes in neurological status, ranging from sickness behavior to delirium to coma. It is associated with changes in EEG background activity and less frequently with electrographic seizures. It has a major impact on outcome, as it increases mortality and causes long-term psychocognitive disorders. Neuroinflammation, ischemia, and excitoxicity are its main pathophysiological mechanisms. Brainstem dysfunction might account for increased mortality while amygdala and hippocampus/frontal cortex dysfunction for psychological and cognitive disorders. There is no specific treatment for SAE, which depends on management of sepsis, delirium, seizure, and coma.
References
Mazeraud A, Pascal Q, Verdonk F, Heming N, Chrétien F, Sharshar T (2016) Neuroanatomy and physiology of brain dysfunction in sepsis. Clin Chest Med 37:333–345
Singer M, Deutschman CS, Seymour CW, et al (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801–810
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–1310
Eidelman LA, Putterman D, Putterman C, Sprung CL (1996) The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 275:470–473
Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA (1990) The encephalopathy associated with septic illness. Clin Investig Med Med Clin Exp 13:297–304
Ely EW, Shintani A, Truman B, Speroff T, Gordon SM, Harrell FE, Inouye SK, Bernard GR, Dittus RS (2004) Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA 291:1753–1762
Seymour CW, Liu VX, Iwashyna TJ, et al (2016) Assessment of clinical criteria for sepsis: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:762–774
Cunningham C, Maclullich AMJ (2013) At the extreme end of the psychoneuroimmunological spectrum: delirium as a maladaptive sickness behaviour response. Brain Behav Immun 28:1–13
Hosokawa K, Gaspard N, Su F, Oddo M, Vincent J-L, Taccone FS (2014) Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Crit Care. https://doi.org/10.1186/s13054-014-0674-y
Oddo M, Carrera E, Claassen J, Mayer SA, Hirsch LJ (2009) Continuous electroencephalography in the medical intensive care unit. Crit Care Med 37:2051–2056
Polito A, Eischwald F, Maho A-L, Polito A, Azabou E, Annane D, Chrétien F, Stevens RD, Carlier R, Sharshar T (2013) Pattern of brain injury in the acute setting of human septic shock. Crit Care Lond Engl 17:R204
Finelli PF, Uphoff DF (2004) Magnetic resonance imaging abnormalities with septic encephalopathy. J Neurol Neurosurg Psychiatry 75:1189–1191
Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB (1990) Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 18:801–806
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304:1787–1794
Jackson JC, Hart RP, Gordon SM, Shintani A, Truman B, May L, Ely EW (2003) Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 31:1226–1234
Hatch R, Young D, Barber V, Griffiths J, Harrison DA, Watkinson P (2018) Anxiety, depression and post traumatic stress disorder after critical illness: a UK-wide prospective cohort study. Crit Care Lond Engl 22:310
Righy C, Rosa RG, da Silva RTA, Kochhann R, Migliavaca CB, Robinson CC, Teche SP, Teixeira C, Bozza FA, Falavigna M (2019) Prevalence of post-traumatic stress disorder symptoms in adult critical care survivors: a systematic review and meta-analysis. Crit Care Lond Engl 23:213
Riker RR, Fugate JE, Participants in the International Multi-disciplinary Consensus Conference on Multimodality Monitoring (2014) Clinical monitoring scales in acute brain injury: assessment of coma, pain, agitation, and delirium. Neurocrit Care 21 Suppl 2:S27-37
Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA (1992) The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol Off Publ Am Electroencephalogr Soc 9:145–152
Azabou E, Magalhaes E, Braconnier A, et al (2015) Early standard electroencephalogram abnormalities predict mortality in septic intensive care unit patients. PloS One 10:e0139969
Semmler A, Widmann CN, Okulla T, et al (2013) Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry 84:62–69
Pandharipande PP, Girard TD, Jackson JC, et al (2013) Long-term cognitive impairment after critical illness. N Engl J Med 369:1306–1316
Nikayin S, Rabiee A, Hashem MD, Huang M, Bienvenu OJ, Turnbull AE, Needham DM (2016) Anxiety symptoms in survivors of critical illness: a systematic review and meta-analysis. Gen Hosp Psychiatry 43:23–29
Lund-Sørensen H, Benros ME, Madsen T, Sørensen HJ, Eaton WW, Postolache TT, Nordentoft M, Erlangsen A (2016) A nationwide cohort study of the association between hospitalization with infection and risk of death by suicide. JAMA Psychiatry 73:912–919
Herridge MS, Cheung AM, Tansey CM, et al (2003) One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 348:683–693
Bozza FA, D’Avila JC, Ritter C, Sonneville R, Sharshar T, Dal-Pizzol F (2013) Bioenergetics, mitochondrial dysfunction, and oxidative stress in the pathophysiology of septic encephalopathy. Shock Augusta Ga 39 Suppl 1:10–16
Adam N, Kandelman S, Mantz J, Chrétien F, Sharshar T (2013) Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther 11:211–221
Flierl MA, Stahel PF, Rittirsch D, et al (2009) Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit Care Lond Engl 13:R12
Taccone FS, Su F, Pierrakos C, He X, James S, Dewitte O, Vincent J-L, De Backer D (2010) Cerebral microcirculation is impaired during sepsis: an experimental study. Crit Care 14:R140
Hennessy E, Gormley S, Lopez-Rodriguez AB, Murray C, Murray C, Cunningham C (2017) Systemic TNF-α produces acute cognitive dysfunction and exaggerated sickness behavior when superimposed upon progressive neurodegeneration. Brain Behav Immun 59:233–244
Skelly DT, Hennessy E, Dansereau M-A, Cunningham C (2013) A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, [corrected] TNF-α and IL-6 challenges in C57BL/6 mice. PloS One 8:e69123
Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D, Raphael J-C, Gajdos P, Annane D (2003) Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet Lond Engl 362:1799–1805
Shechter R, Miller O, Yovel G, et al (2013) Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38:555–569
Cain MD, Salimi H, Diamond MS, Klein RS (2019) Mechanisms of pathogen invasion into the central nervous system. Neuron 103:771–783
McCaffrey G, Davis TP (2012) Physiology and pathophysiology of the blood brain barrier. J Investig Med 60(8):1131–1140. https://doi.org/10.2310/jim.0b013e318276de79
Davies D (2002) Blood–brain barrier breakdown in septic encephalopathy and brain tumours. J Anat 200:639–646
Racchiusa S, Mormina E, Ax A, Musumeci O, Longo M, Granata F (2019) Posterior reversible encephalopathy syndrome (PRES) and infection: a systematic review of the literature. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol 40:915–922
Danielski LG, Giustina AD, Badawy M, Barichello T, Quevedo J, Dal-Pizzol F, Petronilho F (2018) Brain barrier breakdown as a cause and consequence of neuroinflammation in sepsis. Mol Neurobiol 55:1045–1053
Wolf SA, Boddeke HWGM, Kettenmann H (2017) Microglia in physiology and disease. Annu Rev Physiol 79:619–643
Becher B, Spath S, Goverman J (2017) Cytokine networks in neuroinflammation. Nat Rev Immunol 17:49–59
Michels M, Sonai B, Dal-Pizzol F (2017) Polarization of microglia and its role in bacterial sepsis. J Neuroimmunol 303:90–98
Hoogland ICM, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12:114
Lemstra AW, Groen in’t Woud JCM, Hoozemans JJM, van Haastert ES, Rozemuller AJM, Eikelenboom P, van Gool WA (2007) Microglia activation in sepsis: a case-control study. J Neuroinflammation 4:4
Zrzavy T, Höftberger R, Berger T, Rauschka H, Butovsky O, Weiner H, Lassmann H (2019) Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol Appl Neurobiol 45:278–290
Singer BH, Dickson RP, Denstaedt SJ, Newstead MW, Kim K, Falkowski NR, Erb-Downward JR, Schmidt TM, Huffnagle GB, Standiford TJ (2017) Bacterial dissemination to the brain in sepsis. Am J Respir Crit Care Med 197:747–756
Mazeraud A, Bozza FA, Sharshar T (2018) Sepsis-associated encephalopathy is septic. Am J Respir Crit Care Med 197:698–699
Branton WG, Ellestad KK, Maingat F, Wheatley BM, Rud E, Warren RL, Holt RA, Surette MG, Power C (2013) Brain microbial populations in HIV/AIDS: α-proteobacteria predominate independent of host immune status. PloS One 8:e54673
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW (2014) Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87
Murray C, Sanderson DJ, Barkus C, Deacon RMJ, Rawlins JNP, Bannerman DM, Cunningham C (2012) Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol Aging 33:603-616.e3
Moraes CA, Santos G, de Sampaio e Spohr TCL, D’Avila JC, Lima FRS, Benjamim CF, Bozza FA, Gomes FCA (2015) Activated microglia-induced deficits in excitatory synapses through IL-1β: implications for cognitive impairment in sepsis. Mol Neurobiol 52:653–663
Zhu F, Zheng Y, Ding Y, Liu Y, Zhang X, Wu R, Guo X, Zhao J (2014) Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS ONE. https://doi.org/10.1371/journal.pone.0093966
Hofer S, Eisenbach C, Lukic IK, et al (2008) Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit Care Med 36:404–408
Zaghloul N, Addorisio ME, Silverman HA, et al (2017) Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front Immunol. https://doi.org/10.3389/fimmu.2017.01673
Schweighöfer H, Rummel C, Roth J, Rosengarten B (2016) Modulatory effects of vagal stimulation on neurophysiological parameters and the cellular immune response in the rat brain during systemic inflammation. Intensive Care Med Exp. https://doi.org/10.1186/s40635-016-0091-4
van Eijk MMJ, Roes KCB, Honing MLH, et al (2010) Effect of rivastigmine as an adjunct to usual care with haloperidol on duration of delirium and mortality in critically ill patients: a multicentre, double-blind, placebo-controlled randomised trial. Lancet Lond Engl 376:1829–1837
Horti AG, Naik R, Foss CA, et al (2019) PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc Natl Acad Sci 116:1686–1691
Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224
Howarth C (2014) The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci. https://doi.org/10.3389/fnins.2014.00103
Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez PJ, Sáez JC, Giaume C (2007) Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci Off J Soc Neurosci 27:13781–13792
Sharshar T, Annane D, Gradmaison GL de la, Brouland JP, Hopkinson NS, Gray F (2004) The neuropathology of septic shock. Brain Pathol 14:21–33
Taccone FS, Castanares-Zapatero D, Peres-Bota D, Vincent J-L, Berre’ J, Melot C (2010) Cerebral autoregulation is influenced by carbon dioxide levels in patients with septic shock. Neurocrit Care 12:35–42
Brown LS, Foster CG, Courtney J-M, King NE, Howells DW, Sutherland BA (2019) Pericytes and neurovascular function in the healthy and diseased brain. Front Cell Neurosci. https://doi.org/10.3389/fncel.2019.00282
Rosengarten B, Krekel D, Kuhnert S, Schulz R (2012) Early neurovascular uncoupling in the brain during community acquired pneumonia. Crit Care 16:R64
Azevedo LCP (2010) Mitochondrial dysfunction during sepsis. Endocr Metab Immune Disord Drug Targets 10:214–223
Berg RMG, Møller K, Bailey DM (2011) Neuro-oxidative-nitrosative stress in sepsis. J Cereb Blood Flow Metab 31:1532–1544
Bezzi P, Domercq M, Brambilla L, et al (2001) CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710
Darcy CJ, Davis JS, Woodberry T, McNeil YR, Stephens DP, Yeo TW, Anstey NM (2011) An observational cohort study of the kynurenine to tryptophan ratio in sepsis: association with impaired immune and microvascular function. PloS One 6:e21185
Freund H, Atamian S, Holroyde J, Fischer JE (1979) Plasma amino acids as predictors of the severity and outcome of sepsis. Ann Surg 190:571–576
Kadoi Y, Saito S, Kunimoto F, Imai T, Fujita T (1996) Impairment of the brain β-adrenergic system during experimental endotoxemia. J Surg Res 61:496–502
Serantes R, Arnalich F, Figueroa M, et al (2006) Interleukin-1β enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway relevance to sepsis-associated encephalopathy. J Biol Chem 281:14632–14643
Zhang Q-H, Sheng Z-Y, Yao Y-M (2014) Septic encephalopathy: when cytokines interact with acetylcholine in the brain. Mil Med Res. https://doi.org/10.1186/2054-9369-1-20
Bleck TP (2018) Dopamine antagonists in ICU delirium. N Engl J Med 379:2569–2570
Girard TD, Exline MC, Carson SS, et al (2018) Haloperidol and ziprasidone for treatment of delirium in critical illness. N Engl J Med 379:2506–2516
Pandharipande P, Shintani A, Peterson J, Pun BT, Wilkinson GR, Dittus RS, Bernard GR, Ely EW (2006) Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 104:21–26
Shehabi Y, Howe BD, Bellomo R, et al (2019) Early sedation with dexmedetomidine in critically ill patients. N Engl J Med. https://doi.org/10.1056/NEJMoa1904710
Marra A, Ely EW, Pandharipande PP, Patel MB (2017) The ABCDEF bundle in critical care. Crit Care Clin 33:225–243
Muscatell KA, Dedovic K, Slavich GM, Jarcho MR, Breen EC, Bower JE, Irwin MR, Eisenberger NI (2015) Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav Immun 43:46–53
Stare J, Siami S, Trudel E, Prager-Khoutorsky M, Sharshar T, Bourque CW (2015) Effects of peritoneal sepsis on rat central osmoregulatory neurons mediating thirst and vasopressin release. J Neurosci Off J Soc Neurosci 35:12188–12197
Siami S, Bailly-Salin J, Polito A, et al (2010) Osmoregulation of vasopressin secretion is altered in the postacute phase of septic shock. Crit Care Med 38:1962–1969
Sonneville R, Guidoux C, Barrett L, et al (2010) Vasopressin synthesis by the magnocellular neurons is different in the supraoptic nucleus and in the paraventricular nucleus in human and experimental septic shock. Brain Pathol Zurich Switz 20:613–622
Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, Gajdos P (1999) Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med 160:458–465
Annane D, Sébille V, Charpentier C, et al (2002) Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 288:862–871
Reyes E-P, Abarzúa S, Martin A, Rodríguez J, Cortés PP, Fernández R (2012) LPS-induced c-Fos activation in NTS neurons and plasmatic cortisol increases in septic rats are suppressed by bilateral carotid chemodenervation. Adv Exp Med Biol 758:185–190
Carlson DE, Chiu WC, Fiedler SM, Hoffman GE (2007) Central neural distribution of immunoreactive Fos and CRH in relation to plasma ACTH and corticosterone during sepsis in the rat. Exp Neurol 205:485–500
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Peterson JF, Pun BT, Dittus RS, Thomason JWW, Jackson JC, Shintani AK, Ely EW (2006) Delirium and its motoric subtypes: a study of 614 critically ill patients. J Am Geriatr Soc 54:479–484
Andreasson A, Wicksell RK, Lodin K, Karshikoff B, Axelsson J, Lekander M (2018) A global measure of sickness behaviour: development of the sickness questionnaire. J Health Psychol 23:1452–1463
Tracey KJ (2009) Reflex control of immunity. Nat Rev Immunol 9:418–428
Rohaut B, Porcher R, Hissem T, et al (2017) Brainstem response patterns in deeply-sedated critically-ill patients predict 28-day mortality. PloS One 12:e0176012
Azabou E, Navarro V, Kubis N, Gavaret M, Heming N, Cariou A, Annane D, Lofaso F, Naccache L, Sharshar T (2018) Value and mechanisms of EEG reactivity in the prognosis of patients with impaired consciousness: a systematic review. Crit Care Lond Engl 22:184
Azabou E, Rohaut B, Heming N, et al (2017) Early impairment of intracranial conduction time predicts mortality in deeply sedated critically ill patients: a prospective observational pilot study. Ann Intensive Care 7:63
Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D (2002) Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med 30:2371–2375
Tovote P, Fadok JP, Lüthi A (2015) Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16:317–331
Cai H, Haubensak W, Anthony T, Anderson DJ (2014) Central amygdala PKC-δ+ neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci 17:1240–1248
Schelling G, Stoll C, Kapfhammer HP, Rothenhäusler HB, Krauseneck T, Durst K, Haller M, Briegel J (1999) The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder and health-related quality of life in survivors. Crit Care Med 27:2678–2683
Gilmore EJ, Gaspard N, Choi HA, Cohen E, Burkart KM, Chong DH, Claassen J, Hirsch LJ (2015) Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Med 41:686–694
Le Roux P, Menon DK, Citerio G, et al (2014) Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Neurocrit Care 21 Suppl 2:S1-26
Foreman B, Mahulikar A, Tadi P, et al (2016) Generalized periodic discharges and “triphasic waves”: a blinded evaluation of inter-rater agreement and clinical significance. Clin Neurophysiol Off J Int Fed Clin Neurophysiol 127:1073–1080
Zauner C, Gendo A, Kramer L, Funk GC, Bauer E, Schenk P, Ratheiser K, Madl C (2002) Impaired subcortical and cortical sensory evoked potential pathways in septic patients. Crit Care Med 30:1136–1139
Ehler J, Barrett LK, Taylor V, et al (2017) Translational evidence for two distinct patterns of neuroaxonal injury in sepsis: a longitudinal, prospective translational study. Crit Care 21:262
Ehler J, Petzold A, Wittstock M, et al (2019) The prognostic value of neurofilament levels in patients with sepsis-associated encephalopathy – a prospective, pilot observational study. PLoS ONE. https://doi.org/10.1371/journal.pone.0211184
Ehler J, Saller T, Wittstock M, et al (2019) Diagnostic value of NT-proCNP compared to NSE and S100B in cerebrospinal fluid and plasma of patients with sepsis-associated encephalopathy. Neurosci Lett 692:167–173
Needham DM, Colantuoni E, Dinglas VD, Hough CL, Wozniak AW, Jackson JC, Morris PE, Mendez-Tellez PA, Ely EW, Hopkins RO (2016) Rosuvastatin versus placebo for delirium in intensive care and subsequent cognitive impairment in patients with sepsis-associated acute respiratory distress syndrome: an ancillary study to a randomised controlled trial. Lancet Respir Med 4:203–212
Pandharipande PP, Sanders RD, Girard TD, McGrane S, Thompson JL, Shintani AK, Herr DL, Maze M, Ely EW, MENDS investigators (2010) Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: an a priori-designed analysis of the MENDS randomized controlled trial. Crit Care Lond Engl 14:R38
McAuley DF, Laffey JG, O’Kane CM, et al (2014) Simvastatin in the acute respiratory distress syndrome. N Engl J Med 371:1695–1703
Mehta S, Burry L, Cook D, et al (2012) Daily sedation interruption in mechanically ventilated critically ill patients cared for with a sedation protocol: a randomized controlled trial. JAMA 308:1985–1992
Adler J, Malone D (2012) Early mobilization in the intensive care unit: a systematic review. Cardiopulm Phys Ther J 23:5–13
Demoule A, Carreira S, Lavault S, Pallanca O, Morawiec E, Mayaux J, Arnulf I, Similowski T (2017) Impact of earplugs and eye mask on sleep in critically ill patients: a prospective randomized study. Crit Care Lond Engl 21:284
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Invited review for the neurocritical care special issue
Electronic Supplementary Material
ESM 1
(PDF 69 kb)
Rights and permissions
About this article

Cite this article
Mazeraud, A., Righy, C., Bouchereau, E. et al. Septic-Associated Encephalopathy: a Comprehensive Review. Neurotherapeutics 17, 392–403 (2020). https://doi.org/10.1007/s13311-020-00862-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00862-1