
Abstract
Despite continued research efforts, glioblastoma multiforme (GBM) remains the deadliest brain tumor. Immunotherapy offers a novel way to treat this disease, the genetic signature of which is not completely elucidated. Additionally, these tumors are known to induce immunosuppression in the surrounding tumor microenvironment via an array of mechanisms, making effective treatment all the more difficult. The immunotherapeutic strategy of using tumor vaccines offers a way to harness the activity of the host immune system to potentially control tumor progression. GBM vaccines can react to a variety of tumor-specific antigens, which can be harvested from the patient’s unique pathological condition using selected immunotherapy techniques. This article reviews the rationale behind and development of GBM vaccines, the relevant clinical trials, and the challenges involved in this treatment strategy.
Similar content being viewed by others


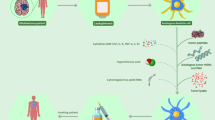
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive primary brain tumor in adults and is notoriously resistant to conventional therapies. Despite aggressive surgery, concurrent radiotherapy and chemotherapy, the median survival time of the 15,000 patients diagnosed each year is only 14.6 months [1]. The difficulty in treatment is centered in the genetic heterogeneity within the tumor itself, heterogeneity among patients, tumor-induced immunosuppression, limited therapeutic penetration of the blood–brain barrier, and inevitable tumor recurrence. Despite these numerous challenges, progress has been made in the development of GBM therapy, especially in the realm of immunotherapy, and several agents have progressed to clinical trials with varying levels of success. The aim of this review is to discuss the vaccine strategies currently under clinical investigation for the treatment of GBM and to consider the challenges facing this therapeutic approach in the setting of this complex disease.
The term immunotherapy is used to describe a spectrum of therapeutic strategies that use the host’s immune system to selectively destroy malignant cells. Passive immunotherapy entails the delivery of monoclonal antibodies directed against specific tumor-associated antigens (TAA) in order to elicit antibody-dependent tumor cell toxicity [2]. Another form of passive immunotherapy is adoptive cell transfer therapy, which consists of the ex vivo culture of effector immune cells followed by their subsequent transfer to the patient. In contrast, active immunotherapy relies upon the body’s own immune response and involves vaccination with a tumor-specific antigen in order to activate a host immune response. Vaccination generates tumor-specific effector T cells and, ideally, confers T-cell immunity, which may control tumor recurrence [3, 4].
Vaccines are categorized based on the way antigens are presented to the immune system. With non-cell-based vaccines, patients are directly inoculated with the specific tumor antigen of choice, which, in turn, is presented to host antigen-presenting cells (APCs) to trigger an immune response. Cell-based vaccines consist of APCs [e.g., dendritic cells (DCs)] that are primed with tumor antigens ex vivo and reintroduced to the host via vaccination, resulting in antigen presentation and amplification of tumor-specific T-cell responses (Fig. 1).

Schematic depicting passive and active immunotherapy strategies. TAA tumor-associated antigen, APC antigen-presenting cell, EGFR epidermal growth factor receptor, IDH1 isocitrate dehydrogenase 1, WT-1 Wilms’ tumor 1, HSP heat shock protein
Non-Cell-Based Vaccination
Peptide Vaccines
Peptide vaccines are constructed from peptides 8 to 25 amino acids long that encompass an epitope within an antigenic target, in order to elicit an immune response against neoplastic cells harboring mutant proteins [5]. These peptides are typically linked with carrier proteins to enhance immunogenicity (e.g., tetanus toxoid or keyhole limpet hemocyanin [KLH]) [6, 7]. Additionally, they are often administered with immune-stimulating agents such as Toll-like receptor (TLR) agonists (e.g., lipopolysaccharide) or cytokines [e.g., granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-2] to enhance the immune response [8, 9]. Gliomagenesis is associated with aberrant production of several TAAs, which can potentially be used for the development of peptide vaccines. However, the challenge lies in identifying antigens that are tumor specific, homogenously expressed, and sufficiently immunogenic [10]. Of the numerous potential GBM antigenic targets, only a few have met these criteria.
Epidermal Growth Factor Receptor vIII
Among peptide vaccines, those directed against epidermal growth factor receptor (EGFR)vIII have received the most attention clinically. Normally, epidermal growth factor or transforming growth factor α-induced activation of the EGFR promotes receptor dimerization, leading to autophosphorylation of the intracellular tyrosine kinase domain and propagation of Ras/mitogen-activated kinase signaling that promotes proliferation [11–13]. EGFRvIII contains a deletion within the extracellular domain that results in autoactivation of the receptor. The fusion of otherwise distant portions of the protein also creates a unique junction with a novel glycine residue, flanked by amino-acid sequences that are not present in the wild-type protein [14, 15]. Peptides derived from this fusion junction, when chemically conjugated to carrier proteins (e.g., KLH), have been shown to direct EGFRvIII-specific immune responses [11]. Importantly, EGFRvIII is expressed in a relevant proportion of patients with GBM (25–30%) and is associated with poor patient prognosis for long-term survivors. Additionally, the presence of this mutation can be determined by immunohistochemical analysis as part of standard pathology work-up [16–19]. EGFRvIII expression has also been documented in glioma stem cells. Even though its expression is heterogeneous within the tumor, EGFRvIII-positive cells can influence adjacent cells and subsequently provide a proliferative signal [20, 21]. In light of these factors, EGFRvIII was deemed a rationale target for immunotherapy.
The efficacy of EGFRvIII vaccination has been evaluated in preclinical models. Heimberger et al. [11] reported that vaccination resulted in a 26% increase in survival of mice bearing intracranial tumors relative to controls. Furthermore, they reported that mice that received vaccination prior to tumor implantation were protected from subsequent tumor formation. To evaluate clinically the safety and efficacy of intradermally administered EGFRvIII vaccine (CDX-110/rindopepimut) in EGFRvIII-positive patients, 3 phase II trials were initiated: ACTIVATE [5], ACT II [22], and ACT III [19]. All 3 trials administered rindopepimut to patients along with GM-CSF as an adjuvant. ACT II and ACT III additionally evaluated CDX-110 administered in conjunction with temozolomide (TMZ) based on earlier studies showing that TMZ induces lymphopenia and paradoxically enhances immunotherapeutic efficacy [16]. Regardless, median survival time and progression-free survival (PFS) time were both significantly increased compared with a matched control cohort receiving standard therapy. ACT II reported a median PFS time (from histological diagnosis) of 15.2 months in vaccine-treated patients (n = 22) versus 6.3 months in the historic control cohort (n = 17). Overall survival times for vaccinated patients and historical controls were 23.6 and 15 months, respectively. These differences remained significant after adjustment for patient age and Karnofsky Performance Scale score (p < 0.05). The larger ACT III trial, consisting of 65 newly diagnosed patients with EGFRvIII-positive GBM, confirmed the promising result of the earlier studies (reported median overall survival 24.6 months from diagnosis) [19]. Notably, upon recurrence, the tumors were noted to be EGFRvIII-negative, indicating antigenic loss [5]. Whether this stemmed from the natural disease course or from immunological elimination is unknown but indicates that targeting heterogeneously expressed antigens is likely to be insufficient for future approaches. Some promise has also emerged for the use of chimeric antigen receptor approaches targeting EGFRvIII. Based on the encouraging results, the ACT IV phase III trial was initiated. In this placebo-controlled, double-blind study, patients received rindopepimut/GM-CSF, or KLH, along with TMZ maintenance therapy (NCT01480479). Unfortunately, on 7 March 2016, Celldex announced that it was halting ACT IV. This decision was based on findings from an interim data analysis showing that patients receiving vaccination were unlikely to have an improvement in overall survival compared with patients in the control arm. Specifically, the treatment arm carried a survival of 20.4 months versus 21.1 months in the control cohort [23]. Despite this result, the ReACT phase II trial is currently ongoing and combines CDX-110 with bevacizumab; however, results of this trial are still pending (NCT01498328).
Another agent targeting EGFRvIII (ADU-623) is currently being evaluated in phase I clinical trials. ADU-623 is a live attenuated Listeria vaccine targeting both EGFRvIII and NY-ESO, which is another known tumor antigen with reportedly high immunogenic capacity [24–27]. This trial is recruiting patients with recurrent high-grade glioma and results are pending (NCT01967758).
Survivin
Although EGFRvIII is the most studied, there are other antigenic candidates under investigation for GBM peptide vaccines. Survivin is a molecule belonging to the inhibitor of apoptosis protein family. Although its mechanism of action is still being fully delineated, studies have shown that survivin is both a regulator of mitosis and a cell death inhibitor [28]. It is upregulated in several cancers, making it a potential target for immunotherapy [29]. Importantly, survivin is expressed to a much greater extent in GBM than in normal tissue, where it is associated a poorer prognosis [30, 31]. The validity of survivin as a target for immunotherapy was evaluated in study using SVN53-67/M57, a peptide vaccine derived from survivin, which can activate an immune response to survivin-expressing cells [32]. This vaccine is conjugated with KLH and has a single cysteine to methionine substitution to enhance immunogenicity and increase antitumor response [32]. SVN53-67/M57 produced cytotoxic T-cell-mediated killing of human glioma cells in an in vitro. Furthermore, injection of SVN53-67/M57 (in combination with GM-CSF) significantly increased survival time and decreased the tumor burden in mice with GL261 gliomas. Based on these results and a completed phase I trial (NCT01250470), a phase II trial of SVN53-67/M57-KLH and TMZ is currently recruiting patients with malignant glioma and has a targeted enrollment of 50 (NCT02455557).
Isocitrate dehydrogenase 1
Isocitrate dehydrogenase 1 (IDH1) is an enzyme that can be mutated in gliomas. Mutations most frequently occur in the arginine 132 residue, resulting in production of the oncometabolite 2-hydroxyglutarate, genomic hypermethylation, and malignant transformation. IDH1 R132H mutation, in particular, is expressed in > 90% of secondary GBMs [33, 34]. IDH1 R132H is an excellent target for cancer immunotherapy given that it is a tumor-specific antigen with reportedly uniform expression in tumor cells [35]. The clinical experience with EGFRvIII highlighted the challenge of GBM antigenic heterogeneity in the development of immunotherapies. It is suggested that targeting driver mutations, such as IDH, may circumvent heterogeneity-induced immune escape [36]. A recent study demonstrated that mice vaccinated with a mutant IDH1 peptide induced a specific T-cell response. Moreover, vaccination suppressed tumor growth in mice bearing an IDH1 R132H-expressing sarcoma. This effect was not seen in sarcomas expressing wild-type IDH, confirming that this response was mutation specific [35]. Further data are required to determine if an IDH vaccine is applicable in glioma. There is currently a phase I trial to evaluate the safety of IDH1 peptide vaccine in patients with IDH1 R132H-mutated gliomas (NCT02454634), with a planned enrollment of 39 patients; results from this are pending.
WT1
The Wilms' tumor gene WT1 is overexpressed in a variety of solid tumors and is recognized as an oncogene [37–39]. WT1 encodes a zinc finger transcription factor, which is involved in cell proliferation and apoptosis [40–43]. Multiple studies have established the expression of WT1 in gliomas [44–46]. Hashiba et al. [47] reported that Wilms’ tumor 1 (WT1) protein was detected in 70 of the 73 examined glial tumors (95.9%) and that expression was correlated with World Health Organization (WHO) grade. In another evaluation of 829 glioma samples, Rauscher et al. [48] reported that WT1 expression in diffuse astrocytomas increased with WHO grade and was associated with negative prognostic factors such as older age and expression of the wild-type IDH [48]. Because WT1 protein has been identified as a TAA, it is postulated that WT1 products may be suitable for the development of peptide-based immunotherapy. A recently published phase I trial tested the safety of WT1-based peptide vaccination combined with TMZ administration in 7 patients newly diagnosed with GBM and reported a PFS time of 5.2 to 49.1 months [49]. In a small nonrandomized phase II trial, 21 patients with WT1/human leukocyte antigen (HLA)-A*2402-positive recurrent GBM were treated with intradermal vaccination of HLA-A*2402-restricted WT1 peptide. Here, the reported 6-month PFS was 33.3%, with minimal treatment side effects; however, none of the patients had a complete radiographic response [50].
Multipeptide Vaccines
In terms of practicality, peptide vaccines offer several benefits, including ease of manufacturing in mass production, straightforward administration (e.g., intradermal), and limited intervaccine variability [51]. One limitation of several of the peptide vaccines (but not the EGFRvIII peptide) is that they are restricted to the HLA-A 02 haplotype; however, patients with GBM present with a variety of haplotypes, restricting the generalizability of this therapy [52]. One of the reasons a 14-mer was selected for the EGFRvIII peptide vaccine was to allow some degree of antigen processing for presentation in a variety of major histocompatibility complex (MHC) interactions [11]. A second limitation is that only a small subset of patients with GBM will ultimately benefit from targeting a single tumor antigen. Lastly, the primary disadvantage of peptide vaccines, particularly single peptides, is the heterogeneity of antigen expression in GBM [20]. Vaccines targeting a single antigen can lead to the development by the tumor of antigenic loss escape variant cells. This phenomenon was demonstrated in the EGFRvIII vaccine trials when tumor recurred post-CDX-110 vaccination, with 82% of cases demonstrating loss of the target antigen (EGFRvIII) [5]. This indicates that recurrence postvaccination requires an alternate treatment or a combination therapy.
In order to circumvent these issues, multipeptide vaccines that target a variety of tumor antigens have been developed. To identify potentially immunogenic tumor antigens, peptides were eluted from the HLA complex on GBM cells. Using liquid chromatography–mass spectrometry, 3000 peptides were identified, from which a panel was selected based on high tumor but low or absent expression in normal tissues [53]. A subsequent 11-peptide panel designated IMA950 encompasses the following proteins: brevican; chondroitin sulfate proteoglycan 4; fatty acid binding protein 7, brain; insulin-like growth factor 2 mRNA binding protein 3; neuroligin 4, X-linked; neuronal cell adhesion molecule; protein tyrosine phosphatase, receptor-type, Z polypeptide 1; tenascin C; Met proto-oncogene; baculoviral inhibitor of apoptosis protein repeat-containing 5; and hepatitis B virus core antigen. The first phase I/II trial of IMA950 (plus GM-CSF) was completed in 45 patients with newly diagnosed GBM who underwent surgical resection followed by standard therapy. The authors reported a PFS of 74% at 6 months and 31% at 9 months (NCT01222221) [54]. A second phase I study of IMA-950 in combination with cyclophosphamide, GM-CSF, and imiquimod at the National Cancer Institute was terminated secondary to poor accrual (NCT01403285). Additionally, there is a phase I/II trial testing the use of this vaccine in combination with polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose, a potent vaccine adjuvant shown to promote T-cell differentiation in glioma [55]; however, no results have been posted yet (NCT01920191). IMA950 in combination with polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose and anti-CD27 is now to be evaluated in patients with low-grade gliomas at University of California, San Francisco (NCT02924038). Of note, it is is also unclear whether the degree of clonotypic T-cell expansion using these types of peptides and adjuvant approaches is sufficient for tumor eradication. SL701 is another recently developed multipeptide vaccine currently being evaluated in clinical trials (NCT07078648). SL-701 consists of 3 peptides corresponding to targets overexpressed in glioma: a highly immunogenic mutant to target survivin, IL-13 receptor α-2, and ephrin A2. Results of this trial are pending.
An alternative approach has been to generate personalized peptide vaccines based on the antigenic profile of individual tumors. One strategy has been to prepare a vaccine from patients’ surgically removed tumor tissue along with pooled allogenic antigens from other resected tumors. ERC-1671 (Gliovac) is a multiple peptide vaccine composed of GBM tumor lysates, autologous tumor cells from the patient (generated from resected tumor), and tumor cells from 3 different GBM donors. This vaccine construction is directed to maximize patient exposure to a heterogeneous set of GBM tumor antigens. With exposure to a larger panel of allogeneic and autologous tumor associated antigens, ideally the chance of immune escape by the tumor is decreased. ERC-1671 has shown promising results in clinical evaluation [56, 57]. Administration of ERC 1671 in 9 patients with recurrent GBM, receiving standard chemoradiation and bevacizumab, resulted in increased survival times compared with controls. Specifically, 6-month survival for the patients on Gliovac was 100% versus 33% in the historical control group [57]. Based on these results, a phase II trial is underway to evaluate the safety and clinical efficacy of ERC1671 (in combination with GM-CSF and cyclophosphamide) plus bevacizumab among patients with recurrent GBM (NCT01903330).
Given the efforts of The Cancer Genome Atlas and the increasing widespread availability of both academic and commercial next-generation sequencing, new antigens may be identified or immunotherapies may be better tailored to specific patients. However, one does need to bear in mind that a genetic alteration does not necessarily give rise to an aberrant protein, and even if there is a resulting antigen it does not necessarily mean it is immunogenic or that it can induce sufficient numbers of clonotypic T cells to mediate tumor clearance. The avidity of an antigen influences the clonal dominance of the effector T-cell population [58], and most tumor antigens are unable to generate sufficient antitumor reactivity in physiological effector to target ratios to exert a therapeutic effect [59].
Certainly, peptide vaccines could be considered in combination with immune checkpoint inhibitors. Although this has obvious theoretical appeal, it may prove challenging to implement. For example, although 30% of GBMs have EGFRvIII target expression for rindopepimut/CDX-110 [16], only 6% also have programmed death 1 (PD-1)+ T cells presents within the tumor and 1% have PD ligand 1 (PD-L1) co-expression [60]. Granted, PD-1+ T cells and/or PD-L1 expression may only be enrichment biomarkers for response to immune checkpoint inhibitors and patients can respond in the absence of these biomarkers [61–63]; nonetheless, these data suggest that this type of combination may only benefit a small minority of patients. High tumor mutational loads have been found to correlate with responses to immune checkpoint inhibitors in other cancers [64–68], but these levels (12 mutations per megabase) are only seen in < 10% of patients with glioblastoma [69]. Finally, no one has demonstrated that the mechanism of peptide vaccine failure is related to enhanced immune checkpoint expression rather than antigen loss.
Heat Shock Protein Vaccines
Heat shock proteins (HSPs) are chaperone proteins that play an important role in the cellular stress response by preventing protein aggregation and guiding the degradation of misfolded proteins [70]. Certain subsets of HSPs, including HSP70 and HSP90, have been shown to bind tumor antigens in GBM and to simulate both innate and adaptive responses [71]. The HSP–antigen complex undergoes receptor-mediated uptake via CD91 expressed on the APCs. Upon internalization, the complex is processed via proteasomes for loading onto MHC class I molecules for presentation to CD8 cytotoxic T cells. Furthermore, HSP–antigen complexes also bind to macrophages via various receptors (i.e., CD36, CD91, CD40, CD14, TLR2, and TLR4) and stimulate the production of proinflammatory cytokines [72]. HSP vaccination entails purification of HSPs from a patient’s resected tumor and subsequent re-administration to the patient in order for HSPs to interact with host APCs and prime effector cells with antigenic peptides [72]. HSPs facilitate the presentation of antigenic peptides to circulating APCs, and an immune response is subsequently elicited when APCs bind to HSPs, endocytose their chaperone proteins, and present these antigenic peptides for targeting T-cell-mediated cytotoxic destruction [73–75].
The majority of HSP vaccine trials have used HSP–peptide complex-96 (HSPPC-96), comprising antigenic peptides chaperoned by HSP glycoprotein-96 [76, 77]. In a phase I trial consisting of 12 patients with recurrent GBM, treatment with HSP vaccine showed no significant toxicities and demonstrated a tumor-specific immune response in 11 of 12 patients (observed both in the tumor microenvironment and peripheral blood) [78]. In a subsequent single-armed, nonrandomized phase II trial, after surgical resection, 41 patients with recurrent GBM received a dose of HSPPC-96 every week for 4 weeks. Median PFS and overall survival were 19.1 and 42.6 weeks, respectively [79]. A phase II trial of HSPPC-96 in patients with newly diagnosed GBM undergoing standard therapy with chemoradiation has recently been completed and is pending publication (NCT00905060). Additionally, another randomized phase II trial combining HSPPC-96 and bevacizumab in patients with recurrent GBM is also actively recruiting, with an estimated enrollment of 222 patients (NCT01814813).
This treatment strategy is strengthened by its ability to present various types of potentially antigenic proteins during vaccination. HSP vaccines are also potentially capable of stimulating both the innate and adaptive immune system responses through multiple mechanisms. In addition to cytotoxic T cells, HSPs induce macrophages to secrete proinflammatory cytokines, such as tumor necrosis factor-α, GM-CSF, and IL-12, and so on. IL-12 subsequently activates cytotoxic activity in lymphocytes and natural killer cells. HSP complexes are also able to increase production and secretion of nitric oxide in DCs and macrophages. Lastly, HSPs induce immature DCs to undergo maturation [80, 81], which may contribute to a more robust immune response postvaccination. It should be noted that HSP vaccines are limited in that a significant amount of tissue is required to generate the vaccine [72]. This criterion restricts the eligibility of the vaccine to patients undergoing a substantial resection and essentially excludes those simply receiving a stereotactic biopsy. Additionally, the basis for HSP vaccination is subject to criticism because its efficacy has not been substantially validated in preclinical glioma studies, and it has shown limited therapeutic benefit in multiple randomized phase III clinical trials for other advanced malignancies [82, 83].
Cell-Based Vaccination
DC Vaccines
DCs are professional APCs and are the most powerful activators of the cell-mediated immune response. DCs present antigens to naïve T cells after phagocytosis, processing, and presentation in the context of the MHC, triggering antigen-specific CD8+ and CD4+ lymphocyte responses. In addition to this, T-cell functionality is augmented by activated DC secretion of proinflammatory cytokines [84]. Because of these capabilities, DCs are ideal for vaccine development. In vaccine preparation, DCs are extracted from the patient, cultured ex vivo, and loaded with TAAs. These primed DCs are subsequently reintroduced into the patient to facilitate antigen-specific T-cell activation (Fig. 2).

Schematic depicting the process of dendritic cell (DC) vaccine generation. TAA tumor-associated antigen
DC vaccines have been studied in GBM [85–87]. Successful preclinical studies and small clinical studies have generated interest in identifying the optimal molecules to load onto DCs to generate the most robust and tumor-specific response. Although the process of constructing DC vaccines is considerably more laborious than that of peptide vaccines, its greatest advantage lies in its flexibility of being able to provide the DCs with a wide range of stimuli, including peptides, tumor lysates, and cancer stem cells, all of which are tailored to the individual make-up of the tumor.
Tumor Peptide-Loaded DCs
At the most basic level, DCs can be stimulated with the same types of peptide epitopes that are the basis of peptide vaccines. The EGFRvIII was one of the earliest peptides loaded into DCs in clinical trial applications. In a phase I study, 12 patients with newly diagnosed GBM underwent inoculation of DCs pulsed with EGFRvIII-specific peptide (PEPvIII)–KLH after surgical resection and radiation therapy [88]. The median time to progression and overall survival time (from vaccination) were reported as 6.8 and 18.7 months, respectively (10.2 months and 22.8 months from diagnosis). No serious adverse reactions or toxicities were observed. DC loading is not limited to single tumor peptides. Phuphanich et al. [89] conducted a phase I trial of ICT-107, an autologous vaccine consisting of patient DCs pulsed with 6 GBM-associated peptide antigens (AIM-2, MAGE1, TRP-2, gp100, HER2/neu, and IL-13 receptor α-2), in combination with traditional chemoradiation [89]. In their preliminary data from 21 patients, vaccination yielded favorable survival with median PFS and overall survival times of 16.9 months and 38.4 months, respectively. Patients are currently being recruited for a phase III trial evaluating this treatment (NCT02546102).
Tumor Lysate-Loaded DCs
In this technique, tumor lysate is prepared from autologous tumor cells obtained during surgical resection. Patient tumor cells are lysed to generate cellular fragments and are subsequently pulsed into DCs. Exposure of DCs to these tumor fragments allows for phagocytosis and subsequent peptide antigen presentation to MHC class I and II [90–92]. The advantage of using tumor lysates is the unbiased stimulation of DCs to the full complement of patients’ unique TAAs. This can include the presentation of thus-far unrecognized tumor antigens as well [93]. Alternatively, this may generate non-tumor-specific immune responses or may dilute clonotypic expansion of T cells to weakly immunogenic antigens. In a phase I study, the safety and bioactivity of autologous tumor lysate-based DC vaccine was determined. Activity of lysate-reactive CD8+ T cells after introduction of these tumor lysate-loaded DCs was observed, confirming the specificity of the resulting immune response. Additionally, the median survival time for patients with recurrent GBM (n = 8) was an impressive 133 weeks [4]. Among the more developed therapies in this subtype of vaccine is DC-VaxL, which consists of autologous DCs pulsed with autologous whole-tumor lysate. DC-VaxL is currently in a phase III clinical trial, and its results are highly anticipated (NCT00045968) [94].
mRNA-Loaded DCs
DCs can also be loaded with tumor-derived mRNA. In contrast to tumor protein, mRNA can be amplified in vitro, giving this technique a practical advantage. Amplification permits a relatively small cell source to be used for the production of a large number of antigen-loaded DCs [95]. Additionally, previous studies have shown that RNA is superior to DNA in the induction of T cells by DCs [96]. There are currently several clinical trials implementing this technique (NCT02649582, NCT02709616).
Glioma Stem Cell-Loaded DCs
Glioma cancer stem cells (GSCs) are a cellular subtype that is unique compared with the rest of the tumor cell mass, and shows self-renewal capability [97]. GSCs are known to be chemoradiation resistant and responsible for recurrence [98, 99]. GSCs also have the capacity to inhibit T-cell proliferation and activation, to induce regulatory T cells, and to trigger T-cell apoptosis [100]. GSCs have been studied as a target for DC immunotherapy, as successful destruction of these drivers of tumor-mediated immunosuppression may allow for greater efficacy [8, 98]. Pellegatta et al. [101] reported that DCs loaded with GL261 neurospheres (enriched with stem cells) cured 80% of GL261 tumor-bearing mice compared with 50% of mice treated with DCs loaded with standard GL261 lysate. Vik-Mo et al. [102] reported on 7 patients treated with a DC vaccine targeting stem cells in a phase I/II (NCT00846456). For this, GBM specimens were dissociated and patient stem cells were isolated and expanded in vitro. After this, stem cell mRNA was amplified and loaded into DCs. In the patients receiving vaccination, no severe toxicities were observed. Moreover, treated patients showed a PFS interval that was 2.9 times longer than that of matched controls (694 days vs 236 days, respectively; p = 0.0018). A current phase I trial is testing the safety of ICT-121, a DC-based vaccine targeting CD133+ (a stem-cell marker) cells in patients with recurrent GBM (NCT02049489). It should be noted that the technique using the GSC generation/harvesting is expensive and difficult, which may limit the generalizability of this treatment.
Viral Antigen-Loaded DCs
The existence of human cytomegalovirus (CMV) and its gene products in GBM was initially identified in 2002, and CMV is deemed to have a relevant role in pro-oncogenic pathways [103, 104]. With this in mind, extensive research has been dedicated to exploiting this feature for development of immunotherapy for GBM. The human CMV virion is enveloped by a matrix referred to as the tegument, which contains most of the viral proteins. The most abundant tegument protein is pp65 [105], and this CMV antigen can be utilized for targeted immunotherapy. A clinical trial is currently evaluating a CMV-based strategy for the treatment of GBM. In this study, autologous DCs are loaded with CMV pp65-lysosomal-associated membrane protein mRNA and administered in combination with the anti-CD25 monoclonal antibody (basiliximab) (NCT00626483). The role of anti-CD25 is to inhibit suppressive T regulatory cells and to enhance CMV-specific T-cell activity. Recent data also indicate that preconditioning of the vaccine site with the recall antigen tetanus/diphtheria (Td) toxoid can facilitate homing of the DCs to the lymph nodes and the efficacy of DC vaccination. In a blinded clinical trial of 12 patients with GBM, participants were randomized to preconditioning with either mature DCs or Td toxoid prior to vaccination with DCs pulsed with pp65 RNA. The authors reported that patients pretreated with Td toxoid had enhanced DC migration and significantly improved survival [106]. ELEVATE is a phase II randomized clinical trial currently recruiting patients newly diagnosed with GBM who have undergone resection and completed standard chemoradiation (NCT02366728). On a larger scale, this trial will further evaluate the impact of preconditioning (with either tetanus or basiliximab) prior to treatment with a CMV-specific DC vaccine, on DC migration and subsequent patient survival.
DCs in Combination Therapies
Owing to the preclinical and clinical promise of DC vaccination, this immunotherapeutic strategy has been combined with other forms of immunotherapy to enhance the desired immune response. Immune system inhibitory pathways may specifically undermine the effect of DC vaccination. Immune checkpoints are well-studied receptors involved in these pathways. A recent preclinical study evaluated the efficacy of tumor lysate-pulsed DCs in combination with immune checkpoint blockade, specifically inhibition of the PD-1/PD-L1 axis. This signaling axis has been implicated in several cancers and has been demonstrated to decrease activation of effector T cells and to promote tumor immunoescape [107, 108]. In glioma-bearing mice, combination treatment with DC vaccination and PD-1 antibody resulted in upregulation in markers of immunologic memory and homing (i.e., L-selectin, integrin α-4 chain) in tumor-infiltrating lymphocytes. Furthermore, animals treated with combination therapy survived significantly longer than those treated with monotherapy. Specifically, there was no therapeutic benefit observed with monotherapy, but there was 40% long-term survival in the combination treatment group [109]. A phase I trial (AVERT) is testing the combination of a CMV-specific DC vaccine with nivolumab (anti-PD-1 antibody) and is currently recruiting patients with recurrent high-grade glioma (NCT02529072).
Limitations of DC Vaccines
One of the primary drawbacks of DC vaccination relates to its practical application. Overall, the cost–benefit ratio of cell-based therapy is considered suboptimal. For DC vaccines specifically, their production is expensive, and the time needed to culture and prime cells can be extensive [110, 111]. Tumor growth in patients with recurrent GBM can at times be rapid and associated with progressive neurological decline, and the time necessary to manufacture a DC vaccine may make this treatment difficult to implement in the clinical setting. Moreover, it has been suggested that the optimal time for vaccination is when tumor burden is minimal, because tumor-induced immunosuppression, which can counteract the desired immune response, has been correlated with amount of tumor present [112, 113]. This makes the time-consuming nature of vaccine preparation a critical issue. Additionally, most DC vaccine studies have used DCs generated from a patient’s monocytes ex vivo. It has been suggested that the process of culturing and differentiating these monocytes may deplete the immunological functional capacity of the cells. Fortunately, this particular issue may be addressed by the use of naturally circulating DCs, such as myeloid or plasmacytoid DCs, as alternative sources. The time required to load naturally occurring DCs with TAAs is significantly shorter than with monocyte-derived DCs. Furthermore, production is highly standardized, and natural DCs have shown a favorable safety profile in addition to inducing antigen-specific T-cell responses [111, 114, 115].
Even though several DC vaccination studies have demonstrated specific immune responses [4, 116], few have actually demonstrated an objective clinical response—defined as a complete or partial response by the World Health Organization or Response Evaluation Criteria In Solid Tumors (RECIST) [117]. A systematic review of all DC vaccine clinical trials reported that in terms of objective response, the clinical benefit of this treatment strategy is very modest. Specifically, objective response rates were reportedly 8.5%, 7.1%, and 15.6% for melanoma, prostate cancer, and glioma trials, respectively [118]. Despite the fact that multiple nonrandomized trials have reported a survival benefit [119, 120], lack of an objective clinical response remains an important area of criticism for DC vaccines.
Conclusion
Tumor vaccines allow the immune system to be directed to a tumor target and, in an appropriate context, could induce a long-term immune response. There are currently multiple clinical trials exploring this treatment platform (Table 1). Based on the cumulative experience thus far with tumor vaccines, future studies should focus ideally on combining vaccination therapy with conventional therapy, combining vaccines targeting different antigens and potentially other forms of immunotherapies (e.g., checkpoint inhibition, adoptive transfer therapy). Combination therapy may decrease the potential for tumor immunoresistance and escape. Additionally, a more precise understanding of the timing of immunotherapy treatment, along with standardization of vaccination protocols, adjuvants, and methods to monitor patient immune responses are needed. Moreover, the issue of frequent concurrent steroid use in this patient population will also need to be addressed because steroid treatment, which is often needed to reduce intracranial pressure and symptomatology, may undermine the desired immune response [93]. The impact of tumor factors on the efficacy of vaccine therapy is also not fully elucidated—especially for low-grade gliomas. Specifically, this includes factors such as tumor site, extent of resection, primary versus secondary GBM, and recurrent disease versus initial treatment [75]. Overall, vaccine therapy is a treatment strategy with potential but which still requires further optimization.
References
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987-996.
Batich KA, Swartz AM, Sampson JH. Enhancing dendritic cell-based vaccination for highly aggressive glioblastoma. Expert Opin Biol Ther 2015;15(1):79-94.
Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 2005;102(27):9571-9576.
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res 2004;64(14):4973-4979.
Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010;28(31):4722-4729.
Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov 2007;6(5):404-414.
Swartz AM, Batich KA, Fecci PE, Sampson JH. Peptide vaccines for the treatment of glioblastoma. J Neurooncol 2015;123(3):433-440.
Mohme M, Neidert MC, Regli L, Weller M, Martin R. Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat Rev 2014;40:248-258.
Xu LW, Chow KK, Lim M, Li G. Current vaccine trials in glioblastoma: a review. J Immunol Res 2014;2014:796856.
Hodges TR, Ferguson SD, Caruso HG, et al. Prioritization schema for immunotherapy clinical trials in glioblastoma. Oncoimmunology 2016;5(6):e1145332.
Heimberger AB, Crotty LE, Archer GE, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res 2003;9(11):4247-4254.
Patel R, Leung HY. Targeting the EGFR-family for therapy: biological challenges and clinical perspective. Curr Pharm Des 2012;18(19):2672-2679.
Prigent SA, Nagane M, Lin H, et al. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J Biol Chem 1996;271(41):25639-25645.
Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 1990;50(24):8017-8022.
Humphrey PA, Wong AJ, Vogelstein B, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A 1990;87(11):4207-4211.
Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 2005;11:1462-1466.
Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997;272(5):2927-2935.
Pelloski CE, Ballman KV, Furth AF, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol 2007;25(16):2288-2294.
Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015;17(6):854-861.
Del Vecchio CA, Giacomini CP, Vogel H, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013;32(21):2670-2681.
Inda MM, Bonavia R, Mukasa A, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 2010;24(16):1731-1745.
Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 2011;13(3):324-333.
Malkki H. Trial Watch: Glioblastoma vaccine therapy disappointment in Phase III trial. Nat Rev Neurol 2016;12(4):190.
Cebon J, Knights A, Ebert L, Jackson H, Chen W. Evaluation of cellular immune responses in cancer vaccine recipients: lessons from NY-ESO-1. Expert Rev Vaccines 2010;9(6):617-629.
Nicholaou T, Ebert L, Davis ID, et al. Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. Immunol Cell Biol 2006;84(3):303-317.
Nicholaou T, Ebert LM, Davis ID, et al. Regulatory T-cell-mediated attenuation of T-cell responses to the NY-ESO-1 ISCOMATRIX vaccine in patients with advanced malignant melanoma. Clin Cancer Res 2009;15(6):2166-2173.
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 2002;188:22-32.
Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003;22(53):8581-8589.
Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: a unique target for tumor therapy. Cancer Cell Int 2016;16:49.
Chakravarti A, Noll E, Black PM, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol 2002;20(4):1063-1068.
Kajiwara Y, Yamasaki F, Hama S, et al. Expression of survivin in astrocytic tumors: correlation with malignant grade and prognosis. Cancer 2003;97(4):1077-1083.
Ciesielski MJ, Ahluwalia MS, Munich SA, et al. Antitumor cytotoxic T-cell response induced by a survivin peptide mimic. Cancer Immunol Immunother 2010;59(8):1211-1221.
Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009;27(25):4150-4154.
Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360(8):765-773.
Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014;512(7514):324-327.
Platten M, Bunse L, Wick W, Bunse T. Concepts in glioma immunotherapy. Cancer Immunol Immunother 2016;65(10):1269-1275.
Nishida S, Hosen N, Shirakata T, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood 2006;107(8):3303-3312.
Oka Y, Udaka K, Tsuboi A, et al. Cancer immunotherapy targeting Wilms' tumor gene WT1 product. J Immunol 2000;164(4):1873-1880.
Sugiyama H. Wilms' tumor gene WT1: its oncogenic function and clinical application. Int J Hematol 2001;73(2):177-187.
Algar EM, Khromykh T, Smith SI, Blackburn DM, Bryson GJ, Smith PJ. A WT1 antisense oligonucleotide inhibits proliferation and induces apoptosis in myeloid leukaemia cell lines. Oncogene 1996;12(5):1005-1014.
Kijima N, Hosen N, Kagawa N, et al. Wilms' tumor 1 is involved in tumorigenicity of glioblastoma by regulating cell proliferation and apoptosis. Anticancer Res 2014;34(1):61-67.
Oji Y, Ogawa H, Tamaki H, et al. Expression of the Wilms' tumor gene WT1 in solid tumors and its involvement in tumor cell growth. Jpn J Cancer Res 1999;90(2):194-204.
Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms' Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene 2005;24(9):1648-1652.
Clark AJ, Dos Santos WG, McCready J, et al. Wilms tumor 1 expression in malignant gliomas and correlation of + KTS isoforms with p53 status. J Neurosurg 2007;107:586-592.
Oji Y, Suzuki T, Nakano Y, et al. Overexpression of the Wilms' tumor gene W T1 in primary astrocytic tumors. Cancer Sci 2004;95(10):822-827.
Rushing EJ, Sandberg GD, Horkayne-Szakaly I. High-grade astrocytomas show increased Nestin and Wilms's tumor gene (WT1) protein expression. Int J Surg Pathol 2010;18(4):255-259.
Hashiba T, Izumoto S, Kagawa N, et al. Expression of WT1 protein and correlation with cellular proliferation in glial tumors. Neurol Med Chir (Tokyo) 2007;47(4):165-170.
Rauscher J, Beschorner R, Gierke M, et al. WT1 expression increases with malignancy and indicates unfavourable outcome in astrocytoma. J Clin Pathol 2014;67(7):556-561.
Hashimoto N, Tsuboi A, Kagawa N, et al. Wilms tumor 1 peptide vaccination combined with temozolomide against newly diagnosed glioblastoma: safety and impact on immunological response. Cancer Immunol Immunother 2015;64(6):707-716.
Izumoto S, Tsuboi A, Oka Y, et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg 2008;108(5):963-971.
Thomas AA, Fisher JL, Ernstoff MS, Fadul CE. Vaccine-based immunotherapy for glioblastoma. CNS Oncol 2013;2(4):331-349.
Jackson C, Ruzevick J, Brem H, Lim M. Vaccine strategies for glioblastoma: progress and future directions. Immunotherapy 2013;5(2):155-167.
Dutoit V, Herold-Mende C, Hilf N, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012;135(Pt 4):1042-1054.
Rampling R, Peoples S, Mulholland PJ, et al. A Cancer Research UK first time in human phase i trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res 2016;22(19):4776-4785.
Zhu X, Fallert-Junecko BA, Fujita M, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother 2010;59(9):1401-1409.
Bota DA, Alexandru-Abrams D, Pretto C, et al. Use of ERC-1671 vaccine in a patient with recurrent glioblastoma multiforme after progression during bevacizumab therapy: first published report. Perm J 2015;19(2):41-46.
Schijns VE, Pretto C, Devillers L, et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine 2015;33(23):2690-2696.
Price DA, Brenchley JM, Ruff LE, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med 2005;202(10):1349-1361.
Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol 1999;162(4):2227-2234.
Garber ST, Hashimoto Y, Weathers SP, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol 2016;18(10):1357-1366.
Daud AI, Loo K, Pauli ML, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest 2016;126(9):3447-3452.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443-2454.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568-571.
Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515(7528):577-581.
Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515(7528):563-567.
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-128.
Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909-1920.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371(23):2189-2199.
Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 2017. In Press.
Craig EA, Weissman JS, Horwich AL. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell 1994;78(3):365-372.
Graner MW, Bigner DD. Chaperone proteins and brain tumors: potential targets and possible therapeutics. Neuro Oncol 2005;7(3):260-278.
Ampie L, Choy W, Lamano JB, Fakurnejad S, Bloch O, Parsa AT. Heat shock protein vaccines against glioblastoma: from bench to bedside. J Neurooncol 2015;123(3):441-448.
Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 2000;6(4):435-442.
Nishikawa M, Takemoto S, Takakura Y. Heat shock protein derivatives for delivery of antigens to antigen presenting cells. Int J Pharm 2008;354(1-2):23-27.
Sayegh ET, Oh T, Fakurnejad S, Bloch O, Parsa AT. Vaccine therapies for patients with glioblastoma. J Neurooncol 2014;119(3):531-546.
Belli F, Testori A, Rivoltini L, et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. J Clin Oncol 2002;20(20):4169-4180.
Srivastava PK. Purification of heat shock protein-peptide complexes for use in vaccination against cancers and intracellular pathogens. Methods 1997;12(2):165-171.
Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, Butowski N, Chang SM, Clarke J, Berger MS, McDermott MW, Prados MD, Parsa AT. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res 2013;19(1):205–14.
Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol 2014;16(2):274-279.
Panjwani NN, Popova L, Srivastava PK. Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol 2002;168(6):2997-3003.
Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2002;2(3):185-194.
Testori A, Richards J, Whitman E, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician's choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol 2008;26(6):955-962.
Wood C, Srivastava P, Bukowski R, et al. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet 2008;372(9633):145-154.
Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med 2000;191(3):411-416.
Finocchiaro G, Pellegatta S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies. Cancer Immunol Immunother 2016;65(1):101-109.
Rutkowski S, De Vleeschouwer S, Kaempgen E, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer 2004;91(9):1656-1662.
Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res 2004;10(16):5316-5326.
Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther 2009;8(10):2773-2779.
Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother 2013;62(1):125-135.
Antonios JP, Everson RG, Liau LM. Dendritic cell immunotherapy for brain tumors. J Neurooncol 2015;123(3):425-432.
Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer 2003;89(7):1172-1179.
Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res 2005;11(11):4160-4167.
Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 2013;10(1):14-26.
Hdeib A, Sloan AE. Dendritic cell immunotherapy for solid tumors: evaluation of the DCVax(R) platform in the treatment of glioblastoma multiforme. CNS Oncol 2015;4(2):63-69.
Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 2000;60(4):1028-1034.
Parajuli P, Mathupala S, Sloan AE. Systematic comparison of dendritic cell-based immunotherapeutic strategies for malignant gliomas: in vitro induction of cytolytic and natural killer-like T cells. Neurosurgery 2004;55(5):1194-1204.
Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010;17(4):362-375.
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756-760.
Fidoamore A, Cristiano L, Antonosante A, et al. Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int 2016;2016:6809105.
Wei J, Wu A, Kong LY, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLOS ONE 2011;6(1):e16195.
Pellegatta S, Poliani PL, Corno D, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 2006;66(21):10247-10252.
Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 2013;62(9):1499-1509.
Cobbs CS, Harkins L, Samanta M, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 2002;62(12):3347-3350.
Dziurzynski K, Chang SM, Heimberger AB, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol 2012;14(3):246-255.
Varnum SM, Streblow DN, Monroe ME, et al. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 2004;78(20):10960-10966.
Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015;519(7543):366-369.
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007;27(1):111-122.
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13(1):84-88.
Antonios JP, Soto H, Everson RG, et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016;1(10).
Sakai K, Shimodaira S, Maejima S, et al. Dendritic cell-based immunotherapy targeting Wilms' tumor 1 in patients with recurrent malignant glioma. J Neurosurg 2015;123(4):989-997.
Schreibelt G, Bol KF, Westdorp H, et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res 2016;22(9):2155-2166.
Gulley JL, Madan RA, Schlom J. Impact of tumour volume on the potential efficacy of therapeutic vaccines. Curr Oncol 2011;18(3):e150-e157.
Widen K, Mozaffari F, Choudhury A, Mellstedt H. Overcoming immunosuppressive mechanisms. Ann Oncol 2008;19(Suppl. 7):vii241-7.
Tel J, Aarntzen EH, Baba T, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res 2013;73(3):1063-1075.
Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJ. Paradigm shift in dendritic cell-based immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front Immunol 2014;5:165.
Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 2005;11(15):5515-5525.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10(9):909-915.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014;15(7):e257-e267.
Cho DY, Yang WK, Lee HC, et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg 2012;77(5-6):736-744.
Oshita C, Takikawa M, Kume A, et al. Dendritic cell-based vaccination in metastatic melanoma patients: phase II clinical trial. Oncol Rep 2012;28(4):1131-1138.
Acknowledgments
We thank David M. Wildrick, PhD, for editorial assistance.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Srinivasan, V.M., Ferguson, S.D., Lee, S. et al. Tumor Vaccines for Malignant Gliomas. Neurotherapeutics 14, 345–357 (2017). https://doi.org/10.1007/s13311-017-0522-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0522-2