
Abstract
Solid biopolymer blend systems consisting of chitosan (CH) and methylcellulose (MC) have been prepared in various compositions through the solution casting method and are characterized by X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA). The XRD data revealed that the blend composed of 30 wt.% of MC is found to be more amorphous. Structural analysis and the extent of interaction between the two polymers, i.e., CH and MC were concluded through FTIR analysis. The dielectric results also show that the blend composed of 70 wt.% of CH shows the highest value of the dielectric constant. The blend system consisting of 70 wt.% of CH and 30 wt.% of MC exhibited good thermal stability and the lowest glass transition temperature (\({T}_{g}\)).
Graphical Abstract

Similar content being viewed by others

1 Introduction
In recent years, interest in agro-polymers has created great interest among researchers and industry alike, as these materials are found to be abundantly available, low cost, biodegradable and eco-friendly. Bio-based polymers are addressing the concerns in some of the impactful areas such as aerospace, automobile, construction, and many more. So, life without these bio-based polymers as an alternative to synthetic polymer materials is unimaginable. The polymer blends are interesting materials for producing hydrogels, composite polymer films, shells for encapsulated drugs, electrolyte membranes for batteries, etc. [1].
Moreover, in batteries and supercapacitors, the main components are cathode, anode, and electrolyte, and parameters such as specific energy, energy density, charge/discharge efficiency, cycle durability, and cell voltage, depending on the electrode and the electrolyte materials incorporated in them. Due to their lightweight, safe, and flexible design, Li-ion/Na-ion blend polymer-based electrolytes are now viewed as viable energy storage devices [2].
Ionic conductivity is a measure of how well polymer electrolytes perform their function of transporting ions across the electrodes. The properties such as \({T}_{g}\) of the polymer, the polymer's dielectric constant, the salt's lattice energy, the size of the ions, the interaction between the ions and the polymer, and the crystallinity influence the ionic conductivity in the polyblend system. The amorphous phase of the polymer electrolyte dominates ion transport, not the crystalline phase [3]. Several approaches are explored to decrease the crystalline nature of the polymer host to increase ionic conductivity. The addition of plasticizer to the polymer host, \(\gamma\)-irradiating the high molecular weight for cross-linking the polymer chains, the addition of nano-fillers into the electrolyte system, copolymerization and blending of polymers [4] are a few approaches. Among these methods, polymer blending is the best and easiest method to achieve this purpose. The goal of the blending technique is to create unique and new materials with improved properties without compromising the original materials' properties. The simplicity of synthesis, cost-effectiveness, and ease of controlling physical properties over compositional changes make polymer blending superior to other techniques [1]. Polyvinyl acetate:polymethyl methacrylate blend system in the ratio of 70:30 wt.% exhibited high mechanical strength and conductivity. In another study blending poly (epichlorohydrin) (PECH) with polyvinyl alcohol (PVA) increased the amorphous nature thereby providing a region for enhanced ionic conductivity. Ahmed et al. [1] prepared a polyblend system of polyethylene oxide (PEO):methylcellulose (MC) with different weight percentages and found that the blend system consisting of 60 wt.% of PEO and 40 wt.% of MC is having the lowest degree of crystallinity via XRD studies and the highest conductivity value of 6.55 × 10–9 Scm−1 without any salt dopants. Reddy et al. [5], stressed the preparation of poly (methyl methacrylate)-poly(ethylene oxide) (PMMA-PEO) polymer blend films with different weight ratios. The addition of PEO to PMMA resulted in shifting of the several bands of PMMA and intensities corresponding to some of the bands got disappeared indicating complexation between the two polymers. However, there were no studies that involved a correlation between conductivity and crystallinity using impedance analysis.
In this paper, a blend of MC and CH biopolymers was prepared wherein CH adds mechanically rigid characteristics to the film. Moreover, CH possesses a hydrophilicity property which makes it used in high-temperature and relatively low-humidity environments [6]. On the other hand, MC has efficient oxygen, and lipid barrier properties and boosts the solubility of the salt in the polymer matrix, which opens a wide range of potential applications [7]. Natural polymers/biopolymers are trending materials in various applications and need to be explored as energy storage material applications [8]. Hamdan et al. used a fixed ratio of 50:50 of CH:MC with different amounts of ammonium triflate salt for polymer electrolyte application [7]. Liu et al. [9], used the same blend system, and structural investigation have been carried out, but thermal analysis particularly DSC analysis was missing, and also it is not clear which is the best blend composition among the prepared blend films. Pornchai Rachtanapun et al. also used the fixed blend composition of CH:MC and studied the mechanical and surface morphology of the film to be used for biodegradable plastic application [10].
In this article, we have carried out the structural, thermal, and electrical analysis of polymer blend systems of CH:MC. It was observed that the blend system composed of 70 wt.% of CH and 30 wt.% of MC is the best composition because it showed the least crystallinity, low Tg, and highest value of dielectric constant which are the desirable properties of a polymer matrix to be used in application such as polymer electrolyte.
2 Experimental details
2.1 Materials
Chitosan and Methylcellulose were purchased from Loba Chemie Pvt. Ltd., Mumbai, India. The analytical grade acetic acid was procured from S.D. Fine-Chem Pvt. Limited., Mumbai, India.
2.2 Preparation of solid polyblend films
Solid polyblend (x wt.% MC + y wt.% CH, where x = 10 to 50 wt.% in steps of 10 wt.% and y = 90 to 50 wt.% in steps of 10 wt.%) films were prepared with total weight components were fixed to 2 g. Composition of CH below 50 wt.% a free-standing film was not obtained. Hence, the study was limited to higher content of CH which provides mechanical support and rigidness to the polyblend system. A homogenous viscous solution was obtained by dissolving CH and MC in 50 ml of 1% acetic acid solution while stirring continuously at 40 °C. Accordingly, the polyblend solution was poured into glass Petri dishes to dry out at ambient temperature. After that, the blended polymer films were kept in a desiccator for further analysis. The films have a thickness between 140 and 160 µm. The sample designation is listed in Table 1.
2.3 Polyblend films characterization
FTIR investigations were performed by utilizing a Shimadzu IR spirit Fourier Transform Infrared Spectrophotometer in the transmittance mode with a resolution of 4 cm−1 to check the interaction between the polymers in the wavenumber range of 4000–400 cm−1. To understand the influence of polymer composition on crystallinity, XRD studies have been carried out using Rigaku Miniflex 600 (fifth gen) copper K- alpha (λ = 1.54 Å, 14 kV, 15 mA) radiation with a scanning angle range of 2θ from 4° to 80°, with a 0.02° step size. Using the SHIMADZU DSC-60 PLUS MODEL, DSC measurements were performed in the RT-300 °C temperature range with a heating scan rate of 10 °C min−1 in a nitrogen atmosphere. The thermal steadiness of the polyblend samples was studied using the instrument SDT Q600 V20.9 build 20. Samples of about 5 mg weight were run in a platinum crucible operated in the range from 20 to 500 °C with nitrogen as purging gas at 80 mL/min, with a heating scan rate of 10 °C min−1. Impedance analyzer model number IM 3570, Hioki corporation, Japan, at a frequency scale of 100 Hz to 5 MHz was used to perform the dielectric study. The polyblend films were sandwiched between the two blocking electrodes of the contact area of 1.13 cm2. The real (Z’), and imaginary (Z′′) part of complex impedance, loss factor (tanδ), and parallel capacitance (C) were all measured using the built-in software.
3 Results and discussion
3.1 FTIR studies
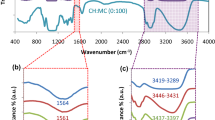
FTIR spectrum of CH, MC & CH: MC blend polymer samples with various weight percentages were shown below in Fig. 1. For CH the \(-\mathrm{OH}\) vibrational band occurs in the wavenumber region from 3600 to 3200 cm−1 [11]. The amine and carboxamide bands occur at 1547 cm−1 and 1641 cm−1, respectively [12]. The \(\mathrm{C}-\mathrm{H}\) stretching vibration band occurs at 2921 cm−1 [13]. The peaks at 1152 cm−1, 1408 cm−1, and 1380 cm−1 correspond to anti-symmetric stretching of the \(\mathrm{C}-\mathrm{O}-\mathrm{C}\) bridge, \({\mathrm{CH}}_{2}\) bending, and \(-\mathrm{CH}\) bending, respectively. For pure MC, the stretching vibrations of hydroxyl groups occur in the wavenumber region from 3400 to 3600 cm−1 wavenumber region. The peak at 2917 cm−1 corresponds to the \(\mathrm{C}-\mathrm{H}\) stretching of \({\mathrm{CH}}_{3}\) group [14]. The \(\mathrm{C}=\mathrm{O}\) stretching vibration occurs at 1644 cm−1, whereas the band at 1408 cm−1 can be attributed to the \(\mathrm{C}-\mathrm{H}\) bending [15, 16]. The asymmetric vibrations of \(\mathrm{CO}\) appear between 1045 and 1055 cm−1 [9]. Compared to pure CH and MC, the FTIR spectra of the CH: MC blend contains no extra distinct peaks, but there was a slight shift in the band. Figure 2 demonstrates the possible interaction scheme between the two polymers. A shift in the band value from region 3435 cm−1 to 3367 cm−1 indicates the development of a new hydrogen bond between the amino group of chitosan and the \(-\mathrm{OH}\) group of MC [18, 19]. The –C–H stretching of –CH3 at 2917 cm−1 shifted to 2883 cm−1 in the blends which indicates a probable hydrogen bonding between -CH3 of chitosan and the hydroxyl group of MC.

FTIR pattern of CH, MC, and CH:MC polyblend systems

Probable interaction between CH and MC through hydrogen bonding (White sphere:H, Gray sphere:C, Blue sphere:N, Red sphere:O)
3.2 XRD analysis
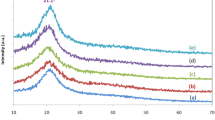
The amorphous and crystalline characteristics of the various blend polymer films have been characterized by an XRD technique. Figure 3 shows the XRD diffractogram of CH-MC blends in various proportions. The broad hump observed indicates the amorphous nature of the polyblend and the peaks observed at 2θ = 11°, 20° [17], and 2θ = 8.5°and 20°[18, 19] correspond to chitosan and methyl cellulose, respectively. Biopolymers exhibit very low crystallinity that is due to intra- and intermolecular hydrogen bonding [20]. From the diffractogram, the intensity of the A3 sample peak is less compared to other samples and it is the most amorphous sample among the prepared samples. The XRD peaks were deconvoluted utilizing Fityk computer software [21] and according to the equation.

XRD pattern of CH: MC polyblend films for different compositions
where Ac and Aa indicate the area under the crystalline peak and amorphous peak, respectively, as shown in Fig. 4. The computed values of the degree of crystallinity of different polyblend systems are shown in Fig. 5. The degree of crystallinity for sample A3 is 6.4%, indicating that this sample is more amorphous compared to other prepared samples and this facilitates the easier segmental motion [22]. The decrease in the crystallinity of a polymer leads to an increase in segmental mobility because crystalline regions of polymers have more ordered and tightly packed structures, which restricts the motion of polymer chains. In contrast, amorphous regions of polymers have a more disordered structure, which allows for more freedom of motion for the polymer chains. Thus, as the degree of crystallinity decreases, the amount of amorphous regions increases, leading to an increase in the segmental mobility of the polymer chains. This increased segmental mobility is beneficial in the case of polymer electrolytes as it can enhance ionic conductivity, which is essential for the functioning of polymer electrolyte systems.

XRD deconvoluted pattern for (a) A1, (b) A2, (c) A3, (d) A4, and (e) A5 polyblend samples

Degree of crystallinity of different polyblend systems versus Methylcellulose content in the polyblend system
3.3 DSC analysis
DSC analysis has been performed to find the \({T}_{g}\) of the polyblend system containing different wt.% of CH and MC. Figure 6 shows the DSC thermograms of the prepared polyblend system. As the concentration of MC increases from 10 to 30 wt.% in the blend, the \({T}_{g}\) value goes on decreasing, i.e., 55 °C for A1, 53 °C for A2, and 48 °C for the A3 sample. The addition of MC promotes a rubbery state in the blend system leading to a drop in the \({T}_{g}\) value. The decrease in the \({T}_{g}\) value may be due to the increase in the segmental motion [23]. The rubbery state contributes to the enhancement of its amorphous nature which was also evident from the XRD studies. Furthermore, as the concentration of MC increased from 40 to 50 wt.%, the \({T}_{g}\) value also increased as of 58 °C for A4 and 61 °C for A5 sample. This may be due to the cross-linkage bonds between the two polymers which hinders the rotation of the polymer segments [24]. Thus, the A3 sample containing 70 wt.% of CH and 30 wt.% of MC have a more amorphous nature than the other prepared blend system causing a reduction in the \({T}_{g}\) value and is the ideal blend composition which can be used in the electrolyte preparation for electrochemical device application.

DSC thermogram of (a) A1, (b) A2, (c) A3, (d) A4, and (e) A5 polyblend samples
3.4 TGA/DTG studies
The thermal steadiness of the SPE films was studied using the instrument SDT Q600 V20.9 build 20. Samples of about 5 mg weight were run in a platinum crucible operated in the range from 25 to 500 °C with nitrogen as purging gas at 80 mL min−1, with a heating scan rate of 10 °C min−1. The thermal steadiness of the polyblend system is a vital property when considering it for energy storage applications (thermal runaway and wide operating temperature range) and therefore the thermal analysis was carried out by TGA/DTG curve. It is observed that in polysaccharides polymer's main thermal degradation process is after 200 °C [25]. Thermographs of polymers CH and MC are represented in Fig. 7, the initial weight loss is due to the dehydration of moisture corresponding to the hydrophilic nature of functional groups \((-\mathrm{OH},\mathrm{ C}-\mathrm{O}-\mathrm{C}, {\mathrm{NH}}_{2})\) of the polymer [26]. The thermal stability of CH is lower than that of the MC polymer as per the thermograph. The main weight loss observed at a maximum of 280 °C in CH is due to thermal degradation by pyrolysis, where splitting of glycosidic linkage takes place (scissoring of \(\mathrm{C}-\mathrm{O}-\mathrm{C})\) [27]. Maximum decomposition temperature (\({T}_{\mathrm{max}})\) of MC is at 360 °C with a sharp DTG curve indicating only one decomposition temperature. In the thermograph of the blended system (CH: MC) two-step degradation was observed (T > 200 °C) initial weight loss corresponds to that of CH and the second one corresponds to MC. No shift in the first degradation step was observed but a variation in temperature related to the second degradation process is observed and may be due to the cross-linking of MC with CH (Fig. 8).

Thermogravimetric analysis/Differential thermogravimetric plots of (a) CS and (b) MC polymers

Thermogravimetric analysis/Differential thermogravimetric plots of (a) A1, (b) A2, (c) A3, (d) A4, and (e) A5 polyblend samples
The thermal activation energy of the polyblend system has been obtained employing the integral Eq. (2) of Coats-Redfern
where \(E\) is the activation energy, which is the minimum energy essential to activate the molecules of polymer to undergo a phase change, \(R\) is the universal gas constant, n is the order of the reaction, \(T\) is the absolute temperature, \(\gamma\) is the fractional weight loss at the particular temperature and calculated by the relation
where \({w}_{i}\) is the initial weight of the polymer sample, \({w}_{f}\) is the final weight of the polymer sample and \({w}_{t}\) is the weight of the polymer sample at a given temperature.
For n = 1, Eq. (2) reduces to
By plotting \(-\mathrm{log}\left[\frac{-\mathrm{log}(1-\upgamma )}{{T}^{2}}\right]\) against \(\frac{1000}{T}\), a straight line is obtained as depicted in Fig. 8, and the activation energy was calculated from the slope obtained from the graph using the relation
A decrease in thermal activation energy was observed due to a decrease in CH content corresponding to the first main degradation followed by an increase in thermal activation energy with an increase in MC content (second degradation) in the polyblend system as per Fig. 9 proving that the availability of the functional group decides the thermal stability.

Variation of \(-\mathrm{log}\left[\frac{-\mathrm{log}(1-\upgamma )}{{T}^{2}}\right]\) Vs \(\frac{1000}{T}\) for (a) chitosan, (b) methylcellulose, and (c) variation of thermal activation energy for the various composition of the CS:MC blend system
3.5 Dielectric studies
The dielectric analysis is one of the methods to quantitively analyze the conductivity of the PE. The dielectric constant of the polyblend decides the ionic species concentration as per the equation \(n={n}_{o }\mathrm{exp}\left[-\frac{U}{kT\varepsilon }\right]\), where n is charged ion concentration, U is the salt dissociation energy and ε is the dielectric constant of the polymer host, ionic conductivity is related to \(n\) as per the equation \(\sigma =\sum_{i}{n}_{i}{q}_{i}{\mu }_{i}\), where i stands for conductivity due to different kinds of conducting species, and q and μ stand for charge and mobility of conducting ions [28]. Real part (\({\varepsilon }^{{\prime}})\) of the complex permittivity \({\varepsilon }^{*}\left(\omega \right)={\varepsilon }^{{\prime}}\left(\omega \right)-{j\varepsilon }^{{{\prime\prime}}{{}}}(\omega )\) is related to the ability of the polymer host to dissociate the ions that in turn decided by the availability of the functional group in the polymer chain and sometimes residual moisture/solvent. The dielectric constant \({\varepsilon }^{{\prime}}\) for the prepared polyblend films were obtained using the formula \({\varepsilon }^{{\prime}}=\frac{Cd}{A\varepsilon_o}\), where C is the measured capacitance and \(\varepsilon_o\) is the permittivity of free space. As per Fig. 10, A3 sample has the highest dielectric constant and therefore is a suitable candidate as polyblend electrolyte host. The room temperature conductivity of blend systems was in the range of 10–7 to 10–9 Scm−1 which was obtained from Nyquist plots which are not shown in this paper.

Frequency dependence of \({\varepsilon }^{{\prime}}\) for the various blended system
The dielectric constant and dielectric loss trend are equally similar in both figures (Figs. 10 and 11), where it rises sharply toward the low frequency and decays at increasing frequency. Dielectric loss (\({\varepsilon }^{{{\prime}}{{\prime}}})\) increases sharply at low frequencies, indicating that electrode polarization and space charge effects have occurred confirming non-Debye dependence [29]. As observed from Fig. 11, there are no relaxation peaks which indicates that the blend system is uniformly distributed polymer chains rather than area-specific entanglement causing packets of air/solvent gap within the matrix. No notable relaxation peaks have been seen in the dielectric loss-frequency spectrum (Fig. 11), indicating that residual water does not contribute to conductivity enhancement [30].

Frequency dependence of \({\varepsilon }^{{{\prime}}{{\prime}}}\) for the various blended system
4 Conclusions
XRD and FTIR were used to illustrate the blend system's ideal composition for electrolyte application. The XRD studies revealed that the blend system consisting of 70 wt.% of CH and 30 wt.% of MC is the most amorphous composition and DSC analysis shows that this blend composition is having the least glass transition temperature of 48 °C which promotes the segmental motion and can be used in the electrolyte application for better ionic conductivity. The thermal activation energy for the A3 sample is neither maximum nor minimum and exhibits a high dielectric constant. The resulting properties of the polyblend system in its facile form are most suitable for application as a polymer electrolyte in energy storage devices. Furthermore, studies on the interaction of salt and plasticizers with a polyblend system would be of great importance in the scientific community. Similarly, the scope of other combinations of bio-based polymers is increasing to satisfy the need for high stability and energy storage capacity in batteries or supercapacitors.
References
H T Ahmed and O G Abdullah Polymers 11 853 (2019)
Y Liang et al. InfoMat 1 6 (2019)
S B Aziz, O G Abdullah and S A Hussein J. Electron. Mater. 47 3800 (2018)
M Abdullah, W Lenggoro and K Okuyama Nanoscience and Nanotechnology 8 731 (2004)
M R Reddy, A R Subrahmanyam, M M Reddy, J S Kumar, V Kamalaker and M J Reddy Mater. Today Proc. 3 3713 (2016)
A Pinotti, M A García, M N Martino and N E Zaritzky Food Hydrocoll. 21 66 (2007)
K Z Hamdan and A S A Khiar Key Eng. Mater. 594 818 (2014)
O G Abdullah, R R Hanna and Y A K Salman J. Mater. Sci. Mater. Electron. 28 10283 (2017)
P T Liu, X M Wei and Z Liu Adv. Mater. Res. 750 802 (2013)
P R and P Wongchaiya Procedia Mater. Sci. 1 499 (2012)
P A Helen, P Perumal, P Sivaraj, M I Diana and P C Selvin Mater. Today Proc. 50 2668 (2020)
M H Buraidah and A K Arof J. Non. Cryst. Solids 357 3261 (2011)
J Wang, S Song, S Gao, R Muchakayala, R Liu and Q Ma Polym. Test. 62 278 (2017)
O G Abdullah, S B Aziz and M A Rasheed Ionics 24 777 (2018)
S B Aziz et al. Ionics 26 1829 (2020)
M Hema, S Selvasekarapandian, D Arunkumar, A Sakunthala and H Nithya J. Non. Cryst. Solids 355 84 (2009)
A S A Khiar Mater. Res. Express 29 055304 (2018)
S B Aziz et al. Polymers (Basel). 12 1 (2020)
A M Abdullah, S B Aziz and S R Saeed Arab. J. Chem. 14 103388 (2021)
E M A Dannoun et al. Polymers (Basel). 12 1 (2020)
M Wojdyr, S Gierlotka and B Palosz Acta Crystallogr. Sect. A Found. Crystallogr. 60 246 (2004)
S B Aziz, M H Hamsan, M F Z Kadir, W O Karim and R M Abdullah Int. J. Mol. Sci. 20 3369 (2019)
S Kiruthika, M Malathi, S Selvasekarapandian, K Tamilarasan and T Maheshwari Polym. Bull. 77 6299 (2020)
S Kiruthika, M Malathi, S Selvasekarapandian, K Tamilarasan, V Moniha and R Manjuladevi J. Solid State Electrochem. 23 2181 (2019)
M Sara, T Rekha, R Koshy, S K Mary, S Thomas and L A Pothan Starch, Chitin and Chitosan Based Composites and Nanocomposites, (Berlin: Springer) p Navard 19 (2019)
E Raphael, C O Avellaneda, B Manzolli and A Pawlicka Electrochim. Acta 55 1455 (2010)
C G T Neto, J A Giacometti, A E Job, F C Ferreira, J L C Fonseca and M R Pereira Carbohydr. Polym. 62 97 (2005)
S B Aziz, T J Woo, M F Z Kadir and H M Ahmed J. Sci. Adv. Mater. Devices 3 1 (2018)
X Qian, N Gu, Z Cheng, X Yang, E Wang and S Dong Electrochim. Acta 46 1829 (2001)
Y N Sudhakar and M Selvakumar Electrochim. Acta 78 398 (2012)
Acknowledgements
Pradeep Nayak is thankful to the University Grants Commission (UGC), New Delhi, Government of India, for providing financial assistance in the form of a Junior Research Fellowship (JRF) UGC-Ref. No. 1237/ (CSIR-UGC NET DEC. 2017) dated 21-01-2019.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nayak, P., Sudhakar, Y.N., De, S. et al. Optimization of Chitosan: Methylcellulose polyblend to obtain highly amorphous polymer matrix useful for ion transportation. Indian J Phys 97, 3483–3493 (2023). https://doi.org/10.1007/s12648-023-02684-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12648-023-02684-1