
Abstract
Background
The spectrum of neuroendocrine neoplasia (NEN) of the head and neck region is wide-ranging and diverse, including a variety of diagnoses stretching from benign and low-malignant tumor forms to highly proliferative, poor prognosis neuroendocrine carcinoma (NEC). Moreover, there are several non-neuroendocrine differential diagnoses to keep in mind as well, displaying various degree of morphological and/or immunohistochemical overlap with bona fide neuroendocrine lesions.
Methods
Review.
Results
While the growth patterns may vary, well-differentiated NEN usually display a stippled “salt and pepper” chromatin, a granular cytoplasm, and unequivocal expression of neuroendocrine markers such as chromogranin A and synaptophysin. However, these features are often less pronounced in NEC, which may cause diagnostic confusion—not the least since several non-NEC head and neck tumors may exhibit morphological similarities and focal neuroendocrine differentiation.
Conclusion
As patients with NEC may require specific adjuvant treatment and follow-up, knowledge regarding differential diagnoses and potential pitfalls is therefore clinically relevant. In this review, the top ten morphological and/or immunohistochemical mimics of NEC are detailed in terms of histology, immunohistochemistry, and molecular genetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroendocrine neoplasia (NEN) of the head and neck region are spectacular lesions, often accompanied by unique morphological, immunohistochemical, hormonal, and/or genetic features. While most of these lesions mainly occur sporadically in adult patients, subsets are intimately coupled to genetically inherit syndromic disease and may therefore present in younger patients [1, 2]. In addition to the demographic diversity, neuroendocrine tumors (NETs) may be notoriously difficult to prognosticate—as the metastatic potential may be challenging to assess by morphology alone. Consequently, pathologists have developed risk stratification algorithms for various NENs in order to assess the risk of disease progression, with the proliferation index (as estimated by mitotic index and/or the Ki-67 labeling index) proving particularly important [3,4,5]. Indeed, these parameters are nowadays routinely used in grading NENs, with neuroendocrine carcinoma (NEC) displaying the highest proliferation index [3]. Due to the highly proliferative nature of this entity, distant metastases and death due to disease are common outcomes for the NEC patient category [6]. Therefore, it is imperative to correctly identify these lesions in a timely fashion, and care must be taken to not confuse NEC with malignant, non-NEC neoplasms with focal immunoreactivity to neuroendocrine markers, as well as the clinically more indolent well-differentiated NETs.
Neuroendocrine Carcinoma of the Head and Neck Region
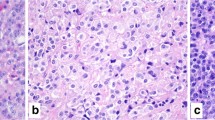
The most clinically urgent NEN subtype is NEC, a poorly differentiated, highly proliferative, malignant tumor of poor prognosis exhibiting significant tumor necrosis and destructively invasive features [3]. In the head and neck region, NEC may develop within the paranasal sinuses, the nasal cavity, the oro- and hypopharynx, the salivary glands, the oral cavity, and the larynx [7,8,9,10,11]. NEC of the head and neck region is further divided into small cell neuroendocrine carcinoma (SCNEC), large cell neuroendocrine carcinoma (LCNEC), and mixed NEC with non-neuroendocrine neoplasms [8]. While SCNEC is composed of diffusely arranged small cells with a high nuclear/cytoplasmic ratio, nuclear molding, and necrosis (Fig. 1A), LCNEC usually exhibits hyperchromatic and pleomorphic tumor cell nuclei (sometimes palisading) with prominent nucleoli (Fig. 1B) arranged in nests, sheets, or trabeculae. Necrosis is often widespread. Immunohistochemistry (IHC) is usually positive for neuroendocrine markers, such as chromogranin A and synaptophysin (Fig. 1C, D), but expression of both markers may be absent in some lesions [12]. Inclusion of second-generation neuroendocrine markers, such as INSM1, may be of value in these instances [13]. Both SCNEC and LCNEC express keratins, and the expression may be faint and dot like in the former entity. This feature can be particularly helpful in distinguishing from paragangliomas. The Ki-67 proliferation index is usually high, always > 20%, and frequently between 55 and 100% (Fig. 1E, F). Subsets of oropharyngeal NECs may be human papillomavirus (HPV)-driven neoplasms, exhibiting strong p16 immunoreactivity [14]. Most head and neck NECs are positive for p53 by immunohistochemistry and negative for retinoblastoma protein (pRb), which is due to frequent somatic mutations in the tumor suppressors TP53 and RB1 [15]. Interestingly, subsets of cases also harbor pathogenic variants in potentially actionable therapeutic target genes associated with the NOTCH and PI3K/AKT/mTOR pathways [15].

Neuroendocrine carcinoma of the head and neck region. A Sinonasal small cell neuroendocrine carcinoma exhibiting solid sheets of tumor cells with high nuclear/cytoplasmic ratio and nuclear molding. B Salivary gland large cell neuroendocrine carcinoma with nuclear pleomorphism and geographic tumor cell necrosis. C, D Immunohistochemistry for synaptophysin and chromogranin A usually reveal diffusely positivity. E, F The Ki-67 proliferation index is always above 20%, here exemplified by 90% and 70%, respectively
Head and Neck Mimics of Neuroendocrine Carcinoma
A plethora of entities of diverse lineages must be excluded when considering a diagnosis of NEC. On one hand, the cellular, closely packed monotonous tumor cells in SCNEC bring nearly all small round blue cell tumors into the differential diagnoses, while on the other hand, miscellaneous head and neck epithelial malignancies remain close mimics of LCNEC. The challenges are particularly heightened when dealing with small tissue volumes in biopsy material. Awareness of the histomorphologic spectrum, immunophenotypic nuances, and molecular traits of the diverse mimics and employing a stepwise algorithmic approach can aid in reaching an accurate diagnosis (Fig. 2, Table 1). Given the possible differential diagnostic dilemmas discussed above, we provide a review of the top ten morphological and immunohistochemical NEC mimics that the practicing pathologist should not miss.

Schematic overview of the top ten differential diagnoses of neuroendocrine carcinoma (NEC) in the head and neck region with key immunohistochemical and molecular attributes. PitNET pituitary neuroendocrine tumor, MTC medullary thyroid carcinoma, PGL paraganglioma, NE neuroendocrine, ME myoepithelial, Sq squamous, SCC basaloid squamous cell carcinoma, ACC adenoid cystic carcinoma, ONB olfactory neuroblastoma, TCS teratocarcinosarcoma, ARMS alveolar rhabdomyosarcoma. Created using BioRender.com
Well-Differentiated Neuroendocrine Tumors
Well-differentiated NETs (WDNETs) encompass well-differentiated neoplasms with neuroendocrine features recognizable on light microscopy, i.e., exhibiting cellular monotony, random anisonucleosis, stippled nuclear chromatin, and varied growth patterns (nests, trabeculae, cords, festoons, rosettes). These tumors are strongly immunoreactive with neuroendocrine markers. WDNETs and NECs are clinically and genetically distinct entities with divergent treatments and outcomes. Therefore, a clear distinction between WDNET and NEC is necessary. This is usually not problematic, however, tends to be challenging in cases with limited or crushed tissue.
Pituitary Neuroendocrine Tumor (PitNET)
Pituitary neuroendocrine tumors (PitNETs) are well-differentiated adenohypophyseal lesions that may cause a wide variety of symptoms depending on its specific hormone production. PitNETs may be encountered in the sinonasal tract, most frequently in the sphenoid sinus, due to invasion from a sellar tumor or rarely as ectopic PitNET. The previous terminology “pituitary adenoma” and “pituitary carcinoma” are no longer recommended [16]. PitNETs should be subtyped in terms of tumor cell lineage and expression of pituitary hormones by the use of IHC, and tumors are derived from a PIT1 lineage (i.e., somatotroph, lactotroph, and thyrotroph), a TPIT lineage (corticotroph tumors), an SF1 lineage (gonadotroph tumors), or tumors without a distinct lineage (plurihormonal lesions and hormonally silent “null cell tumors”) [16]. Most PitNETs are characterized by a hypercellular and well-differentiated mass composed of monomorphic tumor cells (Fig. 3A). Although there are morphological clues regarding the PitNET subtype, the distinction is not entirely reliable without IHC and therefore all PitNETs should be assessed for PIT1, TPIT, and SF1 immunoreactivity [16]. Hormone stains are also recommended, including growth hormone (GH), prolactin (PRL), and beta-thyroid-stimulating hormone (β-TSH) for PIT1 lineage tumors; adrenocorticotropic hormone (ACTH) for TPIT lineage tumors; and beta-follicle-stimulating hormone (β-FSH) and beta-luteinizing hormone (β-LH) for SF1 lineage tumors [17]. Keratin IHC can also be useful (CAM5.2, AE1/AE3, and/or CK18) to subtype somatotroph PitNETs as either densely or sparsely granulated, characterized by a perinuclear or globular cytoplasmic stain, respectively [16]. Metastatic PitNETs are rarely encountered, and when metastatic, most commonly affect liver, bone, lung, and lymph nodes [18].

Differential diagnosis of a neuroendocrine carcinoma: Morphological attributes of different well-differentiated neuroendocrine tumors of the head and neck region. A Somatotroph pituitary neuroendocrine tumor (PitNET) characterized by large, acidophilic and granular cells with little nuclear pleomorphism. B Medullary thyroid carcinoma comes in different forms and shapes, but regularly display cells with amphophilic to basophilic cytoplasm and neuroendocrine-type chromatin and often grow in an amyloid background. C Carotid body paraganglioma exhibiting a nested appearance and cells with abundant, granular cytoplasm. Nuclear pleomorphism may be present but correlate poorly to metastatic behavior. D Neuroendocrine neoplasia of unknown primary (NEN-UP) metastatic to the skin. This lesion required extensive immunohistochemistry and finally led to a diagnosis of a primary pulmonary carcinoid
Medullary Thyroid Carcinoma (MTC)
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor derived from the calcitonin-producing C cells of the thyroid gland. Although the majority of tumors arise sporadically, up to 25% are thought to be associated with multiple endocrine neoplasia type 2 (MEN2A or MEN2B) syndrome, with the affected patient demonstrating activating, constitutional pathogenic variants in the RET proto-oncogene [2, 19]. Sporadic tumors are usually driven by somatic RET mutations or mutually exclusive RAS gene mutations [20]. There are numerous morphological MTC patterns reported, but they are not routinely classified on a histological basis as there is no established correlation to either genotype or clinical outcomes [19]. MTCs are characterized by round (sometimes plasmacytoid, polygonal, or spindle shaped) cells in nests with an interdigitating stroma exhibiting various amounts of amyloid deposition (Fig. 3B). The cytoplasm is usually amphophilic and granular due to their secretory content. MTCs routinely express neuroendocrine markers as well as signs of thyroid differentiation (TTF1), whereas monoclonal PAX8 expression is lacking, as is thyroglobulin (the latter a consequence of the non-follicular cell origin). The hallmark of MTCs is calcitonin immunoreactivity, although the stain can vary in intensity and spatial distribution. In the metastatic setting without an established thyroid lesion, care must be taken not to prematurely assume that a neuroendocrine tumor with focal calcitonin and TTF1 expression is a metastatic MTC—as cases of laryngeal NETs with aberrant expression of these markers have been reported [21]. In terms of prognosis, the 2022 World Health Organization (WHO) classification of endocrine and neuroendocrine tumors recommends that MTCs be graded based on the mitotic index, the presence of tumor necrosis, and the Ki-67 proliferation index, in which high-grade lesions display necrosis, a mitotic count ≥ 5 per 2 mm2, and/or a Ki-67 index of ≥ 5% [5, 19]. In terms of metastatic disease, most MTCs spread regionally to neck lymph nodes, but subsets of cases may also spread to the liver, lungs, and bone. It may be worth noting that rare cases of MTC metastatic to the parotid and pituitary glands have been reported, which potentially could constitute differential diagnostic conundrums [22, 23]. Moreover, subsets of MTCs may display a small cell phenotype, further complicating the histological work-up if NEC is suspected [23].
Paraganglioma (PGL)
Head and neck paragangliomas (PGLs) are usually parasympathetic, non-functioning neuroendocrine tumors, which sets them apart from their infra-diaphragmatic, norepinephrine, and/or epinephrine-producing counterparts [1]. They are collectively the most inheritable of all human neoplasia with approximately 40% of patients carrying an underlying constitutional genetic event, while the metastatic potential of these lesions is usually low [24]. Patients with constitutional pathogenic variants in succinate dehydrogenase (SDH) subunit A, B, C, D, or AF2 (SDHA, SDHB, SDHC, SDHD, SDHAF2) harbor an increased risk of developing head and neck paraganglioma with a low risk of disseminated disease [25]. The underlying molecular biology is complex, with tumors showing a higher risk of metastases often driven by mutations in tricarboxylic acid (TCA) cycle genes (not only restricted to SDH genes) that will lead to TCA cycle arrest and accumulation of early metabolites, which in turn may activate oncogenic hypoxia-inducible factor (HIF) pathways [26]. However, subsets of cases are driven by mutations in various kinase-associated pathways, and these lesions usually tend to be non-metastatic. Therefore, there is a well-developed genotype–phenotype correlation which can be assessed by histopathology and immunohistochemistry: a positive SDHB immunostain strongly argues against mutational inactivation of either SDHB, SDHC, or SDHD genes—in turn arguing against (although not excluding) the risk of metastatic potential [27, 28]. When presenting in characteristic locations, such as the carotid bifurcation or the jugulotympanic area, a head and neck paraganglioma is quite easily distinguished by morphological assessment. The tumor cells are usually round to oval and arranged in small nests (so-called “zellballen”) embedded in a highly vascular stroma (Fig. 3C). The cytoplasm is granular with an amphophilic or basophilic appearance, and nuclear pleomorphism is usually limited to absent. Mitoses and tumor necrosis are rarely detected. Tumor cells are positive for chromogranin A, synaptophysin, and GATA3, while consistently keratin negative [29]. An S100 protein (or SOX10) immunohistochemistry identifies the sustentacular network of cells supporting the tumor cells—but the finding of sustentacular cells is not diagnostic for paraganglioma, as other neuroendocrine tumors also may exhibit this feature [30]. Using functional IHC, most head and neck paragangliomas are positive for choline acetyltransferase, an enzyme in the acetylcholine biosynthesis pathway, while often negative for enzymes responsible for catecholamine production, such as tyrosine hydroxylase [31, 32]. Metastatic head and neck paraganglioma usually spread to regional lymph nodes, while distant site involvement is rare [33].
Metastastic Neuroendocrine Neoplasia of Unknown Primary
NENs of unknown primary (NEN-UPs) are metastatic lesions without a known primary tumor location, a finding reported in 12–22% of NEN patients [34]. The importance of identifying the primary site cannot be underestimated given that the various clinical and prognostic features of NENs depend on the tumor origin site. There are several morphological clues that can be used to properly identity a NEN-UP, including amyloid deposits in MTC, psammoma bodies in somatostatinoma, a hyalinized stroma in insulinoma, as well as the hyaline globules and basophilic cytoplasm of pheochromocytoma [34]. Even so, it is not unusual for a metastatic NEN-UP to be characterized by nested cells with little or no morphological findings unique to the primary tumor site (Fig. 3D). From an immunohistochemical perspective, various combinations of neuroendocrine marker and transcription factors results may be useful. For example, TTF1 may help identify pulmonary carcinoids and MTC, PDX1 may assist in recognizing NENs of the upper gastrointestinal tract including the pancreas, whereas CDX2 and SATB2 may highlight NENs of the lower gastrointestinal tract. In addition, testing for various hormones may be useful, including calcitonin for MTC, serotonin for lower gastrointestinal NENs, islet hormones for pancreatic NENs, and GLP1 for rectal NENs, to name just a few [35, 36]. NECs outside of the head and neck area occasionally may metastasize to the jaws and major salivary glands [36]. Indeed, metastatic neuroendocrine tumors to the parotid gland accounted for 22% of all metastatic tumors to this organ in a recent case series, and most cases were either pulmonary NECs, Merkel cell carcinomas (MCCs), or MTCs [37]. If not previously known, a hypothetical IHC panel for NEN-UPs metastatic to the salivary glands would therefore need at least TTF1, calcitonin, and CK20, in addition to neuroendocrine markers and Ki-67.
Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is a rare neuroendocrine carcinoma of the skin with an estimated incidence of 2.2 cases per million person-years, afflicting predominantly older patients [38]. The tumors are either driven by UV-induced mutations or by a Merkel cell polyoma virus infection, and the exact proportion of these etiologies varies with geographic distribution [39]. From a morphological perspective, MCCs are composed of solid arrangements of monomorphic tumor cells with a high nuclear/cytoplasmic ratio, smudged nuclear chromatin, indistinct nucleoli, and displaying innumerable mitoses (Fig. 4A, B). The IHC profile is characteristically neuroendocrine [40], while a perinuclear, “dot-like” keratin stain (most strikingly with CK20) is characteristic of MCC (Fig. 4C). The Ki-67 proliferation index is usually exceedingly high (> 90%). Moreover, virus-driven MCCs are positive for the Merkel cell polyoma antigen (Fig. 4D). When presenting as a primary tumor, the diagnosis is usually quite straight-forward, but metastatic lesions may cause diagnostic difficulties if the primary tumor is not known. To complicate matters even more, subsets of MCCs have been reported to originate from mucosal linings of the upper respiratory and GI tracts and might be clinically silent [41].

Differential diagnosis of a neuroendocrine carcinoma: Merkel cell carcinoma (MCC). A, B MCC characterized by a diffusely infiltrative tumor within the dermis and subcutaneous space growing in sheets and trabeculae. Cells exhibit a high nuclear-to-cytoplasmic ratio, and mitoses and apoptotic bodies are plentiful. C Cytokeratin 20 (CK20) usually present with a perinuclear or dot-like pattern. D The majority of MCCs are positive for the Merkel cell polyoma virus antigen
NUT Carcinoma
NUT carcinoma is a highly aggressive tumor primarily affecting young patients, often presenting in the midline of the thorax and head and neck regions [42, 43]. On the histological level, NUT carcinoma is composed of small to intermediate cells with an undifferentiated, primitive, and monotonous appearance (Fig. 5A). Mitotic figures and necrosis are easily identified. A significant subset exhibits abrupt keratinization (Fig. 5B). Using IHC, NUT carcinomas are epithelial neoplasms, reacting with keratins and squamous markers, such as CK5/6, p63, and p40 (Fig. 5C). CD34 is also positive in approximately 50% of cases [43]. NUT carcinoma is driven by NUTM1 gene rearrangements [43], and NUT protein immunohistochemistry (Fig. 5D) is useful to highlight this genetic aberrancy—as NUT protein expression is not normally seen in cells outside of the testis and ciliary ganglion [44]. Interestingly, neuroendocrine differentiation has been reported, with a high level of suspicion required when considering NEC in young patients by incorporating NUT immunohistochemistry [45].

Differential diagnosis of a neuroendocrine carcinoma: NUT carcinoma. A, B Primitive cells arranged in solid sheets with areas of abrupt keratinization. C Immunohistochemical positivity is noted for squamous cell markers, such as p40. D Nuclear NUT protein expression is evident, indicating an underlying translocation involving the NUT gene
Sinonasal Undifferentiated Carcinoma
Sinonasal undifferentiated carcinoma (SNUC) is a rare but highly aggressive epithelial neoplasia lacking morphological and immunohistochemical evidence of lineage (including glandular, squamous, neuroendocrine, or mesenchymal differentiation) [46]. Thus, it is a diagnosis of exclusion in which a broad range of possible differential diagnoses must be considered. Usually presenting as a large mass in the sinonasal tract, these tumors are often invasive at diagnosis [47]. SNUC exhibits high-grade histology with uniform tumor cells growing in sheets, lobules, nests or trabeculae, lacking squamous, or glandular differentiation. Tumor cells express keratins, while p40 is negative and p63 may exhibit weak and unspecific staining [48] (Fig. 2). Subsets of SNUC may express patchy chromogranin A and/or synaptophysin immunoreactivity, making them a potential differential diagnosis in the work-up of NEC of the head and neck region [49]. Somatic IDH2 mutations have been identified in large subsets of SNUC and are readily identifiable using sequencing analysis, while IDH immunohistochemistry has proven inconsistent in pinpointing IDH2-mutated SNUC [50, 51].
Basaloid Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is the most common histological type of cancer in the head and neck region. While the diagnosis of a differentiated SCC is not problematic, the basaloid SCC subtype is a common differential diagnosis of SCNEC, particularly in biopsy material. Basaloid SCC remains an uncommon malignancy that is associated with aggressive clinical behavior and poor median survival (18 months) [52]. Closely packed basaloid cells and lack of significant keratinization typically impart a blue cell tumor appearance at low power that resembles SCNEC. Lobules, adenoid/pseudoglandular structures or variably anastomosing islands of tumor cells exhibiting peripheral palisading, thickened basement membrane-like material, and central comedonecrosis are typical histological features (Fig. 6A) [53]. The neoplastic cells show pleomorphic hyperchromatic nuclei with scanty cytoplasm. The presence of carcinoma in situ or areas of abrupt squamous differentiation (keratin pearl formation) are useful clues to the diagnosis. Mitoses are usually easily identified. There is usually strong and diffuse immunoreactivity for pancytokeratin, p40 (Fig. 6B), and p63, while neuroendocrine markers are negative. SOX10, CD117, and carcinoembryonic antigen (CEA) are notably positive in a subset of basaloid SCC [53,54,55], features not seen in SCNEC.

Differential diagnosis of a neuroendocrine carcinoma (NEC): Epithelial morphology. A Basaloid squamous carcinoma (BSC) with lobules of basaloid cells exhibiting peripheral palisading and central comedonecrosis. B Immunohistochemistry for p40 is diffusely and strongly positive in BSC. C Solid adenoid cystic carcinoma (ACC) is composed of solid nests and lobules of basaloid cells with hyperchromatic angulated nuclei and sparse ducts imparting a blue tumor appearance to the tumor. D Immunohistochemistry for myoepithelial marker SOX10 in ACC is helpful in distinguishing it from NEC
Adenoid Cystic Carcinoma Solid Pattern
While histological features of conventional adenoid cystic carcinoma (ACC) (with its typical cribriform architecture, dual-layered tubules lined by epithelial–myoepithelial cells, and luminal basophilic matrix) are quite characteristic and easy to diagnose, ACC with a solid pattern can resemble SCNEC, especially in a limited or small biopsy. Solid ACC is composed of diffuse sheets of basaloid cells that are largely devoid of the hallmark cribriform glands or tubules (Fig. 6C). These tumors commonly have increased mitoses and tumor necrosis. However, distinction from SCNEC can be readily achieved with the use of selected IHC. ACC shows positivity for epithelial (CK7, CEA, EMA) and myoepithelial markers (S100 protein, SOX10, SMA, calponin) (Fig. 6D), while is negative for neuroendocrine markers. CD117 positivity may be seen in both ACC [56] and SCNEC [57], hence lacks specificity. The majority (60–90%) of ACC reveal a diagnostic fusion involving MYB/MYBL1 with NFIB genes, with MYB::NFIB the most common [58]. Molecular testing is not required routinely, but may be performed to establish an ACC diagnosis in challenging cases.
SWI/SNF Complex-Deficient Carcinomas (SMARCB1 & SMARCA4)
The differential diagnoses of NEC have expanded to include SWI/SNF complex-deficient sinonasal carcinomas, whether SMARCB1 or SMARCA4. These tumors predominantly affect adult males [59], typically arise in the paranasal sinuses (particularly the ethmoids) [59, 60], and frequently present at an advanced stage [59]. Both are high-grade malignancies histologically characterized by a monotonous population of undifferentiated cells. Similar to NEC, tumors are cellular and composed of islands and sheets of uniformly high-grade cells with brisk mitotic activity and foci of tumor necrosis.
SMARCB1-deficient sinonasal carcinomas predominantly exhibit a basaloid (~ 2/3) or rhabdoid (~ 1/3) morphology; the latter may be very focal (Fig. 7A, B). Additionally, sharp, punched-out vacuoles within tumor sheets, yolk sac-like morphology, and pagetoid spread along the surface epithelium may be seen in SMARCB1-deficient carcinomas that may aid in diagnosis when present [59].

Differential diagnosis of a neuroendocrine carcinoma: SWI/SNF-deficient carcinomas. A-C SMARCB1-deficient carcinoma: Tumor shows sheets of undifferentiated carcinoma cells with basaloid morphology (A) and rhabdoid morphology (B) in a desmoplastic stroma. C Complete loss of SMARCB1 (INI1) immunoreactivity in the tumor nuclei with retained immunopositivity in the stromal and endothelial cell nuclei is essential for the diagnosis. D SMARCA4-deficient carcinoma showing diffuse sheets of undifferentiated cells with scanty stroma. E SMARCA4-deficient carcinoma on higher magnification reveals epithelioid tumor cells with rhabdoid morphology and conspicuous nucleoli. F SMARCA4 immunohistochemistry shows complete loss of SMARCA4 (BRG1) protein in the tumor nuclei, whereas the stromal cells are positive and serve as internal controls
SMARCA4-deficient carcinoma is composed of large, epithelioid cells lacking overt differentiation (Fig. 7D, E) [59]. Rhabdoid and basaloid cells are less frequent. The cytologic appearance mimics LCNEC, requiring exclusion of the SWI/SNF complex-deficient carcinomas.
Both entities require IHC to confirm the diagnosis. A complete loss of SMARCB1 (testing INI1]) and SMARCA4 (testing BRG1) reactivity in the tumor nuclei is essential for the diagnosis of SMARCB1-deficient (Fig. 7C) and SMARCA4-deficient sinonasal carcinoma (Fig. 7F), respectively [59]. Additionally, the tumor cells are positive for pancytokeratin (AE1/AE3, CAM5.2, OSCAR) and variably positive with CK7. Further, there is frequently reactivity with CK5/6, p63, and p40 in SMARCB1-deficient carcinoma, while these markers are generally negative in SMARCA4-deficient carcinomas. Tumor cells are negative with NUT and there is no HPV or Epstein Barr virus association [59]. It is noteworthy that both tumor types can focally express neuroendocrine markers (synaptophysin, chromogranin, and INSM1) in most SMARCA4-deficient carcinomas and up to 18% of SMARCB1-deficient carcinomas [61, 62]. Thus, INI1 and/or BRG1 must be included in a panel of immunohistochemistry studies when evaluating poorly or undifferentiated carcinomas of the sinonasal tract. Immunohistochemistry is generally sufficient for diagnosis, although FISH or sequencing can be performed to demonstrate biallelic (homozygous) deletions of the SMARCB1 gene [59] or loss-of-function (mostly truncating) mutations in SMARCA4-deficient carcinomas [63].
Tumors with Neuroectodermal Differentiation
Olfactory neuroblastoma (ONB) is a neuroectodermal neoplasm typically arising in the olfactory epithelium centered on the cribriform plate of the ethmoid sinus, composed of lobules of small round cells surrounded by sustentacular cells in a loose fibrovascular stroma. The morphological spectrum of ONB spans from the well-differentiated end (wherein the neoplastic cells display lobular architecture, uniform cells with stippled chromatin, rosettes and/or neurofibrillary stroma, low mitoses, and absence of tumor necrosis) (Fig. 8A) to the poorly differentiated end (which is characterized by limited lobular architecture, pleomorphism, increased mitoses, karyorrhexis, and tumor necrosis) (Fig. 8B). These features of diminishing differentiation are assembled into the Hyams tumor grades [64]. Neurons, melanin pigment, or divergent differentiation (glandular, squamous, or rhabdomyoblastic) may be seen [65,66,67]. A distinction of high-grade ONB from NEC is challenging and requires additional testing. ONB expresses diffuse neuroendocrine markers (synaptophysin, chromogranin, INSM1), neurofilament, and calretinin (Fig. 8C); about a third may show focal keratin reactivity [68]. The peripheral rim of sustentacular cells is highlighted by S100 protein and/or SOX10 (Fig. 8D). Recently, SATB2 and focal GATA3 expression have been demonstrated in grade 1 to 3 ONBs [69]. Tumor cells are negative for CD99, Fli1, NUT, and EBER, while INI1 and BRG1 are retained (intact). In contrast to ONB, NEC is negative for SOX10, S100, calretinin, SATB2, and GATA3 [69].

Differential diagnosis of a neuroendocrine carcinoma: Tumors with neuroectodermal differentiation. A–D Olfactory neuroblastoma (ONB). A Lobules of uniform cells with stippled chromatin separated by fibrovascular stroma in a Hyams low-grade ONB. B Loss of lobular architecture, increasing pleomorphism, and mitotic activity is seen in Hyams high-grade ONB. C Chromogranin is strongly positive in the tumor cells. D S100-positive sustentacular cells are typically seen rimming the periphery of lobules in ONB. E–G Teratocarcinosarcoma (TCS). E Irregular lobules of blue round cells in a variably cellular stroma is a frequent feature of TCS. F Nests of neoplastic cells comprising round primitive neuroectodermal cells intermixed with fetal (clear)-like squamous cells. G Malignant epithelial and sarcomatous components of TCS
Teratocarcinosarcoma (TCS) is a unique sinonasal tumor that is composed of a triad of epithelial, mesenchymal, and primitive neuroectodermal components; the three elements are intermixed and any constituent may predominate in a case. A biopsy with a preponderant primitive neuroectodermal component may be mistaken for NEC if the intimately admixed epithelial or mesenchymal components are either sparse or overlooked. Fetal-like (clear) squamous epithelium, immature mesenchyme, benign and/or carcinomatous epithelium, strap cells, or sarcomatous stroma are features that suggest a diagnosis of TCS (Fig. 8E–G). In contrast to a more uniform histological picture, the varied components of TCS render a very heterogeneous low-power appearance that may serve as an important clue to the diagnosis. Immunohistochemistry can highlight the presence of epithelial and sarcomatous (especially, rhabdomyosarcomatous elements positive for desmin, MyoD1, or Myogenin) apart from neuroendocrine marker expression in the primitive neuroectodermal component. SMARCA4 (BRG1) loss, complete or partial, is identified in up to 80% of the cases [70], while a subset may reveal nuclear ß-catenin immunoreactivity [71].
Mucosal Melanoma
Sometimes referred to as “the great mimicker,” melanoma presents with various morphological appearances and is thus a frequent tumor in many differential categories. Derived from melanocytes of the skin or mucosal linings, up to 25% of melanomas present in the head and neck region, with the scalp and cheek the two most common sites [72], while oral cavity and sinonasal tract may also be primary sites. Melanoma is usually identifiable using IHC targeting S100 protein, SOX10, MART-1 (Melan A), MITF1, and HMB45. From an embryonic perspective, melanocytes and most neuroendocrine cells both derive from the neural crest, and it is therefore not surprising to find expressional evidence of neuroendocrine differentiation in small subsets of melanoma [73, 74]. Indeed, in a retrospective study of > 300 melanomas, immunoreactivity for chromogranin A and synaptophysin was found in 2% and 8.6% of cases, respectively [75]. Focal or faint expression of at least one of these markers was observed in 37.2% of the tumor cohort, thereby highlighting the need for a careful approach when assessing neuroendocrine markers in melanocytic lesions.
Round Cell Sarcomas
Ewing Sarcoma
Ewing sarcoma (ES) is a primitive small round cell tumor that frequently needs to be distinguished from SCNEC. ES is defined by reciprocal translocations between the FET (encompassing EWSR1, FUS, and TAF15 genes) and the ETS (commonly including FLI1, ERG, ETV1, ETV4, or FEV) family of genes [76, 77]. Like SCNEC, it is composed of cellular sheets of monotonous small round cells, 1–2 times the size of lymphocytes, with scant cytoplasm, round to oval nuclei, delicate stippled chromatin, and devoid of conspicuous nucleoli; occasional rosettes are identified (Fig. 9A, B) [78]. Immunohistochemistry can aid in distinguishing ES from SCNEC, in which ES shows diffuse membranous positivity for CD99 (Fig. 9C) and concurrent nuclear reactivity for NKX2.2 (Fig. 9D) [79,80,81,82]. Importantly, NKX2.2 can also be seen in SCNECs [76, 77] as can CD99, but the latter is strong and membranous in ES [83]. Importantly, neuroendocrine marker positivity may be observed in ~ 50% of ES cases and ~ 30% of cases show cytokeratin expression [84, 85]. Fli1 [86] and ERG [87] reactivity are seen in cases with the respective fusions. A subtype of ES, adamantinoma-like Ewing sarcoma (ALES) tends to show a nested/lobular architecture, peripheral palisading, hyalinized stroma, and abrupt squamous differentiation; IHC evidence of squamous differentiation in the form of diffuse cytokeratin and p40/p63 reactivity is noted along with CD99 and NKX2.2 and most commonly the EWSR1::FLI1 fusion [88, 89].

Differential diagnosis of a neuroendocrine carcinoma: Small round cell tumors. A–D Ewing sarcoma (ES). A Typical blue tumor appearance at low power in ES. B Diffuse sheets of monotonous round cells with primitive nuclei and scanty cytoplasm on high power in ES. C Strong and diffuse membranous staining with CD99 is typical of ES; many non-ES tumors also display CD99 positivity albeit usually focal or cytoplasmic. D Nuclear reactivity with NKX2.2 is characteristic although not specific of ES. E, F BCOR-related sarcoma. E Round cells often mixed with focal spindle cells, pale nuclear chromatin, inconspicuous nucleoli, and abundant myxoid stroma with delicate vascularity are seen in BCOR-related sarcoma. F Nuclear immunostaining for BCOR is characteristic. G, H CIC-related sarcoma. G Lobulated growth, undifferentiated round cells with mild pleomorphism, vesicular chromatin and prominent nucleoli, and delicate fibrous septae are features of CIC-related sarcomas. H CIC-related sarcomas display diffuse and strong nuclear WT1 immunoreactivity
Other Undifferentiated Round Cell Sarcomas
Rarely, undifferentiated round cell sarcomas other than ES may be encountered that need to be distinguished from SCNEC. These include (1) round cell sarcomas with EWSR1-non-ETS fusions [90,91,92]; (2) CIC-rearranged sarcomas [93, 94]; and (3) BCOR-rearranged sarcomas [95, 96].
Round Cell Sarcomas with EWSR1–non-ETS Fusions
These are round and spindle cell sarcomas with EWSR1 or FUS fusions involving partners unrelated to the ETS gene family. These mainly comprise EWSR1::NFATC2 and FUS::NFATC2 sarcomas and EWSR1::PATZ1 sarcomas [90,91,92, 97,98,99]. Unlike conventional ES, these tumors exhibit atypical morphological features in the form of scattered enlarged cells, prominent nucleoli, or unusual clinical profiles (older patients). Nonetheless, there is considerable overlap with ES, including membranous CD99 staining. Although the pathologic spectrum is wide, some phenotypic clues to underlying genotypes can be helpful. Sarcomas with NFATC2 fusions tend to exhibit epithelioid features [90, 100, 101], while PATZ1 sarcomas are composed of largely undifferentiated round to ovoid neoplastic cells in a frequently sclerotic background [91, 98, 99, 102]. NFATC2 sarcomas express diffuse CD99 (like ES) in about 50% of cases; NKX2.2, dot-like keratin, and PAX7 positivity may also be observed [103, 104]. PATZ1 sarcomas do not consistently express CD99, however, may variably express CD34, and show a divergent phenotype with both myogenic (desmin, myogenin, MyoD1) and neurogenic (S100 protein, SOX10) markers [91, 98, 99, 102], while neuroendocrine markers are usually absent. Identification of the fusion transcripts on molecular testing is the gold standard.
CIC-rearranged sarcomas are round cell undifferentiated sarcomas that are defined by CIC-related gene fusions, mostly CIC::DUX4 fusion (about 95%) [93, 94]. CIC sarcomas are composed of undifferentiated round cells, however, tend to show lobulated growth (at least focally), and delicate fibrous septae; cells display mild pleomorphism and possess vesicular chromatin and prominent nucleoli (Fig. 9G). At times, epithelioid morphology can predominate [105]. By IHC, WT1 (90–95%) (Fig. 9H) and ETV4 (95–100%) are positive and are extremely useful markers [106,107,108]. CD99 is positive albeit patchy and cytoplasmic [105], rather than membranous. However, NKX2.2 is typically negative [109]. Sarcomas with CIC::NUTM1 fusions are positive for NUT protein [110, 111]. Molecular testing reveals CIC-related fusions.
BCOR-related sarcoma is a primitive round cell sarcoma showing BCOR genetic alterations. These tumors typically affect children with > 90% of patients being < 20 years [95]. Histology typically reveals vague nesting, round cells often admixed with focal spindled cells, pale nuclear chromatin, inconspicuous nucleoli, and abundant myxoid stroma with delicate vascularity (Fig. 9E) [95, 96]. By IHC, tumor cells show diffuse, strong BCOR (Fig. 9F), SATB2, and cyclin D1 positivity. CD99 is seen in about 50% of cases [95], but neuroendocrine markers are usually absent.
Alveolar Rhabdomyosarcoma (Solid Subtype)
Rhabdomyosarcoma (RMS) is a malignant mesenchymal tumor composed of primitive cells exhibiting skeletal muscle differentiation. Head and neck RMS account for about 35–40% of all RMS cases [112]. It encompasses embryonal, alveolar, pleomorphic, and spindle/sclerosing subtypes. Among the subtypes, alveolar rhabdomyosarcoma (ARMS), particularly the solid subtype, most closely mimics SCNEC [113]. In comparison to SCNEC, the patients of ARMS are much younger, with the peak age of ARMS being 10–25-year-old young adults [114,115,116], although cases in adults > 45 years in the sinonasal tract especially are not uncommon [113, 117, 118]. Microscopically, ARMS is characterized by cellular nests of small round cells separated by fibrovascular septae. Toward the center, the tumor cells tend to be dyscohesive conferring an alveolar configuration to the tumor; the latter is a vital diagnostic clue in favor of ARMS. In contrast, the solid subtype of ARMS is composed of diffuse sheets and lacks this nested/alveolar pattern and fibrovascular septae making it morphologically indistinguishable from NEC and small blue round cell tumors (Fig. 10A) [113, 114, 119, 120].

Differential diagnosis of a neuroendocrine carcinoma: Small blue round cell tumors. A–D Solid alveolar rhabdomyosarcoma (ARMS). A Solid sheets and islands of primitive cells with hyperchromatic nuclei, and devoid of a conspicuous alveolar pattern in solid ARMS. Tumor cells are positive for desmin (B) and diffusely positive for Myogenin (C) indicating skeletal muscle differentiation. D Focal expression of chromogranin is seen in ARMS in a subset of cases. This is a potentially serious caveat that can lead to an erroneous diagnosis of NEC. E–G Synovial sarcoma (SS). Blue tumor appearance of SS is typical at low power. Tumor is composed of highly cellular sheets of ovoid cells with hyperchromatic nuclei and often displays hemangiopericytoma-like vascularity. F Poorly differentiated SS is cellular blue tumor with increased pleomorphism and mitotic activity. G Diffuse and strong nuclear expression of TLE1 are seen in nearly all SS, however, is not specific; TLE1 should always be included in a panel of immunohistochemical markers. H, I Lymphoma. H Crushed blue tumor cells with round cell appearance seen within fibrotic stroma in a case of diffuse large B-cell lymphoma. I B-lineage marker, CD20 is positive on the lymphoma cells
By IHC, cytoplasmic desmin (Fig. 10B), diffuse nuclear myogenin (Fig. 10C), and focal nuclear MyoD1 positivity are diagnostic of ARMS. Notably, neuroendocrine markers and keratins can be expressed in some cases of RMS [113]. Specific neuroendocrine markers (chromogranin A and/or synaptophysin) can be seen in up to 43% of cases (Fig. 10D) [121]. About 32% of cases can express both cytokeratins and NE markers [113, 121, 122]. This aberrant keratin and neuroendocrine marker expression in RMS can lead to an erroneous diagnosis of NEC if skeletal muscle markers are not employed. Hence, a panel of markers is essential to avoid diagnostic pitfalls. Molecular testing for ARMS diagnosis and prognostication is recommended although not necessary for distinguishing ARMS from NEC. The majority (~ 70–90%) of ARMSs contain PAX3::FOXO1 fusions with the remaining tumors generally PAX7::FOXO1 [114, 123].
Synovial Sarcoma Poorly Differentiated
Synovial sarcoma (SS) is a soft tissue sarcoma showing variable epithelial differentiation and is characterized by SS18::SSX1, SSX2, or SSX4 fusions [124]. Although SS can occur at any age, the majority of patients are adolescents or young adults and < 2% of patients are older than 50 years at diagnosis [125]. Histologically, SS are cellular monophasic or biphasic tumors composed of dense sheets or vague fascicles of uniform appearing small spindle cells with ovoid, hyperchromatic nuclei with regular granular chromatin and inconspicuous nucleoli, and scant cytoplasm (Fig. 10E, F). A variable proportion of epithelial cells are seen intermixed with spindle components in the biphasic SS [126], yielding a marbled appearance on low power. The high cellularity and monomorphic appearance frequently place SS in the list of small round cell tumors, especially in limited biopsy material. The poorly differentiated subtype of SS particularly needs distinction from SCNEC. Poorly differentiated SS exhibits areas of increased cellularity, greater nuclear pleomorphism, and a high mitotic rate (> 10 mitoses per 2 mm2) (Fig. 10F) [127]. The cells may be spindle to round. The tumors with predominantly round cell morphology especially necessitate segregation from SCNEC [128]. Poorly differentiated tumors also tend to be more common in elderly patients [129]. By IHC, SS shows strong diffuse nuclear positivity for TLE1 in nearly all the cases (Fig. 10G), with variable positivity for CD99 and bcl2, with focal positivity for cytokeratin. Rare reports of neuroendocrine markers (synaptophysin, chromogranin A, and nestin) in FISH-confirmed SS have been reported [130]. Recently, newer antibodies, SS18::SSX fusion-specific antibody (E9X9V, reactive against the breakpoint) and the SSX-specific antibody (E5A2C, reactive against the SSX C-terminus) have shown strong diffuse nuclear staining with excellent sensitivity and specificity (> 95%) for SS [131].
Hematolymphoid Malignancies
Lymphoma is a universal differential diagnosis for all small blue round cell tumors. High-grade diffuse large cell lymphomas, B-cell or T-cell lineage, show diffuse sheets of neoplastic cells with high nuclear-to-cytoplasmic ratio, loose chromatin, conspicuous nucleoli, and scanty cytoplasm (Fig. 10H). Brisk mitoses and apoptotic bodies are frequent. Crushing artifacts are common. Convoluted nuclei, nuclear folds, and grooves are commonly seen in T-cell lineage tumors. Tumor cells infiltrating through fibrotic stroma may simulate clustering similar to carcinomas. Due to overlapping features, distinction from NEC is usually required, especially in a limited biopsy. Immunohistochemistry can readily help in segregating lymphomas from NEC. Lymphomas are positive for hematolymphoid markers including CD45RB, while negative with pancytokeratins and neuroendocrine markers (Fig. 10I).
Conclusion
Pathology is often considered a specialty in which experience is measured in case volume or years of practice—and given the rarity of neuroendocrine neoplasms and their mimics, diagnosticians outside of tertiary diagnostic centers may have difficulties in acquiring adequate experience for some of these entities. Moreover, identification of these lesions in the head and neck region usually requires an integration of clinical information, imaging findings, histomorphology, and immunohistochemical assessments and not all hospital settings may provide the latest antibody panels or molecular platforms to assess some of the key features. Even so, when faced with a head and neck tumor in which NEC is a potential differential diagnosis, careful exclusion of the top ten mimickers as highlighted herein will facilitate narrowing the diagnoses to the correct classification.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Juhlin CC (2021) Challenges in paragangliomas and pheochromocytomas: from histology to molecular immunohistochemistry. Endocr Pathol 32:228–244
Nosé V, Gill A, Teijeiro JMC, Perren A, Erickson L (2022) Overview of the 2022 WHO Classification of familial endocrine tumor syndromes. Endocr Pathol 33:197–227
Rindi G, Mete O, Uccella S, Basturk O, La Rosa S, Brosens LAA et al (2022) Overview of the 2022 WHO Classification of neuroendocrine neoplasms. Endocr Pathol 33:115–154
Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N et al (2014) Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer 21:405–414
Asa SL, Mete O (2022) Medullary thyroid carcinoma in the IARC/WHO neuroendocrine schema. Endocr Pathol 33:346–347
Dasari A, Mehta K, Byers LA, Sorbye H, Yao JC (2018) Comparative study of lung and extrapulmonary poorly differentiated neuroendocrine carcinomas: a SEER database analysis of 162,983 cases. Cancer 124:807–815
Froehlich MH, Shih MC, Shehee L, Kompelli AR, Aylward A, Nguyen SA et al (2022) Systematic review of neuroendocrine carcinomas of the oropharynx. Head Neck 44:1725–1736
Mete O, Wenig BM (2022) Update from the 5th Edition of the World Health Organization Classification of head and neck tumors: overview of the 2022 WHO Classification of head and neck neuroendocrine neoplasms. Head Neck Pathol 16:123–142
Strojan P, Šifrer R, Ferlito A, Grašič-Kuhar C, Lanišnik B, Plavc G et al (2021) Neuroendocrine carcinoma of the larynx and pharynx: a clinical and histopathological study. Cancers (Basel) 13:4813
Mitchell MB, Kimura K, Chapurin N, Saab Chalhoub M, Mehrad M, Langerman A et al (2021) Neuroendocrine carcinomas of the head and neck: a small case series. Am J Otolaryngol 42:102992
Bal M, Sharma A, Rane SU, Mittal N, Chaukar D, Prabhash K et al (2022) Neuroendocrine neoplasms of the larynx: a clinicopathologic analysis of 27 neuroendocrine tumors and neuroendocrine carcinomas. Head Neck Pathol 16:375–387
Juhlin CC (2021) Second-generation neuroendocrine immunohistochemical markers: reflections from clinical implementation. Biology (Basel) 10:874
Rooper LM, Bishop JA, Westra WH (2018) INSM1 is a sensitive and specific marker of neuroendocrine differentiation in head and neck tumors. Am J Surg Pathol 42:665–671
Benzerdjeb N, Traverse-Glehen A, Philouze P, Bishop J, Devouassoux-Shisheboran M (2020) Poorly differentiated neuroendocrine carcinoma of the head and neck: human papillomavirus tumour status/p16 status and impact on overall survival. Histopathology 76:581–591
Ohmoto A, Sato Y, Asaka R, Fukuda N, Wang X, Urasaki T et al (2021) Clinicopathological and genomic features in patients with head and neck neuroendocrine carcinoma. Mod Pathol 34:1979–1989
Asa SL, Mete O, Perry A, Osamura RY (2022) Overview of the 2022 WHO Classification of pituitary tumors. Endocr Pathol 33:6–26
Mete O, Asa SL (2020) Structure, function, and morphology in the classification of pituitary neuroendocrine tumors: the importance of routine analysis of pituitary transcription factors. Endocr Pathol 31:330–336
Xu L, Khaddour K, Chen J, Rich KM, Perrin RJ, Campian JL (2020) Pituitary carcinoma: two case reports and review of literature. World J Clin Oncol 11:91–102
Baloch ZW, Asa SL, Barletta JA, Ghossein RA, Juhlin CC, Jung CK et al (2022) Overview of the 2022 WHO Classification of thyroid neoplasms. Endocr Pathol 33:27
Moura MM, Cavaco BM, Pinto AE, Leite V (2011) High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab 96:E863-868
Porto DP, Wick MR, Ewing SL, Adams GL (1987) Neuroendocrine carcinoma of the larynx. Am J Otolaryngol 8:97–104
Williams MD, Asa SL, Fuller GN (2008) Medullary thyroid carcinoma metastatic to the pituitary gland: an unusual site of metastasis. Ann Diagn Pathol 12:199–203
Canberk S, Thodou E, Bongiovanni M (2022) Small-cell malignancies of thyroid: challenge solved? Acta Cytol 66:307–318
Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A (2022) Overview of the 2022 WHO Classification of paragangliomas and pheochromocytomas. Endocr Pathol 33:90–114
Lin EP, Chin BB, Fishbein L, Moritani T, Montoya SP, Ellika S et al (2022) Head and neck paragangliomas: an update on the molecular classification, state-of-the-art imaging, and management recommendations. Radiol Imaging Cancer 4:e210088
Juhlin CC, Mete O (2022) Advances in adrenal and extra-adrenal paraganglioma: practical synopsis for pathologists. Adv Anat Pathol 30:47
Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY et al (2010) Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 41:805–814
Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS et al (2015) SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol 28:807–821
Williams MD (2017) Paragangliomas of the head and neck: an overview from diagnosis to genetics. Head Neck Pathol 11:278–287
Papaxoinis G, Lamarca A, Quinn AM, Mansoor W, Nonaka D (2018) Clinical and pathologic characteristics of pulmonary carcinoid tumors in central and peripheral locations. Endocr Pathol 29:259–268
Kimura N, Shiga K, Kaneko K-I, Oki Y, Sugisawa C, Saito J et al (2021) Immunohistochemical expression of choline acetyltransferase and catecholamine-synthesizing enzymes in head-and-neck and thoracoabdominal paragangliomas and pheochromocytomas. Endocr Pathol 32:442–451
Konosu-Fukaya S, Omata K, Tezuka Y, Ono Y, Aoyama Y, Satoh F et al (2018) Catecholamine-synthesizing enzymes in pheochromocytoma and extraadrenal paraganglioma. Endocr Pathol 29:302–309
Javidiparsijani S, Brickman A, Lin DM, Rohra P, Ghai R, Bitterman P et al (2021) Is regional lymph node metastasis of head and neck paraganglioma a sign of aggressive clinical behavior: a clinical/pathologic review. Ear Nose Throat J 100:447–453
Juhlin CC, Zedenius J, Höög A (2022) Metastatic neuroendocrine neoplasms of unknown primary: clues from pathology workup. Cancers 14:2210
Yu S, Hornick JL, Gonzalez RS (2021) An algorithmic approach utilizing CK7, TTF1, beta-catenin, CDX2, and SSTR2A can help differentiate between gastrointestinal and pulmonary neuroendocrine carcinomas. Virchows Arch 479:481
Salama AR, Jham BC, Papadimitriou JC, Scheper MA (2009) Metastatic neuroendocrine carcinomas to the head and neck: report of 4 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 108:242–247
Braun A, Cheng L, Reddy S, Gattuso P, Yan L (2022) Metastatic neuroendocrine tumors to parotid gland: where do they come from? Int J Surg Pathol. https://doi.org/10.1177/10668969221095267
Kaae J, Hansen AV, Biggar RJ, Boyd HA, Moore PS, Wohlfahrt J et al (2010) Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst 102:793–801
Dika E, Pellegrini C, Lambertini M, Patrizi A, Ventura A, Baraldi C et al (2021) Merkel cell carcinoma: an updated overview of clinico-pathological aspects, molecular genetics and therapy. Eur J Dermatol 31:691–701
Juhlin CC, Zedenius J, Höög A (2020) Clinical routine application of the second-generation neuroendocrine markers ISL1, INSM1, and secretagogin in neuroendocrine neoplasia: staining outcomes and potential clues for determining tumor origin. Endocr Pathol 31:401–410
Sheldon JD, Lott Limbach AA (2021) Merkel cell carcinoma of the maxillary sinus: an unusual presentation of a common tumor. Head Neck Pathol 15:691–697
Moreno V, Saluja K, Pina-Oviedo S (2022) NUT carcinoma: clinicopathologic features, molecular genetics and epigenetics. Front Oncol 12:860830
French CA, Kutok JL, Faquin WC, Toretsky JA, Antonescu CR, Griffin CA et al (2004) Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol 22:4135–4139
Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB et al (2009) Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am J Surg Pathol 33:984–991
Bishop JA, Westra WH (2012) NUT midline carcinomas of the sinonasal tract. Am J Surg Pathol 36:1216–1221
Agaimy A, Franchi A, Lund VJ, Skálová A, Bishop JA, Triantafyllou A et al (2020) Sinonasal undifferentiated carcinoma (SNUC): from an entity to morphologic pattern and back again-A historical perspective. Adv Anat Pathol 27:51–60
Reiersen DA, Pahilan ME, Devaiah AK (2012) Meta-analysis of treatment outcomes for sinonasal undifferentiated carcinoma. Otolaryngol Head Neck Surg 147:7–14
Singh L, Ranjan R, Arava S, Singh MK (2014) Role of p40 and cytokeratin 5/6 in the differential diagnosis of sinonasal undifferentiated carcinoma. Ann Diagn Pathol 18:261–265
Cerilli LA, Holst VA, Brandwein MS, Stoler MH, Mills SE (2001) Sinonasal undifferentiated carcinoma: immunohistochemical profile and lack of EBV association. Am J Surg Pathol 25:156–163
Jo VY, Chau NG, Hornick JL, Krane JF, Sholl LM (2017) Recurrent IDH2 R172X mutations in sinonasal undifferentiated carcinoma. Mod Pathol 30:650–659
Riobello C, López-Hernández A, Cabal VN, García-Marín R, Suárez-Fernández L, Sánchez-Fernández P et al (2020) IDH2 mutation analysis in undifferentiated and poorly differentiated sinonasal carcinomas for diagnosis and clinical management. Am J Surg Pathol 44:396–405
Winzenburg SM, Niehans GA, George E, Daly K, Adams GL (1998) Basaloid squamous carcinoma: a clinical comparison of two histologic types with poorly differentiated squamous cell carcinoma. Otolaryngol Head Neck Surg 119:471–475
Wieneke JA, Thompson LD, Wenig BM (1999) Basaloid squamous cell carcinoma of the sinonasal tract. Cancer 85:841–854
Rooper LM, McCuiston AM, Westra WH, Bishop JA (2019) SOX10 immunoexpression in basaloid squamous cell carcinomas: a diagnostic pitfall for ruling out salivary differentiation. Head Neck Pathol 13:543–547
Mino M, Pilch BZ, Faquin WC (2003) Expression of KIT (CD117) in neoplasms of the head and neck: an ancillary marker for adenoid cystic carcinoma. Mod Pathol 16:1224–1231
Andreadis D, Epivatianos A, Poulopoulos A, Nomikos A, Papazoglou G, Antoniades D et al (2006) Detection of C-KIT (CD117) molecule in benign and malignant salivary gland tumours. Oral Oncol 42:57–65
Miettinen M, Lasota J (2005) KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol 13:205–220
Brayer KJ, Frerich CA, Kang H, Ness SA (2016) Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov 6:176–187
Agaimy A, Bishop JA (2021) SWI/SNF-deficient head and neck neoplasms: an overview. Semin Diagn Pathol 38:175–182
Shah AA, Jain D, Ababneh E, Agaimy A, Hoschar AP, Griffith CC et al (2020) SMARCB1 (INI-1)-deficient adenocarcinoma of the sinonasal tract: a potentially under-recognized form of sinonasal adenocarcinoma with occasional yolk sac tumor-like features. Head Neck Pathol 14:465–472
Agaimy A, Hartmann A, Antonescu CR, Chiosea SI, El-Mofty SK, Geddert H et al (2017) SMARCB1 (INI-1)-deficient sinonasal carcinoma: a series of 39 cases expanding the morphologic and clinicopathologic spectrum of a recently described entity. Am J Surg Pathol 41:458–471
Kakkar A, Antony VM, Pramanik R, Sakthivel P, Singh CA, Jain D (2019) SMARCB1 (INI1)-deficient sinonasal carcinoma: a series of 13 cases with assessment of histologic patterns. Hum Pathol 83:59–67
Agaimy A, Jain D, Uddin N, Rooper LM, Bishop JA (2020) SMARCA4-deficient sinonasal carcinoma: a series of 10 cases expanding the genetic spectrum of SWI/SNF-driven sinonasal malignancies. Am J Surg Pathol 44:703–710
Hyams VJ, Batsakis JG, Michaels L (1988) Tumors of the upper respiratory tract and ear. In: Atlas of Tumor Pathology, Armed Forces Institute of Pathology
Llombart-Bosch A, Carda C, Peydro-Olaya A, Noguera R, Boix J, Pellin A (1989) Pigmented esthesioneuroblastoma showing dual differentiation following transplantation in nude mice. An immunohistochemical, electron microscopical, and cytogenetic analysis. Virchows Arch A Pathol Anat Histopathol 414:199–208
Faragalla H, Weinreb I (2009) Olfactory neuroblastoma: a review and update. Adv Anat Pathol 16:322–331
Bates T, Plessis DD, Polvikoski T, Sloan P, McQueen A, Meikle D et al (2012) Ganglioneuroblastic transformation in olfactory neuroblastoma. Head Neck Pathol 6:150–155
Mills SE (2002) Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 15:264–278
Uccella S, Facco C, Chiaravalli AM, Pettenon F, La Rosa S, Turri-Zanoni M et al (2022) Transcription factor expression in sinonasal neuroendocrine neoplasms and olfactory neuroblastoma (ONB): hyams’ grades 1–3 ONBs expand the spectrum of SATB2 and GATA3-positive neoplasms. Endocr Pathol 33:264–273
Rooper LM, Uddin N, Gagan J, Brosens LAA, Magliocca KR, Edgar MA et al (2020) Recurrent loss of SMARCA4 in sinonasal teratocarcinosarcoma. Am J Surg Pathol 44:1331–1339
Birkeland AC, Burgin SJ, Yanik M, Scott MV, Bradford CR, McHugh JB et al (2017) Pathogenetic analysis of sinonasal teratocarcinosarcomas reveal actionable β-catenin overexpression and a β-catenin mutation. J Neurol Surg B Skull Base 78:346–352
Zito PM, Scharf R (2022) Melanoma of the head and neck. StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL
Eyden B, Pandit D, Banerjee SS (2005) Malignant melanoma with neuroendocrine differentiation: clinical, histological, immunohistochemical and ultrastructural features of three cases. Histopathology 47:402–409
Juhlin CC, Zedenius J, Haglund F (2020) Metastatic malignant melanoma with neuroendocrine differentiation: a case report and review of the literature. J Med Case Rep 14:44
Wu Y, Lai Y, Zhang M, Li Z (2021) Prognostic significance of the aberrant expression of neuroendocrine markers in melanomas. Diagn Pathol 16:78
OMS (2020) Soft tissue and bone tumours: World health organization classification of tumours, 5th edn. OMS, Geneva
Sbaraglia M, Bellan E, Dei Tos AP (2021) The 2020 WHO Classification of soft tissue tumours: news and perspectives. Pathologica 113:70–84
Llombart-Bosch A, Machado I, Navarro S, Bertoni F, Bacchini P, Alberghini M et al (2009) Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch 455:397–411
Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M (1991) MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer 67:1886–1893
Machado I, Yoshida A, Morales MGN, Abrahão-Machado LF, Navarro S, Cruz J et al (2018) Review with novel markers facilitates precise categorization of 41 cases of diagnostically challenging, “undifferentiated small round cell tumors”. A clinicopathologic, immunophenotypic and molecular analysis. Ann Diagn Pathol 34:1–12
Hung YP, Fletcher CDM, Hornick JL (2016) Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol 29:370–380
Russell-Goldman E, Hornick JL, Qian X, Jo VY (2018) NKX2.2 immunohistochemistry in the distinction of Ewing sarcoma from cytomorphologic mimics: diagnostic utility and pitfalls. Cancer Cytopathol 126:942–949
Zaccarini DJ, Deng X, Tull J, Maciak C, Valente AL, Zhang S (2018) Expression of TLE-1 and CD99 in carcinoma: pitfalls in diagnosis of synovial sarcoma. Appl Immunohistochem Mol Morphol 26:368–373
Machado I, Navarro S, López-Guerrero JA, Verdini L, Picci P, Giner F et al (2021) Neuroendocrine differentiation in a large series of genetically-confirmed Ewing’s sarcoma family tumor: does it provide any diagnostic or prognostic information? Pathol Res Pract 219:153362
Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP et al (2005) Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol 29:1025–1033
Folpe AL, Hill CE, Parham DM, O’Shea PA, Weiss SW (2000) Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumors with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol 24:1657–1662
Wang W-L, Patel NR, Caragea M, Hogendoorn PCW, López-Terrada D, Hornick JL et al (2012) Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol 25:1378–1383
Bal M, Shah A, Rekhi B, Mittal N, Rane SU, Rabade K et al (2022) Adamantinoma-like Ewing sarcoma of the head and neck: a case-series of a rare and challenging diagnosis. Head Neck Pathol 16:679–694
Rooper LM, Bishop JA (2020) Soft tissue special issue: adamantinoma-like Ewing sarcoma of the head and neck: a practical review of a challenging emerging entity. Head Neck Pathol 14:59–69.
Wang GY, Thomas DG, Davis JL, Ng T, Patel RM, Harms PW et al (2019) EWSR1-NFATC2 translocation-associated sarcoma clinicopathologic findings in a rare aggressive primary bone or soft tissue tumor. Am J Surg Pathol 43:1112–1122
Bridge JA, Sumegi J, Druta M, Bui MM, Henderson-Jackson E, Linos K et al (2019) Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol 32:1593–1604
Bode-Lesniewska B, Fritz C, Exner GU, Wagner U, Fuchs B (2019) EWSR1-NFATC2 and FUS-NFATC2 gene fusion-associated mesenchymal tumors: clinicopathologic correlation and literature review. Sarcoma 2019:9386390
Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H et al (2006) Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15:2125–2137
Italiano A, Sung YS, Zhang L, Singer S, Maki RG, Coindre J-M et al (2012) High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosom Cancer 51:207–218
Kao Y-C, Owosho AA, Sung Y-S, Zhang L, Fujisawa Y, Lee J-C et al (2018) BCOR-CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol 42:604–615
Puls F, Niblett A, Marland G, Gaston CLL, Douis H, Mangham DC et al (2014) BCOR-CCNB3 (Ewing-like) sarcoma: a clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol 38:1307–1318
Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PCW (2009) The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 15:2259–2268
Mastrangelo T, Modena P, Tornielli S, Bullrich F, Testi MA, Mezzelani A et al (2000) A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene 19:3799–3804
Chougule A, Taylor MS, Nardi V, Chebib I, Cote GM, Choy E et al (2019) Spindle and round cell sarcoma with EWSR1-PATZ1 gene fusion: a sarcoma with polyphenotypic differentiation. Am J Surg Pathol 43:220–228
Antonescu C (2014) Round cell sarcomas beyond Ewing: emerging entities. Histopathology 64:26–37
Yau DTW, Chan JKC, Bao S, Zheng Z, Lau GTC, Chan ACL (2019) Bone sarcoma with EWSR1-NFATC2 fusion: sarcoma with varied morphology and amplification of fusion gene distinct from Ewing sarcoma. Int J Surg Pathol 27:561–567
Watson S, Perrin V, Guillemot D, Reynaud S, Coindre J-M, Karanian M et al (2018) Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 245:29–40
Toki S, Wakai S, Sekimizu M, Mori T, Ichikawa H, Kawai A et al (2018) PAX7 immunohistochemical evaluation of Ewing sarcoma and other small round cell tumours. Histopathology 73:645–652
Charville GW, Wang W-L, Ingram DR, Roy A, Thomas D, Patel RM et al (2017) EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod Pathol 30:1312–1320
Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM et al (2017) Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol 41:941–949
Hung YP, Fletcher CD, Hornick JL (2016) Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod Pathol 29:1324–1334
Le Guellec S, Velasco V, Pérot G, Watson S, Tirode F, Coindre J-M (2016) ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol 29:1523–1531
Specht K, Sung Y-S, Zhang L, Richter GHS, Fletcher CD, Antonescu CR (2014) Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosom Cancer 53:622–633
Yoshida A, Goto K, Kodaira M, Kobayashi E, Kawamoto H, Mori T et al (2016) CIC-rearranged sarcomas: a study of 20 cases and comparisons with Ewing sarcomas. Am J Surg Pathol 40:313–323
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072
Le Loarer F, Pissaloux D, Watson S, Godfraind C, Galmiche-Rolland L, Silva K et al (2019) Clinicopathologic features of CIC-NUTM1 sarcomas, a new molecular variant of the family of CIC-fused sarcomas. Am J Surg Pathol 43:268–276
Healy JN, Borg MF (2010) Paediatric nasopharyngeal rhabdomyosarcoma: a case series and literature review. J Med Imaging Radiat Oncol 54:388–394
Thompson LDR, Jo VY, Agaimy A, Llombart-Bosch A, Morales GN, Machado I et al (2018) Sinonasal tract alveolar rhabdomyosarcoma in adults: a clinicopathologic and immunophenotypic study of fifty-two cases with emphasis on epithelial immunoreactivity. Head Neck Pathol 12:181–192
Rudzinski ER, Anderson JR, Chi Y-Y, Gastier-Foster JM, Astbury C, Barr FG et al (2017) Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 64:e26645
van der Graaf WTA, Orbach D, Judson IR, Ferrari A (2017) Soft tissue sarcomas in adolescents and young adults: a comparison with their paediatric and adult counterparts. Lancet Oncol 18:e166–e175
Malempati S, Hawkins DS (2012) Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 59:5–10
Yasuda T, Perry KD, Nelson M, Bui MM, Nasir A, Goldschmidt R et al (2009) Alveolar rhabdomyosarcoma of the head and neck region in older adults: genetic characterization and a review of the literature. Hum Pathol 40:341–348
Dumont SN, Lazar AJ, Bridge JA, Benjamin RS, Trent JC (2012) PAX3/7-FOXO1 fusion status in older rhabdomyosarcoma patient population by fluorescent in situ hybridization. J Cancer Res Clin Oncol 138:213–220
Parham DM, Barr FG (2013) Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol 20:387–397
Szablewski V, Neuville A, Terrier P, Laé M, Schaub R, Garrel R et al (2015) Adult sinonasal soft tissue sarcoma: analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 125:615–623
Bahrami A, Gown AM, Baird GS, Hicks MJ, Folpe AL (2008) Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol 21:795–806
Coindre JM, de Mascarel A, Trojani M, de Mascarel I, Pages A (1988) Immunohistochemical study of rhabdomyosarcoma. Unexpected staining with S100 protein and cytokeratin. J Pathol 155:127–132
Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ, Gastier-Foster JM et al (2012) Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosom Cancer 51:662–674
Banito A, Li X, Laporte AN, Roe J-S, Sanchez-Vega F, Huang C-H et al (2018) The SS18-SSX oncoprotein hijacks KDM2B-PRC11 to drive synovial sarcoma. Cancer Cell 33:527–541
Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A (2009) Comparing children and adults with synovial sarcoma in the surveillance, epidemiology, and end results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115:3537–3547
Streich L, Johnson DN, Alexiev BA (2021) Synovial sarcoma with overwhelming glandular (adenocarcinoma-like) component: A case report and review of the literature. Pathol Res Pract 222:153418
Guillou L, Benhattar J, Bonichon F, Gallagher G, Terrier P, Stauffer E et al (2004) Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22:4040–4050
Bergh P, Meis-Kindblom JM, Gherlinzoni F, Berlin O, Bacchini P, Bertoni F et al (1999) Synovial sarcoma: identification of low and high risk groups. Cancer 85:2596–2607
Chan JA, McMenamin ME, Fletcher CDM (2003) Synovial sarcoma in older patients: clinicopathological analysis of 32 cases with emphasis on unusual histological features. Histopathology 43:72–83
Chen Y, Zhou N, Guo D, Wang X, He X, Xu Y (2022) Predominantly epithelial-type synovial sarcoma with overwhelming neuroendocrine differentiation: a potential diagnostic pitfall. Diagn Pathol 17:59
Baranov E, McBride MJ, Bellizzi AM, Ligon AH, Fletcher CDM, Kadoch C et al (2020) A novel SS18-SSX fusion-specific antibody for the diagnosis of synovial sarcoma. Am J Surg Pathol 44:922–933
Funding
Research reported in this publication was supported in part by the Swedish Cancer Society (Dr CC Juhlin; Junior Clinical Investigator Award).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Both authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Ethical Approval
No human participants were included in this invited review, but all materials were treated in accordance with the ethical standards of the institutional and/or the national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to Participate
Consent to participate was waived due to the retrospective nature of this review work.
Consent for Publication
Consent for publication was obtained when personally identifiable information was included.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Juhlin, C.C., Bal, M. Top 10 Histological Mimics of Neuroendocrine Carcinoma You Should Not Miss in the Head and Neck. Head and Neck Pathol 17, 66–84 (2023). https://doi.org/10.1007/s12105-022-01521-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-022-01521-x