
Abstract
Alzheimer’s disease (AD) is a major cause of age-related dementia, which is becoming a global health crisis. However, the pathogenesis and etiology of AD are still not fully understood. And there are no valid treatment methods or precise diagnostic tools for AD. There is increasing evidence that P2X7R expression is upregulated in AD and is involved in multiple related pathological processes such as Aβ plaques, neurogenic fiber tangles, oxidative stress, and chronic neuroinflammation. This suggests that P2X7R may be a key player in the development of AD. P2X7R is a member of the ligand-gated purinergic receptor (P2X) family. It has received attention in neuroscience due to its role in a wide range of aging and age-related neurological disorders. In this review, we summarize current information on the roles of P2X7R in AD and suggest potential pharmacological interventions to slow down AD progression.
Similar content being viewed by others


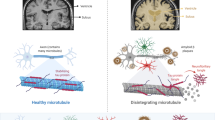
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disease characterized by overall cognitive decline, including progressive loss of memory, orientation, and reasoning skills. In line with increasing life expectancy, the 2020 census found that 18.67% of the total population of China (approximately 264 million) is over the age of 60 and that number will grow, projected to reach 500 million by 2050. The risk of developing AD increases exponentially with age, and society will bear an increasing burden due to AD unless effective prevention and treatment strategies are developed [1].
In the past decades, the Aβ hypothesis and the tau hypothesis were promulgated as the main explanations for the pathogenesis of Alzheimer’s disease [2]. However, the pathological features of AD are not only characterized by Aβ plaques, neurogenic fiber tangles (NFT). Studies now find that AD is always accompanied by synaptic loss and inflammation [3, 4]. Not only that, studies have shown that mitochondrial dysfunction and oxidative stress are involved in the pathogenesis of AD [5,6,7]. Even in the absence of Aβ plaques and tau tangles, mitochondrial dysfunction is one of the earliest prominent features of AD. Therefore, it is now generally accepted that AD is a multifactorial neurodegenerative disease involving different pathological processes. However, the pathogenesis and etiology of AD are still not fully understood. And there are no valid treatment methods or precise diagnostic tools for AD.
Therefore, there is an urgent need to find new treatment pathways to reduce the progression of the disease. The purinergic receptors have been of interest for a long time in neurodegenerative diseases [8, 9]. The purinergic receptor family can be divided into two main types, P2X and P2Y, respectively. P2X7R is a family member of P2X. It is widely expressed in the nervous system and is involved in a variety of neurological functions [10]. Not only that, P2X7R is involved in the progression of multiple diseases including Parkinson’s disease, multiple sclerosis, and Huntington’s disease [11,12,13]. These had intrigued researchers about the role of P2X7R in AD. Researchers find P2X7R involved in multiple processes in the progression of AD. Firstly, P2X7R can regulate Aβ formation and the researchers found that the use of P2X7R antagonists reduced plaques in J20 mice [14]. P2X7R is inextricably linked to tau protein phosphorylation, oxidative stress, or chronic neuroinflammatory pathological processes in AD [15,16,17]. These indicate that this P2X7R may become a drug target for a new AD therapeutic approach.
Here, we summarize the molecular characteristics, structure, and features of P2X7R. Subsequently, we discuss the significance of P2X7R in the pathogenesis of AD. Finally, a brief review of the research progress on P2X7R antagonists is presented.
The Structure and Molecular Physiology of P2X7R
Adenosine 5-triphosphate (ATP) is the main energy carrying molecule in cells. However, studies now show that ATP can also play an important role in the nervous system as a neurotransmitter. Purinergic receptors were first described in 1976, and as research progressed, researchers have so far identified multiple purinergic receptors, including seven P2X receptor subtypes and eight P2Y receptor subtypes [18]. Ionotropic P2X7R belongs to the P2X purinoceptor family [19]. Similar to other P2XR types, P2X7R is an ATP-gated, non-selective homotrimeric cation channel [20]. However, P2X7R also has several features that are significantly different from other members of the P2X receptor family and deserve special attention. P2X1-7 receptors are homo-trimeric forms formed from three identical P2X subunits. In addition, some P2X receptors exist as heterotrimeric [21]. P2X7R acts via the homotrimeric form. The common structural motifs of P2X7R are two transmembrane structural domains (TM1, TM2) [22]. It contains a large, glycosylated, cysteine-rich extracellular loop, and a short intracellular N-terminal domain. Moreover, an intracellular C-terminal structural domain is longer than in other P2X receptor subunits [23]. The molecular structure of a single P2X7R subunit resembles a jumping dolphin. When co-assembled as a homotrimer, P2X7R has a cup-like structure [24]. Notably, P2X7R is only activated by high concentrations of ATP in the millimolar range, which is significantly higher than that required to activate other purinoceptor channels [19]. In addition, it is a non-selective cation channel. It shows different reactions to the agonist based on its concentration and time of application. A brief activation, resulting in the opening of cation channels, allows K+ efflux as well as Ca2+ and Na+ influx into the cell, leading to an inward current/depolarization at the resting membrane potential (Fig. 1). By contrast, prolonged activation of P2X7R by agonists leads to the formation of a large aqueous pore, which is permeable to molecules with molecular masses up to 900 Da [25,26,27,28]. This ultimately leads to membrane blebbing, cytokine release, and cell death [29, 30]. However, the underlying molecular mechanism is still a hotly debated issue. There are two main possible mechanism hypotheses. The first hypothesis suggests an asymptotic expansion of P2X7R-gated channels, in which a second transmembrane structural domain in P2X7R is thought to be critical for pore formation [31]. Another piece of evidence in favor of this hypothesis is that negatively charged fluorescent dyes with molecular diameters up to 1.4 nm pass directly through the P2X7R channel [32]. Another hypothesis suggests an association with a pore-forming protein, pannexin-1 hemichannel. It was shown that in the absence of pannexin-1 hemichannel, the ability of astrocytes to take up macromolecular dyes was significantly reduced [33]. But some studies have found the opposite [34]. One possible explanation is that the P2X7R splice variant shows different pore-forming properties [35, 36].

The P2X7R functions as a homo-trimer, forming a chalice-like structure, while the individual P2X7R subunit is akin to a leaping dolphin. P2X7R is a non-selective cation channel that is activated by high concentrations of ATP. Transient activation leads to the opening of the cation channel, allowing K+ efflux and Na+ influx into the cell, resulting in an inward current/depolarization at the resting membrane potential
The Function of P2X7R in the CNS
P2X7R in Microglia
P2X7R was early found to be expressed in immune cells. Notably, P2X7R mediates multiple physiological aspects of microglia. A recent study unexpectedly found that P2X7R activation promotes the migratory capacity and phagocytosis of microglia [37]. Another study showed that the key enzyme for microglia migration is glycogen synthase-3 [38]. Indeed, P2X7R antagonists were found to reduce the number of amyloid plaques in the rat hippocampus, which is also thought to be associated with reduced GSK3 activity in microglia [14]. The large number of microglia with high P2X7R expression clustered around senile plaques in postmortem brain samples of AD patients and in AD mouse models may also be closely related to this phenomenon [39, 40]. In addition, activation of P2X7R drives microglial activation and is a key factor in microglial proliferation [41]. Experiments in rat primary hippocampal neurons show that P2X7R drives microglial activation and can promote microglial proliferation [42]. More importantly, P2X7R is an important regulator of microglial secretion of pro-inflammatory cytokines and chemokines. For example, P2X7R is not only involved in the maturation of IL-1β in microglia but also plays an important role in its release [16, 43].
P2X7R in Neurons
The localization of P2X7R on neurons has been the subject of a long-standing debate, and even now there is no universal consensus. At first, researchers found that P2X7R immunoreactivity was significantly expressed on excitatory nerve terminals [44]. Yu et al. used isotope in situ hybridization to examine the distribution of P2X7R mRNAs in the brain and found that P2X7 mRNA signals were also detected on NeuN-positive neurons cells [45]. During brain growth, both neural precursor cells and neuroblastoma cells can express P2X7R. Many studies have reported the presence of functional P2X7R, including embryonic stem cells, neuron-like human embryonic stem cells-derived neural progenitor cells, human neural progenitor cells, as well as NPCs isolated from the subventricular zone of adult mice or the striatum of embryonic mice [46,47,48,49]. However, immunohistochemical studies showed that P2X7R was expressed at the cell membrane of microglia and NPC but not on neurons [50]. In contrast to the contradictory results of immunohistochemistry, the function of the P2X7 receptor in the nervous system was demonstrated. P2X7R is involved in the growth of neuronal axons [51], and further experiments demonstrated that alkaline phosphatase regulates axonal growth via P2X7R. A study that exposed cultured hippocampal neurons to ATP reported slow axon growth. By contrast, hippocampal neurons cultured with P2X7R antagonists or P2X7R deficiency had faster axon growth and formed more branches [52]. In addition, presynaptic P2X7R regulates the release of neurotransmitters [51]. P2X7R is also involved in the process of neuronal differentiation [48, 53]. However, P2X7R is harmful in pathological states. For example, P2X7R is involved in ATP-induced neuronal death [54]. ATP was found to induce neuronal death in pure culture, and neuronal activity was restored after the use of the P2X7R-specific antagonists A438079 and KN-62, suggesting that P2X7R is involved in ATP-induced neuronal death. In a later study, ATP-induced neuronal death was found to be correlated with high expression levels of P2X7R [55].
P2X7R in Astrocytes and Oligodendrocytes
In addition to microglia, P2X7R is also expressed by astrocytes [56, 57] and oligodendrocytes [58]. P2X7R mediates multiple physiological functions of astrocytes. For example, the release of glutamate from astrocytes can be involved in signaling between brain cells, and glutamate can also regulate synaptic activity, while P2X7R receptors are involved in the regulation of glutamate release from astrocytes [59]. In addition, P2X7R is also involved in the regulation of ATP release from astrocytes, which participate in intercellular communication through ATP-mediated Ca2+ waves [60, 61]. Another important role of P2X7R activation in astrocytes is the upregulation of MCP1/CCL2 expression via the p-38MAPK and ERK1/2 pathways [62, 63]. Functional P2X7 receptors that can mediate cell death in vitro and in vivo are expressed by Schwann cells and oligodendrocytes [58]. More intuitively, P2X7R labeled by functional EGFP was observed in oligodendrocytes of P2X7 BAC transgenic mice [64]. Another study also showed that P2X7R may be involved in oligodendrocyte migration under pathological conditions [65].
Mechanism of P2X7R Upregulation in Alzheimer’s Disease
Initially, researchers discovered that P2X7R is upregulated in a transgenic mouse model of Alzheimer’s disease [17]. Subsequently, semi-quantitative reverse transcriptase-polymerase chain reaction also revealed enhanced expression of P2X7R in the microglia of AD patients [40]. However, due to the complexity of P2X7 function, the exact mechanism of P2X7 upregulation remains unclear. In general, the regulatory mechanisms of P2X7 expression can be divided into transcriptional and post-translational regulation [66]. The P2X7 promoter contains putative binding sites for several transcription factors, among which specificity protein 1 (Sp1) has been particularly noted as a potential P2X7 transcriptional regulator. Sp1 is a damage-activated transcription factor highly expressed in the brain [67]. Sp1 is involved in the transcriptional regulation of receptors in the central nervous system, and it has been demonstrated that Sp1 is a key component in the transcriptional regulation of P2X7 [68]. It was found that in a mouse model of epilepsy, the transcription factor Sp1 induced P2X7R expression [69]. A recent study reported that progerin 2 deficiency promotes Aβ-induced injury and neuroinflammation by upregulating P2X7R expression via the Sp1 pathway [70]. Inhibition of SP1 may therefore contribute to the upregulation of P2X7R in AD.
The Relationship Between P2X7R and Aβ
One pathological feature of AD is the extracellular aggregation of Aβ plaques [71, 72]. Aβ peptide is produced by the hydrolytic cleavage of amyloid precursor protein (APP), which is sequentially cleaved by aspartic proteases through amyloidogenic and non-amyloidogenic pathways [73]. In the amyloidogenic pathway, APP is successively hydrolyzed by β-secretase and γ-secretase to produce Aβ, while in the anti-amyloidogenic pathway, APP is cleaved by α-secretase and γ-secretase, ultimately producing p3 and sAPP-alpha, the latter of which has a well-established neuroprotective effect [74, 75].
Notably, both APP processing modalities can be found in the same cells of the central nervous system [76]. The balance between normal and pathological APP processing is still an active area of research on Aβ accumulation as a characteristic hallmark of AD. The process of Aβ production can be regulated by various signaling pathways. Glycogen synthase kinase-3β (GSK3-β) is considered an important enzyme in AD pathophysiology [77]. In fact, an increase in GSK-3 activity can directly lead to increased accumulation of Aβ [78]. It was shown that inhibition of P2X7R activity in J20 mice reduced Aβ, which was associated with increased α-secretase activity by decreased GSK-3β activity [14]. GSK-3β also interferes with APP cleavage by affecting presenilin1(PS1) activity [79]. Furthermore GSK-3β can mediate β-site APP cleaving enzyme 1(BACE1) expression through nuclear factor kappa-B(NF-kB) [80]. When this balance is upset, the accumulation of Aβ will increase, but P2X7R receptor antagonists can reverse this effect (Fig. 2). It has been shown that the use of P2X7R antagonists reduces GSK-3β activity, which in turn reduces Aβ [14].

The relationship between P2X7R and Aβ. P2X7R activation promotes Aβ formation by adjusting Glycogen synthase kinase-3β (GSK3-β) activity. (1) GSK3-β induces β-site APP cleaving enzyme 1(BACE1) gene expression through upregulation of nuclear factor kappa-B(NF-kB) signaling; (2) regulates γ-secretase activity by modulating presenilin1((PS1) activity; (3) directly regulates α-secretase activity
In addition, P2X7R activation can also regulate the function of microglia, affecting Aβ. First, P2X7R regulates microglial cell migration, causing microglia to accumulate near senile plaques [37]. In addition, P2X7R also regulates the ability of microglia to phagocytize Aβ [37]. In another study, researchers using BBG, an antagonist of P2X7R, in a mouse model of AD could observe smaller plaques [14].
The Connection Between P2X7R and Tau
Another characteristic pathological feature of AD is the intracellular aggregation of neurofibrillary tangles (NFT), mainly composed of highly phosphorylated tau protein [81]. Tau is a microtubule-associated protein (MAPT) that polymerizes tubulin into microtubules and participates in sustaining the complex neuronal cell microarchitecture, with roles such as stabilization and microtubule assembly, especially in axons [82].Under pathological conditions, tau proteins dissociate from microtubule-binding proteins and form NFTS [83]. This process is regulated by a variety of enzymes including, but not limited to, C-kinase, A-kinase, cell cycle protein-dependent kinase-5, glycogen synthase kinase-3β [84, 85]. As a phosphoprotein, tau is directly regulated by its phosphorylation state, and NFTs are induced by abnormal Tau phosphorylation, which is modulated by glycogen synthase kinase-3 β (GSK-3β) and cyclin-dependent kinase 5 (CDK5) [86]. GSK-3β is arguably the most widely investigated kinase associated with abnormally high tau phosphorylation [87]. GSK-3β phosphorylates tau mainly by using the PI3K/ AKT/GSK-3β pathway [87]. Studies have shown that P2X7R promotes neuronal tau phosphorylation via GSK3 kinase [52]. What’s more, the tau-related pathology of AD is also affected by inflammation, and tau propagation is suppressed by the depletion of microglia [88]. P2X7R plays an important role in this process. Elevated P2X7R expression was found in the brains of transgenic mice and patients with tauopathies. Moreover, hippocampus-dependent spatial memory and long-term synaptic plasticity were improved by P2X7R deletion in a mouse model of tauopathy [15, 89]. In a tauopathy mouse model, researchers found that oral administration of GSK1482160, a P2X7R-specific antagonist, significantly improved cognitive performance in rats [90]. Similarly, blockade of IL-1β signaling leads to reduced tau pathology [91]. Conversely, tau pathology was exacerbated by persistent interleukin-1β overexpression [92]. Microglial activation has been shown to precede tau pathology in the P301S mouse model [93]. Studies have demonstrated that microglial activation significantly accelerated tau pathology and behavioral abnormalities in model mice [94]. Reactive microglia drive tau pathology and it help spread abnormal tau in the brain [95]. Pharmacological blockade of P2X7R decreased the accumulation of misfolded tau aggregates and restored cognitive function in P301S mice, likely by suppressing exosome secretion [90]. Studies have shown that the use of P2X7R antagonists reduces cell death by altering the balance of tau phosphorylation inside and outside the cells [96]. The same study also found that P2X7R inhibition affected extracellular tau phosphorylation by reducing tissue-nonspecific alkaline phosphatase (TNAP) expression.
P2X7R and Neuroinflammation in AD
Neuroinflammation is considered to be the third core pathology of AD, and inflammation is involved in the onset as well as the progression of the disease. Moreover, the neuroinflammation hypothesis also links the two other hypotheses of AD pathogenesis [97]. As resident immune cells of the CNS, microglia are key regulators of neuroinflammation [98]. As such, microglia are involved in the maintenance of the inflammatory and immune response in the entire brain [99]. In response to different environmental factors and stimuli, microglia can usually be activated into two polarized states, called the M1 phenotype and M2 phenotype [100, 101]. M1 microglia are thought to enhance the inflammatory response, while M2 microglia exert neuroprotective effects and promote tissue repair by inhibiting neuroinflammation. M1 microglia can secrete a variety of pro-inflammatory cytokines and chemokines, such as IL-1β, IL-6, IL-18, and TNF-α [102, 103]. IL-1β plays an important role in inflammation. The activation of the NLRP3 inflammasome in microglia is the basis for the maturation and release of IL-1, IL-6,IL-18, and TNF-α [104], and P2X7R is involved in regulating NLRP3. Therefore, P2X7R is essential for the release of pro-IL-1β from microglia [16, 43]. The production of IL-1β is a two-step process. First, a large amount of Pro-IL-1βis synthesized in the cytoplasm and is then proteolytically processed into IL-1B before it is finally released. Even LPS-mediated synthesis of large amounts of pro-IL-1β requires the activation of the P2X7 receptor to release mature IL-1β [105]. This also illustrates the importance of P2X7R in the maturation process of pro-inflammatory cytokines. The release of IL-1 β by microglia is mainly dependent on NLRP3 activation. P2X7R activation leads to K+ efflux, after which reduced intracellular K+ initiates NLRP3 inflammasome assembly and activation [106, 107]. The NLRP3 inflammasome consists of the adapter protein ASC (apoptosis-associated speck-like protein containing a CARD) and the sensor protein NLRP3. ASC can recruit and activate pro-caspase-1, which can be cleaved by the induced complex to produce active caspase-1, which in turn cleaves pro-IL-1β and IL-18, eventually leading to the release of IL-1 [108]. A defective NLRP3 inflammasome was found to result in reduced Aβ deposition in the APP/PS1 model of Alzheimer’s disease [109]. All these studies suggest that the NLRP3/caspase-1 axis may be a target for the treatment of Alzheimer’s disease.
P2X7R and Mitochondrial Dysfunction in Alzheimer’s Disease
Alterations in energy metabolism occur in their brains during the early stages of AD, and the researchers used fluorodeoxyglucose positron emission tomography to monitor hypometabolism of glucose in the brain [110]. This implies that mitochondrial dysfunction is extremely important in the development of the disease. In amyotrophic lateral sclerosis, P2X7R was shown to be involved in autophagy in microglia [111]. Not only that, the P2X7R signaling pathway mediates impairment of lysosomal function [112]. The adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway is closely related to mitochondrial division [113]. Researchers find that P2X7R activation can induce mitochondrial fission and affect mitochondrial autophagy through an AMPK-dependent pathway [114]. Mitochondrial autophagy maintains a balance between mitochondrial production and mitochondrial death. However, in AD, mitochondrial autophagy is impaired, which in turn affects energy metabolism [115, 116]. Impaired mitochondrial autophagy impairs mitochondrial autophagy by increasing oxidative damage and cellular energy deficits cognitive deficits, which in turn impair mitochondrial autophagy [117].
Oxidative stress is a condition caused by the increased production of reactive oxygen species (ROS) that is greater than the capacity of cellular antioxidant mechanisms. ROS are normally produced during physiological processes and have both beneficial and harmful effects in biological systems [118]. In AD, the apparent oxidative imbalance and increase of ROS have been widely noted [119]. A study comparing the levels of isoprostane 8,12-iso-iPF(2alpha)-VI in cerebrospinal fluid, plasma, and urine of cognitively normal elderly and probands with mild cognitive impairment (MCI) found significantly elevated levels of these substances in MCI. The isoprostane 8,12-iso-iPF(2alpha)-VI is a specific marker of in vivo lipid peroxidation. This also suggests a significant oxidative imbalance in the early stages of AD when there is no significant accumulation of senile plaques and NFTs [120, 121]. This suggests that the onset of oxidative stress precedes the development of AD-related pathology and may contribute to its development. In addition, high levels of ROS are often detected in the brains of patients with different types of neurodegenerative diseases [122]. In addition, a variety of blood markers of oxidative stress, such as protein carbonyl and 3-nitrotyrosine, are frequently detected in AD patients or corresponding animal models [123, 124]. Although ROS are mainly produced by mitochondria, which have strong innate oxidation resistance, excessive ROS accumulation can also cause mitochondrial dysfunction, which is also one of the prominent pathological features of AD [125].
Notably, a recent study demonstrated the mitochondrial localization of the P2X7R ionotropic purinergic receptor [126]. In addition, the basal respiration rate, ATP-coupled respiration, maximal uncoupled respiration, resting mitochondrial potential, and mitochondrial matrix Ca2+ levels were affected when there was no P2X7R in the mitochondria. This may indicate that P2X7R is also an improtant regulator of mitochondrial energy metabolism. The researchers also found reduced proteasome activity in the brains of AD patients as well [127]. The latest study found that sustained activation of P2X7R can induce ubiquitin–proteasome system dysfunction and ultimately lead to neuronal death [128]. This implies that P2X7R is involved in multiple metabolic mechanisms in the brains of AD patients.
In addition, it was shown that P2X7R expression in the APPswe/PS1dE9 mouse model of Alzheimer’s disease may mediate neuronal injury through the generation of ROS [39]. It was also confirmed in another study that Aβ can induce mitochondrial toxicity, but this process requires the involvement of P2X7R in microglia [129]. Furthermore, activation of P2X7R in the microglial cell membrane by ATP may induce the generation of hydrogen peroxide [130]. Recent research has demonstrated that P2X7R activation following stimulation with BzATP or ATP can induce ROS generation in microglia and macrophages. And the researchers found that the use of P2X7R inhibitors reduced the production of ROS [17, 131, 132]. Mitochondrial dysfunction along with oxidative stress appears to be an important early event in disease development.
P2X7R and Synaptic Dysfunction in AD
AD is also characterized by synaptic loss and dysfunction. Synaptic loss and dysfunction in the brain can be detected by researchers early in the progression of AD disease and is strongly associated with cognitive decline in AD patients [133]. By directly observing neurons in AD transgenic mice, the researchers found a significant reduction in spine density [134]. Reduced synaptic density and fewer synaptic connections per neuron can also be observed in the AD brain [135]. In addition, synaptic dysfunction is one of the earliest hallmarks of AD. In the early stages of AD, the decrease in synaptic proteins precedes all other neurodegenerative markers in the cerebrospinal fluid [136].
P2X7R is likely to be involved in synaptic changes in AD progression. In a mouse model of AD, researchers show that synaptic dysfunction may be linked to ROS production by P2X7R [39]. Meanwhile, the inhibition of P2X7R favors the reduction of aβ and Tau. aβ and Tau play a role in synaptic damage [137]. P2X7R is also involved in regulating synaptic neurotransmission. P2X7R can excite synapses by mediating the release of glutamate from astrocytes [59]. Further studies, the researchers found that the use of BBG, an antagonist of P2X7R, improved the development of dendritic spines in hippocampal neurons of AD model mice [138].
P2X7R as a Potential Target in AD
AD is now increasingly recognized as a multifactorial neurodegenerative disease with different pathological processes as possible contributors, including amyloid deposition, tau protein phosphorylation, oxidative stress, or chronic neuroinflammation. Notably, P2X7R is involved in all these processes (Fig. 3) and consistently appears to play an important role in the development of Alzheimer’s disease. The fact that P2X7R is antagonistic only when activated at high ATP concentrations is a significant advantage for a drug target [19]. In this way, pharmacological antagonism of P2X7R will not affect the function of P2X7R in normal physiological states. It was reported that in vivo P2X7R inhibition reduced amyloid plaques, providing the first evidence of the potential of P2X7R antagonists as therapeutic agents for Alzheimer’s disease [14]. Similarly, P2X7R antagonist treatment was also found to prevent the development of amyloid plaques in a mouse model of AD. Moreover, cognitive decline was rescued by P2X7R knockout in the APP/PS1 mouse model of AD [139].

This figure suggests that P2X7R is involved in different physiopathological processes in Alzheimer’s disease. As the figure demonstrates, P2X7R regulates the processing of amyloid APP, promotes Tau phosphorylation, and is also involved in synaptic changes, ROS, microglia activation, and promotes inflammatory factor release which are all processes that contribute to AD progression
The potential of P2X7R as a therapeutic target is supported by reports of improved symptoms and neuropathology in animal models of AD through pharmacological blockade or gene deletion [138,139,140]. For example, one study showed that the P2X7R antagonist BBG rescued spatial memory, learning, and cognitive deficits in a mouse model of AD [138]. BBG is a derivative of Brilliant Blue FCF, which has been proven safe in healthy animals and is approved for use as a food additive in the USA under various brand names, such as FD&C Blue No. 1 or Acid Blue 9 [141]. Notably, BBG also has the advantage of high blood–brain barrier permeability [142]. In the brain, BBG not only reduces the level of purinoceptor expression but also attenuates gliosis [143]. Moreover, BZ-ATP treatment increased IL-1β secretion in human microglia that had been preactivated with Aβ(1–42), while pretreatment with P2X7 receptor antagonists had the opposite effects [144]. In addition to treatment with P2X7R antagonists, inhibition of the P2X7R pathway may be a new therapeutic approach for the treatment of AD. Thus, P2X7R has great therapeutic potential.
Advances in Drug Research
The potential of P2X7R as a drug target has long been noted in other diseases, such as neuropsychiatric disorders, and cancer [145, 146]. Surprisingly, P2X7R antagonists also have neuroprotective effects in central nervous system disorders. It has shown great potential in both multiple sclerosis and ALS [147, 148]. BzATP is a P2X7R agonist. It elicits pore formation, IL-1β release, and calcium influx in rats, human receptors, and mice. Researchers have investigated a variety of P2X7R antagonists. The species-dependent differences in receptor sensitivity have been well summarized for the various P2X7R antagonists [149]. Although most P2X7R antagonists do not cross the blood–brain barrier and are unable to act in vivo, recent studies have made significant progress in identifying brain-permeable P2X7R antagonists. GlaxoSmithKline has developed the P2X7 receptor antagonist GSK1482160 with good CNS penetration [150]. The amide GSK1370319A also showed good brain penetration [151]. It inhibits inflammasome-induced cell death and neurodegeneration [152]. JNJ-54175446 and JNJ-55308942 are two new brain-penetrant P2X7R antagonists [153, 154]. More brain penetrant P2X7R antagonists are listed in Table 1. For more details see Table 1.
P2X7R as a New Diagnostic Tool for AD
PET imaging is a recognized technique commonly used to diagnose brain diseases including Alzheimer’s disease [158]. PET studies of Alzheimer’s disease have the advantage of identifying different subtypes of the disease through neuropathology and sometimes genetic causation, which may have implications for guiding treatment. PET imaging of Alzheimer’s disease has so far mainly used the radiotracer [18F]FDG to image glucose metabolism [159]. However several radiotracers have been developed for the detection of other molecules, interestingly including several P2X7R radiotracers [146]0.11C-JNJ-54173717 was developed as a high-affinity P2X7R antagonist. In animal rat models, it has demonstrated its advantage as a PET radioligand for visualizing the expression and distribution of P2X7R in vivo. And it can also be used in monkey brain to selectively display P2X7RX expression and distribution [160]. 18F-JNJ-64413739 is also considered a suitable PET ligand for quantifying P2X7R expression in the human brain, which can be used for P2X7R expression in health and Alzheimer’s disease to provide ideas for therapy [161]. However, whether P2X7R-PET has the potential to stratify Alzheimer’s disease such as disease severity needs to be analysed in a larger cohort of patients, but P2X7R-based PET imaging may be a promising tool.
P2X7R is not only expressed in brain tissue but also in the peripheral immune system, where it can be found in macrophages and T cells [162, 163]. A recent study has shown that the expression of P2X7R is elevated in the blood of patients with Alzheimer’s disease, so it can be assumed that the plasma level of P2X7R is a biomarker that can differentiate between patients with Alzheimer’s disease and non-Alzheimer’s disease [164]. Although we should not use a single biomarker as a diagnostic tool, elevated plasma levels of P2X7R in patients with Alzheimer’s disease suggest that combined plasma levels of P2X7R are promising as a diagnostic tool.
Conclusions and Prospects
We summarized the roles of P2X7R in the central nervous system and its significance in the pathogenesis of AD, after which we discussed the various effects of P2X7R activation. As described in this review, the success of P2X7R antagonists in preclinical models indicates that P2X7R should be a focus of future research on targeted therapies for AD. A detailed understanding of the roles of P2X7R is essential for the discovery of new therapeutic approaches for the treatment of neurological disorders. A great potential advantage may lie in the absence of P2X7R activation or low P2X7R expression in healthy tissues, which may limit the side effects of drug treatment.
Although many P2X7R antagonists have now been developed, many challenges remain. Highly selective and effective P2X7R agonists and antagonists that can penetrate the CNS need to be further explored. Importantly, the clinical use of these drugs will first require extensive further study of their safety. In addition to treatment with P2X7R antagonists, inhibition of the P2X7R pathway may also be a new therapeutic approach for the treatment of AD. With further exploration of AD pathogenesis and further drug development, P2X7R-targeted therapies are likely to become a promising new treatment modality in the future.
Data Availability
Not applicable for that section.
References
(2021) 2021 Alzheimer's disease facts and figures. Alzheimers Dement 17:327–406. https://doi.org/10.1002/alz.12328
Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y (2020) Molecular and cellular mechanisms underlying the pathogenesis of alzheimer’s disease. Mol Neurodegener 15:40. https://doi.org/10.1186/s13024-020-00391-7
Dansokho C, Heneka MT (2018) Neuroinflammatory responses in alzheimer’s disease. J Neural Transm (Vienna) 125:771–779. https://doi.org/10.1007/s00702-017-1831-7
Tönnies E, Trushina E (2017) Oxidative stress, synaptic dysfunction, and alzheimer’s disease. J Alzheimers Dis 57:1105–1121. https://doi.org/10.3233/jad-161088
Moreira PI, Cardoso SM, Santos MS, Oliveira CR (2006) The key role of mitochondria in alzheimer’s disease. J Alzheimers Dis 9:101–110. https://doi.org/10.3233/jad-2006-9202
Moreira PI, Santos MS, Oliveira CR (2007) Alzheimer’s disease: a lesson from mitochondrial dysfunction. Antioxid Redox Signal 9:1621–1630. https://doi.org/10.1089/ars.2007.1703
Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, Smith MA, Zhu X (2008) Oxidative stress signaling in alzheimer’s disease. Curr Alzheimer Res 5:525–532. https://doi.org/10.2174/156720508786898451
Apolloni S, Montilli C, Finocchi P, Amadio S (2009) Membrane compartments and purinergic signalling: P2X receptors in neurodegenerative and neuroinflammatory events. Febs J 276:354–364. https://doi.org/10.1111/j.1742-4658.2008.06796.x
Burnstock G (2016) An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104:4–17. https://doi.org/10.1016/j.neuropharm.2015.05.031
Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS (2011) Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 63:641–683. https://doi.org/10.1124/pr.110.003129
Oliveira-Giacomelli Á, Albino CM, de Souza HDN, Corrêa-Velloso J, de Jesus Santos AP, Baranova J, Ulrich H (2019) P2Y6 and P2X7 receptor antagonism exerts neuroprotective/neuroregenerative effects in an animal model of Parkinson’s disease. Front Cell Neurosci 13:476. https://doi.org/10.3389/fncel.2019.00476
Díaz-Hernández M, Díez-Zaera M, Sánchez-Nogueiro J, Gómez-Villafuertes R, Canals JM, Alberch J, Miras-Portugal MT, Lucas JJ (2009) Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. Faseb J 23:1893–1906. https://doi.org/10.1096/fj.08-122275
Domercq M, Matute C (2019) Targeting P2X4 and P2X7 receptors in multiple sclerosis. Curr Opin Pharmacol 47:119–125. https://doi.org/10.1016/j.coph.2019.03.010
Diaz-Hernandez JI, Gomez-Villafuertes R, León-Otegui M, Hontecillas-Prieto L, Del Puerto A, Trejo JL, Lucas JJ, Garrido JJ et al (2012) In vivo P2X7 inhibition reduces amyloid plaques in alzheimer’s disease through GSK3β and secretases. Neurobiol Aging 33:1816–1828. https://doi.org/10.1016/j.neurobiolaging.2011.09.040
Di Lauro C, Bianchi C, Sebastián-Serrano Á, Soria-Tobar L, Alvarez-Castelao B, Nicke A, Díaz-Hernández M (2022) P2X7 receptor blockade reduces tau induced toxicity, therapeutic implications in tauopathies. Prog Neurobiol 208:102173. https://doi.org/10.1016/j.pneurobio.2021.102173
Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F (2006) The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176:3877–3883. https://doi.org/10.4049/jimmunol.176.7.3877
Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R (2003) P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of alzheimer’s disease. J Biol Chem 278:13309–13317. https://doi.org/10.1074/jbc.M209478200
Burnstock G (1976) Purinergic receptors. J Theor Biol 62:491–503. https://doi.org/10.1016/0022-5193(76)90133-8
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272:735–738. https://doi.org/10.1126/science.272.5262.735
Nicke A (2008) Homotrimeric complexes are the dominant assembly state of native P2X7 subunits. Biochem Biophys Res Commun 377:803–808. https://doi.org/10.1016/j.bbrc.2008.10.042
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067. https://doi.org/10.1152/physrev.00015.2002
Jiang LH, Baldwin JM, Roger S, Baldwin SA (2013) Insights into the molecular mechanisms underlying mammalian P2X7 receptor functions and contributions in diseases, revealed by structural modeling and single nucleotide polymorphisms. Front Pharmacol 4:55. https://doi.org/10.3389/fphar.2013.00055
Sperlágh B, Illes P (2014) P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol Sci 35:537–547. https://doi.org/10.1016/j.tips.2014.08.002
Hattori M, Gouaux E (2012) Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485:207–212. https://doi.org/10.1038/nature11010
Yan Z, Li S, Liang Z, Tomić M, Stojilkovic SS (2008) The P2X7 receptor channel pore dilates under physiological ion conditions. J Gen Physiol 132:563–573. https://doi.org/10.1085/jgp.200810059
Yan Z, Khadra A, Li S, Tomic M, Sherman A, Stojilkovic SS (2010) Experimental characterization and mathematical modeling of P2X7 receptor channel gating. J Neurosci 30:14213–14224. https://doi.org/10.1523/jneurosci.2390-10.2010
Khakh BS, Bao XR, Labarca C, Lester HA (1999) Neuronal P2X transmitter-gated cation channels change their ion selectivity in seconds. Nat Neurosci 2:322–330. https://doi.org/10.1038/7233
Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A (1999) Pore dilation of neuronal P2X receptor channels. Nat Neurosci 2:315–321. https://doi.org/10.1038/7225
Roger S, Pelegrin P, Surprenant A (2008) Facilitation of P2X7 receptor currents and membrane blebbing via constitutive and dynamic calmodulin binding. J Neurosci 28:6393–6401. https://doi.org/10.1523/jneurosci.0696-08.2008
Verhoef PA, Estacion M, Schilling W, Dubyak GR (2003) P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1 beta release. J Immunol 170:5728–5738. https://doi.org/10.4049/jimmunol.170.11.5728
Sun C, Heid ME, Keyel PA, Salter RD (2013) The second transmembrane domain of P2X7 contributes to dilated pore formation. PLoS ONE 8:e61886. https://doi.org/10.1371/journal.pone.0061886
Browne LE, Compan V, Bragg L, North RA (2013) P2X7 receptor channels allow direct permeation of nanometer-sized dyes. J Neurosci 33:3557–3566. https://doi.org/10.1523/jneurosci.2235-12.2013
Suadicani SO, Iglesias R, Wang J, Dahl G, Spray DC, Scemes E (2012) ATP signaling is deficient in cultured Pannexin1-null mouse astrocytes. Glia 60:1106–1116. https://doi.org/10.1002/glia.22338
Alberto AV, Faria RX, Couto CG, Ferreira LG, Souza CA, Teixeira PC, Fróes MM, Alves LA (2013) Is pannexin the pore associated with the P2X7 receptor? Naunyn Schmiedebergs Arch Pharmacol 386:775–787. https://doi.org/10.1007/s00210-013-0868-x
Nicke A, Kuan YH, Masin M, Rettinger J, Marquez-Klaka B, Bender O, Górecki DC, Murrell-Lagnado RD et al (2009) A functional P2X7 splice variant with an alternative transmembrane domain 1 escapes gene inactivation in P2X7 knock-out mice. J Biol Chem 284:25813–25822. https://doi.org/10.1074/jbc.M109.033134
Xu XJ, Boumechache M, Robinson LE, Marschall V, Gorecki DC, Masin M, Murrell-Lagnado RD (2012) Splice variants of the P2X7 receptor reveal differential agonist dependence and functional coupling with pannexin-1. J Cell Sci 125:3776–3789. https://doi.org/10.1242/jcs.099374
Martínez-Frailes C, Di Lauro C, Bianchi C, de Diego-García L, Sebastián-Serrano Á, Boscá L, Díaz-Hernández M (2019) Amyloid peptide induced neuroinflammation increases the P2X7 receptor expression in microglial cells, impacting on its functionality. Front Cell Neurosci 13:143. https://doi.org/10.3389/fncel.2019.00143
Yuskaitis CJ, Jope RS (2009) Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal 21:264–273. https://doi.org/10.1016/j.cellsig.2008.10.014
Lee HG, Won SM, Gwag BJ, Lee YB (2011) Microglial P2X7 receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of alzheimer’s disease. Exp Mol Med 43:7–14. https://doi.org/10.3858/emm.2011.43.1.001
McLarnon JG, Ryu JK, Walker DG, Choi HB (2006) Upregulated expression of purinergic P2X(7) receptor in alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol 65:1090–1097. https://doi.org/10.1097/01.jnen.0000240470.97295.d3
Monif M, Burnstock G, Williams DA (2010) Microglia: proliferation and activation driven by the P2X7 receptor. Int J Biochem Cell Biol 42:1753–1756. https://doi.org/10.1016/j.biocel.2010.06.021
Monif M, Reid CA, Powell KL, Smart ML, Williams DA (2009) The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci 29:3781–3791. https://doi.org/10.1523/jneurosci.5512-08.2009
Takenouchi T, Sugama S, Iwamaru Y, Hashimoto M, Kitani H (2009) Modulation of the ATP-induced release and processing of IL-1beta in microglial cells. Crit Rev Immunol 29:335–345. https://doi.org/10.1615/critrevimmunol.v29.i4.40
Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TF, Buckley NJ, Parson SH et al (2001) Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci 21:7143–7152. https://doi.org/10.1523/jneurosci.21-18-07143.2001
Yu Y, Ugawa S, Ueda T, Ishida Y, Inoue K, Kyaw Nyunt A, Umemura A, Mase M et al (2008) Cellular localization of P2X7 receptor mRNA in the rat brain. Brain Res 1194:45–55. https://doi.org/10.1016/j.brainres.2007.11.064
Delarasse C, Gonnord P, Galante M, Auger R, Daniel H, Motta I, Kanellopoulos JM (2009) Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. J Neurochem 109:846–857. https://doi.org/10.1111/j.1471-4159.2009.06008.x
Lovelace MD, Gu BJ, Eamegdool SS, Weible MW 2nd, Wiley JS, Allen DG, Chan-Ling T (2015) P2X7 receptors mediate innate phagocytosis by human neural precursor cells and neuroblasts. Stem Cells 33:526–541. https://doi.org/10.1002/stem.1864
Glaser T, de Oliveira SL, Cheffer A, Beco R, Martins P, Fornazari M, Lameu C, Junior HM et al (2014) Modulation of mouse embryonic stem cell proliferation and neural differentiation by the P2X7 receptor. PLoS ONE 9:e96281. https://doi.org/10.1371/journal.pone.0096281
Messemer N, Kunert C, Grohmann M, Sobottka H, Nieber K, Zimmermann H, Franke H, Nörenberg W et al (2013) P2X7 receptors at adult neural progenitor cells of the mouse subventricular zone. Neuropharmacology 73:122–137. https://doi.org/10.1016/j.neuropharm.2013.05.017
Francistiová L, Vörös K, Lovász Z, Dinnyés A, Kobolák J (2021) Detection and functional evaluation of the P2X7 receptor in hiPSC derived neurons and microglia-like cells. Front Mol Neurosci 14:793769. https://doi.org/10.3389/fnmol.2021.793769
Miras-Portugal MT, Sebastián-Serrano Á, de Diego GL, Díaz-Hernández M (2017) Neuronal P2X7 receptor: involvement in neuronal physiology and pathology. J Neurosci 37:7063–7072. https://doi.org/10.1523/jneurosci.3104-16.2017
Díaz-Hernandez M, del Puerto A, Díaz-Hernandez JI, Diez-Zaera M, Lucas JJ, Garrido JJ, Miras-Portugal MT (2008) Inhibition of the ATP-gated P2X7 receptor promotes axonal growth and branching in cultured hippocampal neurons. J Cell Sci 121:3717–3728. https://doi.org/10.1242/jcs.034082
Tsao HK, Chiu PH, Sun SH (2013) PKC-dependent ERK phosphorylation is essential for P2X7 receptor-mediated neuronal differentiation of neural progenitor cells. Cell Death Dis 4:e751. https://doi.org/10.1038/cddis.2013.274
Nishida K, Nakatani T, Ohishi A, Okuda H, Higashi Y, Matsuo T, Fujimoto S, Nagasawa K (2012) Mitochondrial dysfunction is involved in P2X7 receptor-mediated neuronal cell death. J Neurochem 122:1118–1128. https://doi.org/10.1111/j.1471-4159.2012.07868.x
Ohishi A, Keno Y, Marumiya A, Sudo Y, Uda Y, Matsuda K, Morita Y, Furuta T et al (2016) Expression level of P2X7 receptor is a determinant of ATP-induced death of mouse cultured neurons. Neuroscience 319:35–45. https://doi.org/10.1016/j.neuroscience.2016.01.048
Carrasquero LM, Delicado EG, Bustillo D, Gutiérrez-Martín Y, Artalejo AR, Miras-Portugal MT (2009) P2X7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J Neurochem 110:879–889. https://doi.org/10.1111/j.1471-4159.2009.06179.x
Nobile M, Monaldi I, Alloisio S, Cugnoli C, Ferroni S (2003) ATP-induced, sustained calcium signalling in cultured rat cortical astrocytes: evidence for a non-capacitative, P2X7-like-mediated calcium entry. FEBS Lett 538:71–76. https://doi.org/10.1016/s0014-5793(03)00129-7
Matute C, Torre I, Pérez-Cerdá F, Pérez-Samartín A, Alberdi E, Etxebarria E, Arranz AM, Ravid R et al (2007) P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 27:9525–9533. https://doi.org/10.1523/jneurosci.0579-07.2007
Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23:1320–1328. https://doi.org/10.1523/jneurosci.23-04-01320.2003
Ballerini P, Rathbone MP, Di Iorio P, Renzetti A, Giuliani P, D’Alimonte I, Trubiani O, Caciagli F et al (1996) Rat astroglial P2Z (P2X7) receptors regulate intracellular calcium and purine release. NeuroReport 7:2533–2537. https://doi.org/10.1097/00001756-199611040-00026
Suadicani SO, Brosnan CF, Scemes E (2006) P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci 26:1378–1385. https://doi.org/10.1523/jneurosci.3902-05.2006
Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, Yong VW, Ransohoff RM et al (2001) P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci 21:7135–7142. https://doi.org/10.1523/jneurosci.21-18-07135.2001
Tewari M, Monika VRK, Menon M, Seth P (2015) Astrocytes mediate HIV-1 Tat-induced neuronal damage via ligand-gated ion channel P2X7R. J Neurochem 132:464–476. https://doi.org/10.1111/jnc.12953
Kaczmarek-Hajek K, Zhang J, Kopp R, Grosche A, Rissiek B, Saul A, Bruzzone S, Engel T et al (2018) Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife 7. https://doi.org/10.7554/eLife.36217
Feng JF, Gao XF, Pu YY, Burnstock G, Xiang Z, He C (2015) P2X7 receptors and Fyn kinase mediate ATP-induced oligodendrocyte progenitor cell migration. Purinergic Signal 11:361–369. https://doi.org/10.1007/s11302-015-9458-3
Jimenez-Mateos EM, Smith J, Nicke A, Engel T (2019) Regulation of P2X7 receptor expression and function in the brain. Brain Res Bull 151:153–163. https://doi.org/10.1016/j.brainresbull.2018.12.008
Tan NY, Khachigian LM (2009) Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol 29:2483–2488. https://doi.org/10.1128/mcb.01828-08
García-Huerta P, Díaz-Hernandez M, Delicado EG, Pimentel-Santillana M, Miras-Portugal MT, Gómez-Villafuertes R (2012) The specificity protein factor Sp1 mediates transcriptional regulation of P2X7 receptors in the nervous system. J Biol Chem 287:44628–44644. https://doi.org/10.1074/jbc.M112.390971
Engel T, Brennan GP, Sanz-Rodriguez A, Alves M, Beamer E, Watters O, Henshall DC, Jimenez-Mateos EM (2017) A calcium-sensitive feed-forward loop regulating the expression of the ATP-gated purinergic P2X7 receptor via specificity protein 1 and microRNA-22. Biochim Biophys Acta Mol Cell Res 1864:255–266. https://doi.org/10.1016/j.bbamcr.2016.11.007
Qin J, Zhang X, Wang Z, Li J, Zhang Z, Gao L, Ren H, Qian M et al (2017) Presenilin 2 deficiency facilitates Aβ-induced neuroinflammation and injury by upregulating P2X7 expression. Sci China Life Sci 60:189–201. https://doi.org/10.1007/s11427-016-0347-4
Gouras GK, Olsson TT, Hansson O (2015) β-Amyloid peptides and amyloid plaques in alzheimer’s disease. Neurotherapeutics 12:3–11. https://doi.org/10.1007/s13311-014-0313-y
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766. https://doi.org/10.1152/physrev.2001.81.2.741
Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2:a006270. https://doi.org/10.1101/cshperspect.a006270
Han P, Dou F, Li F, Zhang X, Zhang YW, Zheng H, Lipton SA, Xu H et al (2005) Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation. J Neurosci 25:11542–11552. https://doi.org/10.1523/jneurosci.3831-05.2005
Ma T, Zhao Y, Kwak YD, Yang Z, Thompson R, Luo Z, Xu H, Liao FF (2009) Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J Neurosci 29:11226–11236. https://doi.org/10.1523/jneurosci.6150-08.2009
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356. https://doi.org/10.1126/science.1072994
Lauretti E, Dincer O, Praticò D (2020) Glycogen synthase kinase-3 signaling in alzheimer’s disease. Biochim Biophys Acta Mol Cell Res 1867:118664. https://doi.org/10.1016/j.bbamcr.2020.118664
Aplin AE, Gibb GM, Jacobsen JS, Gallo JM, Anderton BH (1996) In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J Neurochem 67:699–707. https://doi.org/10.1046/j.1471-4159.1996.67020699.x
Cai Z, Zhao Y, Zhao B (2012) Roles of glycogen synthase kinase 3 in alzheimer’s disease. Curr Alzheimer Res 9:864–879. https://doi.org/10.2174/156720512802455386
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y et al (2013) Inhibition of GSK3β-mediated BACE1 expression reduces alzheimer-associated phenotypes. J Clin Invest 123:224–235. https://doi.org/10.1172/jci64516
Gao Y, Tan L, Yu JT, Tan L (2018) Tau in alzheimer’s disease: mechanisms and therapeutic strategies. Curr Alzheimer Res 15:283–300. https://doi.org/10.2174/1567205014666170417111859
Goedert M, Spillantini MG (2006) A century of alzheimer’s disease. Science 314:777–781. https://doi.org/10.1126/science.1132814
Iqbal K, Liu F, Gong CX (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12:15–27. https://doi.org/10.1038/nrneurol.2015.225
Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S (2002) Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115:201–211. https://doi.org/10.1016/s0306-4522(02)00404-9
Singh TJ, Haque N, Grundke-Iqbal I, Iqbal K (1995) Rapid alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS Lett 358:267–272. https://doi.org/10.1016/0014-5793(94)01445-7
Iqbal K, Liu F, Gong CX, Grundke-Iqbal I (2010) Tau in alzheimer disease and related tauopathies. Curr Alzheimer Res 7:656–664. https://doi.org/10.2174/156720510793611592
Lopes da Silva S, Vellas B, Elemans S, Luchsinger J, Kamphuis P, Yaffe K, Sijben J, Groenendijk M et al (2014) Plasma nutrient status of patients with alzheimer’s disease: systematic review and meta-analysis. Alzheimers Dement 10:485–502. https://doi.org/10.1016/j.jalz.2013.05.1771
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. https://doi.org/10.1038/nn.4132
Carvalho K, Martin E, Ces A, Sarrazin N, Lagouge-Roussey P, Nous C, Boucherit L, Youssef I et al (2021) P2X7-deficiency improves plasticity and cognitive abilities in a mouse model of tauopathy. Prog Neurobiol 206:102139. https://doi.org/10.1016/j.pneurobio.2021.102139
Ruan Z, Delpech JC, Venkatesan Kalavai S, Van Enoo AA, Hu J, Ikezu S, Ikezu T (2020) P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol Neurodegener 15:47. https://doi.org/10.1186/s13024-020-00396-2
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an alzheimer’s disease model. J Immunol 187:6539–6549. https://doi.org/10.4049/jimmunol.1100620
Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK (2013) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an alzheimer’s mouse model. J Neurosci 33:5053–5064. https://doi.org/10.1523/jneurosci.4361-12.2013
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T et al (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. https://doi.org/10.1016/j.neuron.2007.01.010
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68:19–31. https://doi.org/10.1016/j.neuron.2010.08.023
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138:1738–1755. https://doi.org/10.1093/brain/awv081
Di Lauro C, Bianchi C, Sebastián-Serrano Á, Soria-Tobar L, Alvarez-Castelao B, Nicke A, Díaz-Hernández M (2021) P2X7 receptor blockade reduces tau induced toxicity, therapeutic implications in tauopathies. Prog Neurobiol:102173. https://doi.org/10.1016/j.pneurobio.2021.102173
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT (2018) Inflammation as a central mechanism in alzheimer’s disease. Alzheimers Dement (N Y) 4:575–590. https://doi.org/10.1016/j.trci.2018.06.014
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845. https://doi.org/10.1126/science.1194637
Gehrmann J, Matsumoto Y, Kreutzberg GW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20:269–287. https://doi.org/10.1016/0165-0173(94)00015-h
Colton C, Wilcock DM (2010) Assessing activation states in microglia. CNS Neurol Disord Drug Targets 9:174–191. https://doi.org/10.2174/187152710791012053
Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 4:399–418. https://doi.org/10.1007/s11481-009-9164-4
He Y, Taylor N, Fourgeaud L, Bhattacharya A (2017) The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflammation 14:135. https://doi.org/10.1186/s12974-017-0904-8
Shieh CH, Heinrich A, Serchov T, van Calker D, Biber K (2014) P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 62:592–607. https://doi.org/10.1002/glia.22628
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865. https://doi.org/10.1038/ni.1636
Colomar A, Marty V, Médina C, Combe C, Parnet P, Amédée T (2003) Maturation and release of interleukin-1beta by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J Biol Chem 278:30732–30740. https://doi.org/10.1074/jbc.M304534200
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153. https://doi.org/10.1016/j.immuni.2013.05.016
Wang W, Hu D, Feng Y, Wu C, Song Y, Liu W, Li A, Wang Y et al (2020) Paxillin mediates ATP-induced activation of P2X7 receptor and NLRP3 inflammasome. BMC Biol 18:182. https://doi.org/10.1186/s12915-020-00918-w
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426. https://doi.org/10.1016/s1097-2765(02)00599-3
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. https://doi.org/10.1038/nature11729
Sörensen A, Blazhenets G, Rücker G, Schiller F, Meyer PT, Frings L (2019) Prognosis of conversion of mild cognitive impairment to alzheimer’s dementia by voxel-wise Cox regression based on FDG PET data. Neuroimage Clin 21:101637. https://doi.org/10.1016/j.nicl.2018.101637
Fabbrizio P, Amadio S, Apolloni S, Volonté C (2017) P2X7 receptor activation modulates autophagy in SOD1-G93A mouse microglia. Front Cell Neurosci 11:249. https://doi.org/10.3389/fncel.2017.00249
Takenouchi T, Nakai M, Iwamaru Y, Sugama S, Tsukimoto M, Fujita M, Wei J, Sekigawa A et al (2009) The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J Immunol 182:2051–2062. https://doi.org/10.4049/jimmunol.0802577
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H et al (2016) Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351:275–281. https://doi.org/10.1126/science.aab4138
Sekar P, Huang DY, Hsieh SL, Chang SF, Lin WW (2018) AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun Signal 16:83. https://doi.org/10.1186/s12964-018-0293-3
Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF (2020) Mitophagy and neuroprotection. Trends Mol Med 26:8–20. https://doi.org/10.1016/j.molmed.2019.07.002
Chu CT (2019) Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis 122:23–34. https://doi.org/10.1016/j.nbd.2018.07.015
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF (2017) Mitophagy and alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40:151–166. https://doi.org/10.1016/j.tins.2017.01.002
Ferrer MD, Sureda A, Mestre A, Tur JA, Pons A (2010) The double edge of reactive oxygen species as damaging and signaling molecules in HL60 cell culture. Cell Physiol Biochem 25:241–252. https://doi.org/10.1159/000276558
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in alzheimer’s disease. Biochim Biophys Acta 1842:1240–1247. https://doi.org/10.1016/j.bbadis.2013.10.015
Price JL, McKeel DW Jr, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC et al (2009) Neuropathology of nondemented aging: presumptive evidence for preclinical alzheimer disease. Neurobiol Aging 30:1026–1036. https://doi.org/10.1016/j.neurobiolaging.2009.04.002
Praticò D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ (2002) Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of alzheimer disease. Arch Neurol 59:972–976. https://doi.org/10.1001/archneur.59.6.972
Albers DS, Beal MF (2000) Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl 59:133–154. https://doi.org/10.1007/978-3-7091-6781-6_16
Beal MF (2002) Oxidatively modified proteins in aging and disease. Free Radic Biol Med 32:797–803. https://doi.org/10.1016/s0891-5849(02)00780-3
Butterfield DA, Kanski J (2001) Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech Ageing Dev 122:945–962. https://doi.org/10.1016/s0047-6374(01)00249-4
Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X (2009) The role of abnormal mitochondrial dynamics in the pathogenesis of alzheimer’s disease. J Neurochem 109(Suppl 1):153–159. https://doi.org/10.1111/j.1471-4159.2009.05867.x
Sarti AC, Vultaggio-Poma V, Falzoni S, Missiroli S, Giuliani AL, Boldrini P, Bonora M, Faita F et al (2021) Mitochondrial P2X7 receptor localization modulates energy metabolism enhancing physical performance. Function (Oxf) 2:zqab005. https://doi.org/10.1093/function/zqab005
Keck S, Nitsch R, Grune T, Ullrich O (2003) Proteasome inhibition by paired helical filament-tau in brains of patients with alzheimer’s disease. J Neurochem 85:115–122. https://doi.org/10.1046/j.1471-4159.2003.01642.x
Bianchi C, Alvarez-Castelao B, Sebastián-Serrano Á, Di Lauro C, Soria-Tobar L, Nicke A, Engel T, Díaz-Hernández M (2023) P2X7 receptor inhibition ameliorates ubiquitin-proteasome system dysfunction associated with alzheimer’s disease. Alzheimers Res Ther 15:105. https://doi.org/10.1186/s13195-023-01258-x
Chiozzi P, Sarti AC, Sanz JM, Giuliani AL, Adinolfi E, Vultaggio-Poma V, Falzoni S, Di Virgilio F (2019) Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci Rep 9:6475. https://doi.org/10.1038/s41598-019-42931-2
Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ, Pocock JM (2012) Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J Neurochem 121:287–301. https://doi.org/10.1111/j.1471-4159.2012.07659.x
Skaper SD, Facci L, Culbert AA, Evans NA, Chessell I, Davis JB, Richardson JC (2006) P2X(7) receptors on microglial cells mediate injury to cortical neurons in vitro. Glia 54:234–242. https://doi.org/10.1002/glia.20379
Pfeiffer ZA, Guerra AN, Hill LM, Gavala ML, Prabhu U, Aga M, Hall DJ, Bertics PJ (2007) Nucleotide receptor signaling in murine macrophages is linked to reactive oxygen species generation. Free Radic Biol Med 42:1506–1516. https://doi.org/10.1016/j.freeradbiomed.2007.02.010
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. https://doi.org/10.1002/ana.410300410
Kashyap G, Bapat D, Das D, Gowaikar R, Amritkar RE, Rangarajan G, Ravindranath V, Ambika G (2019) Synapse loss and progress of alzheimer’s disease-a network model. Sci Rep 9:6555. https://doi.org/10.1038/s41598-019-43076-y
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791. https://doi.org/10.1126/science.1074069
Lleó A, Núñez-Llaves R, Alcolea D, Chiva C, Balateu-Paños D, Colom-Cadena M, Gomez-Giro G, Muñoz L et al (2019) Changes in synaptic proteins precede neurodegeneration markers in preclinical alzheimer’s disease cerebrospinal fluid. Mol Cell Proteomics 18:546–560. https://doi.org/10.1074/mcp.RA118.001290
John A, Reddy PH (2021) Synaptic basis of alzheimer’s disease: focus on synaptic amyloid beta. P-tau and mitochondria Ageing Res Rev 65:101208. https://doi.org/10.1016/j.arr.2020.101208
Chen X, Hu J, Jiang L, Xu S, Zheng B, Wang C, Zhang J, Wei X et al (2014) Brilliant blue G improves cognition in an animal model of alzheimer’s disease and inhibits amyloid-β-induced loss of filopodia and dendrite spines in hippocampal neurons. Neuroscience 279:94–101. https://doi.org/10.1016/j.neuroscience.2014.08.036
Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, Brückner M, Prigent A et al (2019) New role of P2X7 receptor in an alzheimer’s disease mouse model. Mol Psychiatry 24:108–125. https://doi.org/10.1038/s41380-018-0108-3
Sanz JM, Falzoni S, Rizzo R, Cipollone F, Zuliani G, Di Virgilio F (2014) Possible protective role of the 489C>T P2X7R polymorphism in alzheimer’s disease. Exp Gerontol 60:117–119. https://doi.org/10.1016/j.exger.2014.10.009
Borzelleca JF, Depukat K, Hallagan JB (1990) Lifetime toxicity/carcinogenicity studies of FD & C Blue no. 1 (brilliant blue FCF) in rats and mice. Food Chem Toxicol 28:221–234. https://doi.org/10.1016/0278-6915(90)90034-k
Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF et al (2009) Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A 106:12489–12493. https://doi.org/10.1073/pnas.0902531106
Ryu JK, McLarnon JG (2008) Block of purinergic P2X(7) receptor is neuroprotective in an animal model of alzheimer’s disease. NeuroReport 19:1715–1719. https://doi.org/10.1097/WNR.0b013e3283179333
Rampe D, Wang L, Ringheim GE (2004) P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol 147:56–61. https://doi.org/10.1016/j.jneuroim.2003.10.014
Burnstock G, Knight GE (2018) The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal 14:1–18. https://doi.org/10.1007/s11302-017-9593-0
Bhattacharya A (2018) Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front Pharmacol 9:30. https://doi.org/10.3389/fphar.2018.00030
Sadovnick AD, Gu BJ, Traboulsee AL, Bernales CQ, Encarnacion M, Yee IM, Criscuoli MG, Huang X et al (2017) Purinergic receptors P2RX4 and P2RX7 in familial multiple sclerosis. Hum Mutat 38:736–744. https://doi.org/10.1002/humu.23218
Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M, Popoli P, D’Ambrosi N et al (2014) Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis Model Mech 7:1101–1109. https://doi.org/10.1242/dmm.017038
Tewari M, Seth P (2015) Emerging role of P2X7 receptors in CNS health and disease. Ageing Res Rev 24:328–342. https://doi.org/10.1016/j.arr.2015.10.001
Gao M, Wang M, Green MA, Hutchins GD, Zheng QH (2015) Synthesis of [(11)C]GSK1482160 as a new PET agent for targeting P2X(7) receptor. Bioorg Med Chem Lett 25:1965–1970. https://doi.org/10.1016/j.bmcl.2015.03.021
Baudelet D, Lipka E, Millet R, Ghinet A (2015) Involvement of the P2X7 purinergic receptor in inflammation: an update of antagonists series since 2009 and their promising therapeutic potential. Curr Med Chem 22:713–729. https://doi.org/10.2174/0929867322666141212120926
Murphy N, Cowley TR, Richardson JC, Virley D, Upton N, Walter D, Lynch MA (2012) The neuroprotective effect of a specific P2X7 receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathol 22:295–306. https://doi.org/10.1111/j.1750-3639.2011.00531.x
Chrovian CC, Soyode-Johnson A, Peterson AA, Gelin CF, Deng X, Dvorak CA, Carruthers NI, Lord B et al (2018) A dipolar cycloaddition reaction to access 6-methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines enables the discovery synthesis and preclinical profiling of a P2X7 antagonist clinical candidate. J Med Chem 61:207–223. https://doi.org/10.1021/acs.jmedchem.7b01279
Letavic MA, Savall BM, Allison BD, Aluisio L, Andres JI, De Angelis M, Ao H, Beauchamp DA et al (2017) 4-methyl-6,7-dihydro-4H-triazolo[4,5-c]pyridine-based P2X7 receptor antagonists: optimization of pharmacokinetic properties leading to the identification of a clinical candidate. J Med Chem 60:4559–4572. https://doi.org/10.1021/acs.jmedchem.7b00408
Bhattacharya A, Wang Q, Ao H, Shoblock JR, Lord B, Aluisio L, Fraser I, Nepomuceno D et al (2013) Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br J Pharmacol 170:624–640. https://doi.org/10.1111/bph.12314
Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, Lovenberg TW, Carruthers N et al (2014) Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J Pharmacol Exp Ther 351:628–641. https://doi.org/10.1124/jpet.114.218487
Timmers M, Ravenstijn P, Xi L, Triana-Baltzer G, Furey M, Van Hemelryck S, Biewenga J, Ceusters M et al (2018) Clinical pharmacokinetics, pharmacodynamics, safety, and tolerability of JNJ-54175446, a brain permeable P2X7 antagonist, in a randomised single-ascending dose study in healthy participants. J Psychopharmacol 32:1341–1350. https://doi.org/10.1177/0269881118800067
Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P et al (2016) Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 8:338ra66. https://doi.org/10.1126/scitranslmed.aaf2362
Quispialaya KM, Therriault J, Aliaga A, Zimmermann M, Fernandez-Arias J, Lussier F, Massarweh G, Pascoal T et al (2022) Discordance and concordance between cerebrospinal and [(18)F]FDG-PET biomarkers in assessing atypical and early-onset AD dementia cases. Neurology 99:e2428–e2436. https://doi.org/10.1212/wnl.0000000000201198
Ory D, Celen S, Gijsbers R, Van Den Haute C, Postnov A, Koole M, Vandeputte C, Andrés JI et al (2016) Preclinical evaluation of a P2X7 receptor-selective radiotracer: PET studies in a rat model with local overexpression of the human P2X7 receptor and in nonhuman primates. J Nucl Med 57:1436–1441. https://doi.org/10.2967/jnumed.115.169995
Koole M, Schmidt ME, Hijzen A, Ravenstijn P, Vandermeulen C, Van Weehaeghe D, Serdons K, Celen S et al (2019) (18)F-JNJ-64413739, a novel PET ligand for the P2X7 ion channel: radiation dosimetry, kinetic modeling, test-retest variability, and occupancy of the P2X7 antagonist JNJ-54175446. J Nucl Med 60:683–690. https://doi.org/10.2967/jnumed.118.216747
Ren W, Rubini P, Tang Y, Engel T, Illes P (2021) Inherent P2X7 receptors regulate macrophage functions during inflammatory diseases. Int J Mol Sci 23. https://doi.org/10.3390/ijms23010232
Vultaggio-Poma V, Di Virgilio F (2022) P2 Receptors: Novel disease markers and metabolic checkpoints in immune Cells. Biomolecules 12. https://doi.org/10.3390/biom12070983
Aivar P, Bianchi C, Di Lauro C, Soria-Tobar L, Alvarez-Castelao B, Calero M, Medina M, Diaz-Hernandez M (2023) TNAP and P2X7R: new plasma biomarkers for Alzheimer's Disease. Int J Mol Sci 24. https://doi.org/10.3390/ijms241310897
Acknowledgements
I would like to express my sincere gratitude to my teachers and classmates for their continuous support and encouragement, and especially to my teacher, Mr. Hua, for his guidance on my writing.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82060219 and 81860259); Natural Science Foundation of Jiangxi Province (20212ACB216009; 20202BAB206033 and 20212BAB216048); Jiangxi Province “Double Thousand Plan” (jxsq2019201023); Youth Team Project of the Second Affiliated Hospital of Nanchang University (2019YNTD12003); and Postgraduate Innovation Special Foundation of Jiangxi Province (YC2022-S225).
Author information
Authors and Affiliations
Contributions
All authors were involved in the conception and design of the study. Material preparation, data collection, and analysis. Qiang Huang was primarily responsible for helping to conceptualize, write the original manuscript, and review and edit the manuscript. Jun Ying, Wen Yu, and Jie Liu helped to conceptualize, write the original manuscript, and review and edit the manuscript. Xiong Hao, Dong Yao, and Zhang Yiping helped to review and edit the manuscript. Wang Xifeng and Hua Fuzhou contributed to the study design, literature search, and manuscript preparation. All authors have commented on previous versions of the manuscript. All authors have read and agreed to the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable for that section.
Consent to Participate
Not applicable for that section.
Consent for Publication
Not applicable for that section.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Huang Qiang is the first author of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, Q., Ying, J., Yu, W. et al. P2X7 Receptor: an Emerging Target in Alzheimer’s Disease. Mol Neurobiol 61, 2866–2880 (2024). https://doi.org/10.1007/s12035-023-03699-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03699-9