
Abstract
Ergothioneine (ET) is a naturally occurring antioxidant that is synthesized by non-yeast fungi and certain bacteria. ET is not synthesized by animals, including humans, but is avidly taken up from the diet, especially from mushrooms. In the current study, we elucidated the effect of ET on the hCMEC/D3 human brain endothelial cell line. Endothelial cells are exposed to high levels of the cholesterol oxidation product, 7-ketocholesterol (7KC), in patients with cardiovascular disease and diabetes, and this process is thought to mediate pathological inflammation. 7KC induces a dose-dependent loss of cell viability and an increase in apoptosis and necrosis in the endothelial cells. A relocalization of the tight junction proteins, zonula occludens-1 (ZO-1) and claudin-5, towards the nucleus of the cells was also observed. These effects were significantly attenuated by ET. In addition, 7KC induces marked increases in the mRNA expression of pro-inflammatory cytokines, IL-1β IL-6, IL-8, TNF-α and cyclooxygenase-2 (COX2), as well as COX2 enzymatic activity, and these were significantly reduced by ET. Moreover, the cytoprotective and anti-inflammatory effects of ET were significantly reduced by co-incubation with an inhibitor of the ET transporter, OCTN1 (VHCL). This shows that ET needs to enter the endothelial cells to have a protective effect and is unlikely to act via extracellular neutralizing of 7KC. The protective effect on inflammation in brain endothelial cells suggests that ET might be useful as a nutraceutical for the prevention or management of neurovascular diseases, such as stroke and vascular dementia. Moreover, the ability of ET to cross the blood-brain barrier could point to its usefulness in combatting 7KC that is produced in the CNS during neuroinflammation, e.g. after excitotoxicity, in chronic neurodegenerative diseases, and possibly COVID-19-related neurologic complications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pathological accumulation of cholesterol oxidation products or oxysterols occurs in many major chronic diseases. One of the main oxysterols is 7-ketocholesterol (7KC), which is formed by oxygen radical attack on cholesterol. 7KC can be derived from the diet or via endogenous production within the body and is associated with many diseases and disabilities of ageing (Anderson et al. 2020). Serum 7KC concentrations are significantly higher in subjects with coronary artery disease compared to healthy controls, and their levels are correlated with the inflammatory marker, C-reactive protein (Hitsumoto et al. 2009). In another study, serum 7KC levels were associated with increased risk of cardiovascular events and mortality in patients with stable coronary artery disease (Song et al. 2017). Elevated plasma 7KC levels are associated with the incidence of cardiovascular disease in a population-based cohort (Wang et al. 2017), and increased 7KC level is found in patients with hypercholesterolemia or diabetes (Murakami et al. 2000). Serum 7KC levels are reduced by statins, oral hypoglycaemic agents and antihypertensive agents in patients with type 2 diabetes (Samadi et al. 2019). Levels of 7KC are also elevated in cerebral cortical tissue from Alzheimer’s’ Disease (AD) patients (Testa et al. 2016) and in the rat hippocampus after kainate-induced excitotoxicity (W. Y. Ong et al. 2003; He et al. 2006; Kim et al. 2010).
There is much interest in the effects of 7KC on different cell types in the cardiovascular system, including endothelial cells, smooth muscle cells, cardiac myocytes, red blood cells and macrophages. Treatment of human umbilical vein endothelial cells (HUVEC) with 7KC results in increased expression of the cell adhesion molecules E-selectin, ICAM-1 and VCAM-1, and increased interactions between the endothelial cells and human monocytic THP-1 cells (Tani et al. 2018). 7KC also increases the expression of endoglin, resulting in increased adhesion and transmigration of monocytes across human aortic endothelial cells (HAECs) (Vicen et al. 2019). 7KC damages endothelial cells and increases the expression and release of clotting protein, von Willebrand factor (W. Li et al. 2011). It also increases the expression of the proatherogenic protein profilin-1 in aortic endothelial cells (Romeo and Kazlauskas 2008). In addition, 7KC blocks the release of nitric oxide from vascular endothelial cells, which leads to inhibition of arterial relaxation (Deckert et al. 2002). Moreover, 7KC increases the number of pores or fenestrations in liver sinusoidal endothelial cells (Svistounov et al. 2012; Hunt et al. 2019). High concentrations of 7KC induce ROS production, ATM/Chk2, ATR-Chk1 and p53 signalling pathways, G0/G1 cell cycle arrest and apoptosis in endothelial cells (Chang et al. 2016). There is also evidence that 7KC could induce death of endothelial cells by necrosis (Ghelli et al. 2002; Hans et al. 2008).
Ergothioneine (ET) is a naturally occurring antioxidant that is found in non-yeast fungi and certain bacteria (reviewed by (Halliwell et al. 2018)). ET is not synthesized by animals but is avidly taken up from the diet (Cheah et al. 2017). The major dietary source of ET is from mushrooms. However, other foods, such as asparagus and tempeh (fermented soya bean), may contain high levels of ET, which is most likely due to a symbiotic relationship between plants and soil fungi or bacteria (Halliwell et al. 2018). ET enters cells via the organic cation transporter novel type-1 (OCTN1). This transporter, which has a high degree of specificity for ET (Grigat et al. 2007; Grundemann et al. 2005; Tucker et al. 2019), is also present on the blood–brain barrier (BBB). It facilitates the entry of ET into the CNS where the compound has been detected in human cerebrospinal fluid and post-mortem brain samples (Halliwell et al. 2018). ET can accumulate in the body at up to millimolar concentrations and is believed to function as a physiological cytoprotectant. Studies show that it can accumulate in organs, cells and secretions that are exposed to high levels of oxidative stress and inflammation (Tang et al. 2018). In vitro assays show that ET is a powerful scavenger of hydroxyl radicals (OH), hypochlorous acid (HOCl) and peroxynitrite (ONOO-) (Akanmu et al. 1991). Moreover, silencing of OCTN1 in HeLa cells leads to increased oxidative burden on mitochondria and mitochondrial DNA damage (Paul and Snyder 2010). OCTN1 knockout mice are more susceptible to oxidative stress and inflammation, as demonstrated by higher lethality in OCTN1 knockout mice to intestinal reperfusion injury (Kato et al. 2010). ET has also been shown to inhibit IL-1β-induced expression of vascular and intracellular adhesion molecules in endothelial cells (Anticoli et al. 2010).
The ability of ET to cross the BBB and to act as an antioxidant and cytoprotectant makes it a potential therapeutic option for neurodegenerative diseases, where disease progression is often driven by oxidative stress (Halliwell 2006; Butterfield and Halliwell 2019). There is also a possibility that it could modulate neurovascular diseases by reducing damage to brain endothelial cells. Thus far, however, little is known about the effect of ET on 7KC-induced injury in endothelial cells. The purpose of this study is to examine the effect of ET on 7KC-induced damage in hCMEC/D3 brain endothelial cells, in the hope of reducing damage to these cells when they are exposed to 7KC.
Materials and Methods
Ergothioneine/Chemicals
ET was kindly provided by Tetrahedron (14 Avenue de l’Opera, Paris, France). Based on dose response studies of the effect of 7KC, endothelial cells were pre-treated with 0.1, 1.0 or 10 mM ET for 1 h followed by co-incubation with 30 μM or 50 μM 7KC for 24 h before analyses. These concentrations of ET are comparable to that used for other cell culture studies (G. Li et al. 2010). In some experiments, the OCTN1 inhibitor Verapamil Hydrochloride (VHCl) was purchased from Sigma-Aldrich (St. Louis, USA) and used as an inhibitor of ET entry into Human Cerebral Microvascular Endothelial Cells (hCMEC/D3), at a concentration of 100 µM.
Cells and Culture
hCMEC/D3 brain endothelial cells were obtained from EMD Millipore (Temecula, CA, USA), and cultured as described previously (B. B. Weksler et al. 2005). This cell line was isolated from human temporal lobe microvessels and was shown to preserve typical brain endothelial cell properties, such as tight junction protein expression and localization, receptor expression and selective permeability, establishing its suitability as a simple blood–brain barrier model (B. Weksler et al. 2013). All experiments were carried out on hCMEC/D3 cells from passages 5 to 15. Cells were seeded in flasks coated with collagen and maintained humidified at 37 °C in 5% CO2 atmosphere in Endothelial Basal Medium (EBM-2, Lonza, USA) supplemented with HyClone™ Antibiotic/Antimycotic Solution (1x, Thermal Fisher Scientific, USA) and EGM-2MV kit from Lonza (USA), which includes foetal bovine serum (5%), hydrocortisone, human fibroblast growth factor-beta (hFGF-β), vascular endothelial growth factor (VEGF), R3-Insulin-like Growth Factor-1 (R3-IGF-1), ascorbic acid, human Epidermal Growth Factor (hEGF) and Gentamicin/Amphotericin-B (GA). Cells were grown to confluence and pre-treated with ET alone or ET with VHCl for 1 h, followed by addition of 7KC or ethanol (vehicle control). Cells were assessed 24 h after treatment for gene expression, immunocytochemistry or enzymatic assays, or in real time for cell barrier function assay.
Trypan Blue Cell Viability Assay
Trypan blue exclusion method was employed to assess the effects of 7KC and neuroprotective agents on cell viability. Viable cells exclude the dye while nonviable ones do not, differentiating live from dead cells. After washing twice with 1 × PBS, cells were detached with 0.25% trypsin/EDTA (Omega Scientific, USA) for 5 min. An equivalent amount of EBM-2 was added, followed by centrifuging at 1000 × g for 5 min and re-suspending in culture media. 0.4% trypan blue (Thermo Fisher Scientific, USA) was mixed with the cell suspension in a ratio of 1:1 and loaded into a cell counting chamber for visualization. Live cell counts with diameter gated at 10 μm were determined using an automated cell counter (TC10, Bio-Rad, USA).
MTS Assay
Cells were pre-treated with vehicle control or ET for 1 h, then treated with 7KC at different concentrations (0 μM, 1 μM, 10 μM, 30 μM, 50 μM or 100 μM 7KC). After 24 h, 20 μL CellTiter AQueous One Solution (MTS cell proliferation assay, Promega, USA) was added to each well. Absorbance was read at 490 nm using the Tecan Microplate Reader (Tecan, Switzerland).
Immunocytochemistry
Immunofluorescence staining of tight junction proteins was carried out to determine possible changes in cellular distribution of these proteins after 7KC treatment. Wells in duplicate were briefly washed with 1 × PBS, fixed with 4% paraformaldehyde for 15 min and washed again with 1 × PBS. Fixed cells were permeabilized with 0.3% Triton-X-100 in PBS and blocked with 6% horse or goat serum in PBS-Tx for 1 h before incubating in primary antibodies for 4 h at room temperature. Mouse anti-ZO1 antibody (1:500, Cat# ab61357, Abcam, Cambridge, UK) was used in combination with rabbit anti-claudin-5 antibody (1:100, Cat# 34–1600, Invitrogen, USA) or rabbit anti-occludin antibody (10 μg/mL, Cat# PA5-20,755, Invitrogen, USA) and stained with anti-mouse and anti-rabbit secondary antibodies conjugated with CY3 and FITC, respectively, (1:200, Thermo Fisher Scientific, USA) for 30 min with PBS washes in between and after. Immunostained coverslips were mounted on glass slides using Fluoroshield Mounting Medium with DAPI (Cat# ab104139, Abcam, UK). Images were captured with a 40X objective using excitation wavelengths of 405 nm (DAPI), 488 nm (FITC) and 543 nm (CY3) using a confocal microscope (Olympus, Tokyo, Japan).
Apoptosis/necrosis Assay by Flow Cytometry
An apoptosis/ necrosis assay (ab176749, Abcam, Cambridge, UK) was performed using flow cytometry to quantify apoptotic and necrotic cells. Cells were first detached, and cell pellets were washed with PBS. They were incubated for 1 h with the stain in the absence of light before conducting flow cytometry analysis. Apoptosis was quantified using a phosphatidylserine sensor which emits green fluorescence upon binding to membrane phosphatidylserine. Necrosis was quantified using 7-AAD, a red membrane-impermeable dye labelling the nucleus, demonstrating late-stage apoptosis or early necrosis. Flow cytometry measurements were performed using a BD Accuri™ C6 personal flow cytometer (BD Biosciences, USA) with 20,000 cells per sample for analysis. The FL1 channel was used to quantify apoptosis (PS sensor, Ex/Em = 490/525 nm), and the FL2 channel was used to quantify necrosis (7-AAD, Ex/Em = 546/647 nm).
Quantitative RT-PCR
Cells were collected and stored as cell pellets. Total RNA was isolated using the RNeasy® Mini kit (Qiagen, Germany) as per the manufacturer’s instructions. 1000 ng RNA was reverse transcribed to produce cDNA (High-Capacity cDNA Reverse Transcription Kit; Applied Biosystems, USA) at 25 °C for 10 min, 37 °C for 30 min and then 85 °C for 5 min, using a T-Personal Thermocycler (Biometra, Germany). qPCR was carried out to quantify gene expression of tight junction proteins (ZO-1: Hs01551861_m1, claudin-5: Hs01561351_m1, occludin: Hs01561351_m1), Nitric Oxide Synthase (NOS) (inducible NOS: Hs01075529_m1, endothelial NOS: Hs01574665_m1) and pro-inflammatory genes (IL-1β: Hs01555410_m1, IL-6: Hs00174131_m1, IL-8: Hs00174103_m1, COX-2: Hs00153133_m1, TNFα: Hs00174128_m1, Nuclear Factor Kappa B (NFκB): Hs00765730_m1, p65: Hs01042014_m1). TaqMan Universal PCR Master Mix (Applied Biosystems, USA), with TaqMan Gene Expression Assay Probes obtained from Applied Biosystems (USA), was used for qPCR. The housekeeping gene used was β-Actin (Hs99999903_m1). Analysis was done using the 7500 Real-time PCR System (Applied Biosystems, USA). The PCR reaction conditions were as follows: 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The comparative CT (ΔΔCT) method was used to determine the relative mRNA expression of genes of interest.
COX-2 Enzymatic Activity Assay
Cyclooxygenase 2 (COX2) activity was measured using a cyclooxygenase activity assay kit (Fluorometric) (ab204699) from Abcam (Cambridge, UK). Cells were detached and washed, before addition of lysis buffer (1% NP-40 containing EDTA-free protease–phosphatase inhibitor cocktail). Cell lysates were centrifuged at 12,000 × g, 4 °C for 3 min, and the supernatant was collected for the assay. After the preparation of reagents and samples on a 96-well black clear bottom plate, an arachidonic acid/NaOH solution was added to each well using a multichannel pipette to start the reaction in each well at the same time. Fluorescence (Ex/Em = 535/587 nm) was measured in a kinetic mode using the Tecan Microplate Reader (Tecan, Switzerland) reading every minute for > 1 h.
Statistical Analysis
Results are presented as mean ± standard error of mean (SEM) for all groups. Comparisons made in the MTS cell proliferation assay were made using two-way ANOVA with Bonferroni post hoc correction. Comparisons between groups for gene expression, COX2 activity and apoptosis/necrosis were made using one-way ANOVA with Bonferroni post hoc correction. P < 0.05 was considered significant.
Results
7KC Induces Cell Death and Loss of Viability in hCMEC/D3 Cells and Protective Effect of ET
The toxicity of 7KC manifested as loss of viable cells by the Trypan Blue assay. Increasing concentrations of 7KC were found to cause a dose-dependent loss of cell viability in endothelial cells with an approximate IC50 of 10 µM (Fig. 1a). This effect was significantly attenuated by co-treatment of cells with ET (Fig. 1b).

a Effect of 7KC on endothelial cells in culture, determined using the trypan blue dye exclusion assay. a dose-dependent cytotoxicity of 7KC. hCMEC/D3 cells were treated with at 0–50 μM 7KC for 24 h. Cell viability is expressed as percentage viability from Vehicle Control. The oxysterol induced significant loss in viable cells at 30-100 μM. Data are represented as mean ± S.E.M. (n = 4). b protective effect of ET on 7KC cytotoxicity. hCMEC/D3 cells were pre-treated with different concentrations of ET for 1 h, followed by co-treatment with 7KC for 24 h. Co-treatment of cells with ET results in modulation of 7KC-induced loss of viable cells. Data are represented as mean ± S.E.M. (n = 4). * Significant difference compared to 7KC (p < 0.01)
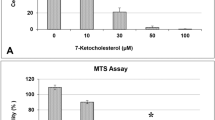
Likewise, 7KC was found to significantly reduce cell viability in endothelial cells by the MTS assay with an approximate IC50 of 30 µM (Fig. 2a). This effect was significantly attenuated by co-treatment of cells with ET (Fig. 2b).

a Effect of 7KC on endothelial cells in culture, determined using the MTS assay. a dose-dependent cytotoxicity of 7KC. hCMEC/D3 cells were treated with at 0–100 μM 7KC for 24 h. Cell viability is expressed as percentage viability from Vehicle Control. The oxysterol induced significant loss in viable cells at 30–100 μM. Data are represented as mean ± S.E.M. (n = 3). b protective effect of ET on 7KC cytotoxicity. hCMEC/D3 cells were pre-treated with different concentrations of ET for 1 h, followed by co-treatment with 7KC for 24 h. Co-treatment of cells with ET results in modulation of 7KC-induced loss of viable cells. Data is represented as mean ± S.E.M. (n = 3). * Significant difference compared to 7KC (p < 0.01)
Apoptosis/Necrosis Assay by Flow Cytometry
7KC was found to significantly increase the number of phosphatidylserine-positive or 7-AAD-positive cells, indicating induction of apoptosis and necrosis in these cells, respectively. ET by itself had no significant effect on apoptosis or necrosis. However, it significantly modulated the induction of apoptosis and necrosis after addition of 7KC (Fig. 3).

a Mode of cell death of hCMEC/D3 human brain endothelial cells after 30 μM 7KC and ET treatment, determined by flow cytometry. a Plot showing effect of 7KC. The oxysterol induced significant increase in percentage of cells that are labelled for phosphatidylserine, indicating apoptosis. At the same time, the oxysterol increased another population of cells that were labelled with 7-AAD, indicating necrosis. Yet a third population of cells showed neither the apoptotic nor necrotic marker. b ET by itself had no significant effect on apoptosis or necrosis; however, it significantly modulated the increase in apoptosis and necrosis after addition of 7KC. Data are represented as mean ± S.E.M. (n = 3). ^ Significant difference compared to controls. *Significant difference compared to 7KC (both p < 0.01)
Quantitative RT-PCR and Immunocytochemistry of ZO-1, Claudin-5 and Occludin
No significant changes in mRNA expression of endothelial tight junction proteins, ZO-1, claudin-5 or occludin, were detected after 7KC or ET treatment (Fig. 4). However, a relocalization of these proteins towards the nucleus of the cell was found after treatment with 7KC by immunocytochemistry.

Quantitative RT-PCR of Zonula occludens-1 (ZO-1), claudin-5 and occludin (OCLN) mRNA expression after 7KC and or ET treatment. hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 30 μM 7KC for 24 h. No significant changes in mRNA expression of endothelial tight junction proteins, ZO-1, claudin-5 or occludin were detected. Data are represented as mean ± S.E.M (n = 5)
ET by itself had no significant effect on mRNA expression of ZO-1, claudin-5 or occludin. However, it modulated the central concentration of the tight junction proteins ZO-1 and claudin-5 after addition of 7KC (Fig. 5).

Effect of 7KC and ET on cellular localization of blood–brain barrier tight junction proteins, determined by immunocytochemistry. hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 50 μM 7KC for 24 h. 7KC induced a movement of ZO-1 and claudin-5 towards the nucleus, but this change of distribution was abolished by ET. Scale bar = 40 μm
Quantitative RT-PCR of eNOS and iNOS
No significant changes in mRNA expression of endothelial nitric oxide synthase (eNOS) or inducible nitric oxide synthase (iNOS) were detected after treatment with 7KC alone. Treatment with 7KC and ET resulted in a significant increase in eNOS expression (Fig. 6).

Quantitative RT-PCR of eNOS a or iNOS b mRNA expression after 7KC and /or ET treatment. hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 30 μM 7KC for 24 h. No significant changes in mRNA expression of these two NOS isoforms were detected. Data are represented as mean ± S.E.M (n = 4). * Significant difference compared to 7KC (p < 0.01)
Quantitative RT-PCR of Inflammatory Genes
7KC was found to significantly increase the expression of the pro-inflammatory genes IL-1β, IL-6, IL-8, TNF-α, NF-kB and COX-2 by quantitative RT-PCR (Figs. 7 and 8). ET by itself had no significant effect on mRNA expression of these pro-inflammatory genes. However, ET significantly attenuated the increase in pro-inflammatory gene expression after addition of 7KC. The effect of ET was blocked by the ET transporter inhibitor VHCl, indicating that this compound needs to be taken up into the endothelial cells to have its anti-inflammatory effect (Fig. 8).

Effect of ET on 7KC-induced increase in mRNA expression of pro-inflammatory genes, determined by real-time RT-PCR. hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 30 μM 7KC for 24 h. Co-treatment of cells with ET results in modulation of 7KC-induced increase in IL-1β (a), IL-6 (b), IL-8 (c) and TNFα (d) mRNA expression in endothelial cells. Data are represented as mean ± S.E.M. ^ Significant difference compared to controls. * Significant difference compared to 7KC (both p < 0.01) (n = 6)

Effect of ET on 7KC-induced increase in mRNA expression of pro-inflammatory genes, determined by real-time RT-PCR. hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 50 μM 7KC for 24 h. Co-treatment of cells with ET results in modulation of 7KC-induced increase in NFkB and component genes (a), as well as COX-2 (b) mRNA expression in endothelial cells. This effect was abolished by concurrent treatment with an inhibitor of ET transporter, VHCl. Data are represented as mean ± S.E.M. (n = 4). ^ Significant difference compared to controls. * Significant difference compared to 7KC. # Significant difference compared to ET + 7KC (all p < 0.01)
COX-2 Enzymatic Activity Assay
7KC induced significant increase in the COX-2 activity. ET by itself had no significant effect on COX-2 activity. However, it significantly attenuated the increase in COX-2 activity induced by 7KC (Fig. 9).

Effect of ET on 7KC-induced increase in COX-2 mRNA expression (A) and COX-2 enzymatic activity (B) hCMEC/D3 cells were pre-treated with 1 mM ET for 1 h, followed by co-treatment with 50 μM 7KC for 24 h. Co-treatment of cells with ET results in modulation of 7KC-induced increase in COX-2 expression and activity. Data are represented as mean ± S.E.M. (n = 4). ^ Significant difference compared to controls. * Significant difference compared to 7KC (all p < 0.01)
Discussion
This study was carried out to elucidate the possible effects of 7KC on brain endothelial cells, and effects of ET on such damage. 7KC was found to be toxic to brain endothelial cells in culture. The oxysterol induced significant reduction in number of viable cells as shown by cell counts and MTS assay at 30 μM and above. It is possible for 7KC to be accumulated to concentrations above 100 μM in atherosclerotic lesions (Brown et al. 2000); hence, the 7KC concentrations tested in this study are reflective of potential physiological 7KC levels. The observed 7KC-induced reduced cell viability are also supported by findings in other endothelial cell lines, including Eahy926 (EAHY) endothelial cells (Chang et al. 2016) and human aortic endothelial cells (Chalubinski et al. 2013). A similar trend was observed in a previous study, where treatment for 24 h with a 7KC concentration of 25 μM (with serum restriction) led to a decrease in cell viability by 55%, and at 50 μM, cell viability dropped by nearly 75% (Rosa-Fernandes et al. 2017). Our results show that this loss of cell viability can be attenuated by ET, and co-treatment of cells with ET and 7KC resulted in significant increase in percentage of surviving cells. No changes in mRNA expression of ZO-1, claudin-5 or occludin, which are key proteins involved in tight junction formation of brain endothelial cells, were found, after 7KC treatment. Rather, there was a concentration of the tight junction proteins ZO-1 and claudin-5 towards the nucleus of the cells as observed by immunocytochemistry. These results are similar to a previous study in smooth muscle cells, which reported that ZO-1 was translocated to the nucleus following treatment with 7KC (Cho et al. 2018). 7KC and other oxysterols 5α,6α-and 5β,6β-epoxycholesterol, 7α-and 7β-hydroxycholesterol also induced perturbation of spatial localization of the tight junction proteins, such as zonula occludens-1 (ZO-1), occludin and Junction Adhesion Molecule-A (JAM-A) in differentiated CaCo-2 cells (Deiana et al. 2017). Disturbance of the proper localization of tight junction proteins in brain endothelial cells might affect BBB function.
The mode of cell death, after 7KC treatment was then determined. This is in view of potentially conflicting reports that 7KC causes endothelial cell death by apoptosis (Zhou et al. 2006; W. Li et al. 2011), or necrosis (Ghelli et al. 2002; Hans et al. 2008). In this study, we directly compared the mechanism(s) of cell death in the same experiment by flow cytometry. An increase in apoptosis, as seen by an increase in the population of phosphatidylserine-positive endothelial cells, was found after 7KC treatment. This is consistent with previous studies which showed that 7KC induces ROS production, ATM/Chk2, ATR-Chk1 and p53 signalling pathways, G0/G1 cell cycle arrest and apoptosis in endothelial cells (Chang et al. 2016) and human vascular smooth muscle cells (Pedruzzi et al. 2004; Ohtsuka et al. 2006). Interestingly, however, an almost equal number of cells were found to be positive for 7-AAD, which is a marker for necrosis. Increase in both these markers therefore indicate that 7KC induces both apoptosis and necrosis in brain endothelial cells. ET had not effect by itself but significantly reduced 7KC-induced cell death by both apoptosis and necrosis. It is nonetheless possible that apoptosis and necrosis are not the only forms of cell death induced by 7KC.
Since 7KC is thought to be an inflammatory mediator in cardiovascular disease, we sought to determine whether 7KC could induce inflammation. A marked increase in the pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α gene expression was observed after treatment with 30 μM and 50 μM 7KC, indicating that 7KC induces inflammation in hCMEC/D3 brain endothelial cells. These results are consistent with an observed induction of inflammation by 7KC in other cell types. For example, 7KC was reported to induce apoptosis and release pro-inflammatory cytokines IL-1β and TNFα, and increase expression of adhesion molecules ICAM-1, VCAM-1 and E-selectin in HUVECs (Lemaire et al. 1998). In another study, it was demonstrated that IL-8 expression was increased with the increasing concentrations of 7KC in HUVECs (Chang et al. 2016). 7KC has been found to induce the expression of IL-1β, IL-6 and IL-8 in human retinal pigment epithelial cells with similar expression fold changes as that reported in this study (4- to 18-fold increases) (Huang et al. 2014). Increased NF-κB translocation is found during 7KC-induced inflammation (Larrayoz et al. 2010) which could give rise to an upregulation of cytokines, chemokines and adhesion molecules (Lawrence 2009). 7KC has also been shown to increase inflammation by activating Nlrp3 inflammasomes in endothelial cells (X. Li et al. 2014; Koka et al. 2017; Yuan et al. 2018).
We next examined whether the 7KC-induced increase in inflammation could be modulated by ET. Co-treatment with ET significantly attenuated 7KC-induced induction of pro-inflammatory genes, IL-1β, IL-6, IL-8, TNFα, NF-kB and COX2. The cyclooxygenase (COX) family of enzymes (COX1 and COX2) catalyse the rate-limiting step in the synthesis of prostaglandins. COX1 is constitutively expressed and involved in homeostasis, whereas COX2 is the inducible cyclooxygenase that is upregulated during inflammation, and plays a critical role in generation of proinflammatory eicosanoids. COX2 gene expression is induced by cytokines (Seibert and Masferrer 1994), including IL-1β (Dinarello 2002) and TNFα (Qiu et al. 2016). Since an upregulation of TNF-α gene expression was found after 7KC treatment, we postulate that this could lead to increased COX2 expression and activity. It was found that 7KC induces not only an increase in COX2 mRNA expression but also an increase in COX2 enzymatic activity. Importantly, ET significantly attenuated the 7KC-induced increase in both COX2 mRNA expression and activity. Moreover, the protective effect of ET was significantly reduced by co-incubation with an inhibitor of OCTN1 (VHCL). This shows that ET needs to enter the endothelial cells to have a protective effect, and not perhaps act via extracellular neutralizing of 7KC. Further studies are necessary to determine whether ET inhibits COX-2 directly.
Since 7KC is mainly formed by free radical attack on cholesterol, its ability to produce inflammation may be a way in which the oxysterol could propagate oxidative stress. COX2 metabolizes arachidonic acid to produce eicosanoids which are proinflammatory in nature. Moreover, free radicals, such as superoxide radicals, are formed by the metabolism of arachidonic acid (Marks and Furstenberger 2000). Although this study focusses on endothelial cells, it is likely that this process could also occur in other cell types of the body. The findings of a potent anti-inflammatory properties of ET could position it as a therapeutic candidate for treatment of inflammatory diseases. Besides ET, phytochemicals from a large number of plants around the world have been reported to have anti-inflammatory effects (Reviewed in (W.Y. Ong et al. 2017).
7KC can be taken up through the diet, e.g. meat products that are exposed to sunlight and heat (Hernandez Becerra et al. 2014). However, more recent studies indicate that much of the dietary 7KC is metabolized by the liver (Lyons et al. 1999), and that most circulating 7KC is endogenously produced by oxidation of cholesterol within the body, e.g. in hypercholesterolemia or diabetes mellitus (Murakami et al. 2000). A protective effect on brain endothelial cells suggests that ET might be useful as a nutraceutical for the prevention or management of neurovascular diseases, such as stroke or vascular dementia. Moreover, the ability of ET to cross the BBB could point to its usefulness in combatting 7KC that is produced in the CNS during neuroinflammation (W. Y. Ong et al. 2003; He et al. 2006; Kim et al. 2010), e.g. in chronic neurodegenerative diseases (Testa et al. 2016) and possibly COVID-19-induced neurologic complications (Cheah and Halliwell 2020; W. Y. Ong et al. 2020).
Further studies are necessary to elucidate the detailed mechanism of the protective effect of ET against 7KC-induced inflammation.
References
Akanmu, D., Cecchini, R., Aruoma, O. I., & Halliwell, B. (1991). The antioxidant action of ergothioneine. Archives of Biochemistry and Biophysics, 288(1), 10–16. https://doi.org/10.1016/0003-9861(91)90158-f
Anderson, A., Campo, A., Fulton, E., Corwin, A., Jerome, W. G., 3rd., & O’Connor, M. S. (2020). 7-Ketocholesterol in disease and aging. Redox Biol, 29, 101380. https://doi.org/10.1016/j.redox.2019.101380
Anticoli, S., Arciello, M., Mancinetti, A., De Martinis, M., Ginaldi, L., Iuliano, L., et al. (2010). 7-ketocholesterol and 5,6-secosterol modulate differently the stress-activated mitogen-activated protein kinases (MAPKs) in liver cells. Journal of Cellular Physiology, 222(3), 586–595. https://doi.org/10.1002/jcp.21972
Brown, A. J., Watts, G. F., Burnett, J. R., Dean, R. T., & Jessup, W. (2000). Sterol 27-hydroxylase acts on 7-ketocholesterol in human atherosclerotic lesions and macrophages in culture. Journal of Biological Chemistry, 275(36), 27627–27633. https://doi.org/10.1074/jbc.M004060200
Butterfield, D. A., & Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience, 20(3), 148–160. https://doi.org/10.1038/s41583-019-0132-6
Chalubinski, M., Zemanek, K., Skowron, W., Wojdan, K., Gorzelak, P., & Broncel, M. (2013). The effect of 7-ketocholesterol and 25-hydroxycholesterol on the integrity of the human aortic endothelial and intestinal epithelial barriers. Inflammation Research, 62(12), 1015–1023. https://doi.org/10.1007/s00011-013-0660-x
Chang, M. C., Chen, Y. J., Liou, E. J., Tseng, W. Y., Chan, C. P., Lin, H. J., et al. (2016). 7-Ketocholesterol induces ATM/ATR, Chk1/Chk2, PI3K/Akt signalings, cytotoxicity and IL-8 production in endothelial cells. Oncotarget, 7(46), 74473–74483. https://doi.org/10.18632/oncotarget.12578
Cheah, I. K., & Halliwell, B. (2020). Could Ergothioneine Aid in the Treatment of Coronavirus Patients? Antioxidants (Basel). https://doi.org/10.3390/antiox9070595
Cheah, I. K., Tang, R. M., Yew, T. S., Lim, K. H., & Halliwell, B. (2017). Administration of Pure Ergothioneine to Healthy Human Subjects: Uptake, Metabolism, and Effects on Biomarkers of Oxidative Damage and Inflammation. Antioxidants & Redox Signaling, 26(5), 193–206. https://doi.org/10.1089/ars.2016.6778
Cho, H. R., Kim, K., An, W. G., Eo, S. K., Bae, S. S., Kim, C. D., et al. (2018). 7-Oxygenated cholesterol molecules differentially affect the expression of zonula occludens-1 in vascular smooth muscle cells and monocyte/macrophage cells. Biochemical and Biophysical Research Communications, 497(2), 521–526. https://doi.org/10.1016/j.bbrc.2018.02.064
Deckert, V., Duverneuil, L., Poupon, S., Monier, S., Le Guern, N., Lizard, G., et al. (2002). The impairment of endothelium-dependent arterial relaxation by 7-ketocholesterol is associated with an early activation of protein kinase C. British Journal of Pharmacology, 137(5), 655–662. https://doi.org/10.1038/sj.bjp.0704920
Deiana, M., Calfapietra, S., Incani, A., Atzeri, A., Rossin, D., Loi, R., et al. (2017). Derangement of intestinal epithelial cell monolayer by dietary cholesterol oxidation products. Free Radic Biol Med, 113, 539–550. https://doi.org/10.1016/j.freeradbiomed.2017.10.390
Dinarello, C. A. (2002). The IL-1 family and inflammatory diseases. Clinical and Experimental Rheumatology, 20(5 Suppl 27), S1-13.
Ghelli, A., Porcelli, A. M., Zanna, C., & Rugolo, M. (2002). 7-Ketocholesterol and staurosporine induce opposite changes in intracellular pH, associated with distinct types of cell death in ECV304 cells. Archives of Biochemistry and Biophysics, 402(2), 208–217. https://doi.org/10.1016/S0003-9861(02)00085-1
Grigat, S., Harlfinger, S., Pal, S., Striebinger, R., Golz, S., Geerts, A., et al. (2007). Probing the substrate specificity of the ergothioneine transporter with methimazole, hercynine, and organic cations. Biochemical Pharmacology, 74(2), 309–316. https://doi.org/10.1016/j.bcp.2007.04.015
Grundemann, D., Harlfinger, S., Golz, S., Geerts, A., Lazar, A., Berkels, R., et al. (2005). Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A, 102(14), 5256–5261. https://doi.org/10.1073/pnas.0408624102
Halliwell, B. (2006). Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry, 97(6), 1634–1658. https://doi.org/10.1111/j.1471-4159.2006.03907.x
Halliwell, B., Cheah, I. K., & Tang, R. M. Y. (2018). Ergothioneine - a diet-derived antioxidant with therapeutic potential. FEBS Letters, 592(20), 3357–3366. https://doi.org/10.1002/1873-3468.13123
Hans, C. P., Zerfaoui, M., Naura, A. S., Catling, A., & Boulares, A. H. (2008). Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovascular Research, 78(3), 429–439. https://doi.org/10.1093/cvr/cvn018
He, X., Jenner, A. M., Ong, W. Y., Farooqui, A. A., & Patel, S. C. (2006). Lovastatin modulates increased cholesterol and oxysterol levels and has a neuroprotective effect on rat hippocampal neurons after kainate injury. Journal of Neuropathology and Experimental Neurology, 65(7), 652–663. https://doi.org/10.1097/01.jnen.0000225906.82428.69
Hernandez Becerra, J. A., Ochoa Flores, A. A., Valerio-Alfaro, G., Soto-Rodriguez, I., Rodriguez-Estrada, M. T., & Garcia, H. S. (2014). Cholesterol oxidation and astaxanthin degradation in shrimp during sun drying and storage. Food Chemistry, 145, 832–839. https://doi.org/10.1016/j.foodchem.2013.08.098
Hitsumoto, T., Takahashi, M., Iizuka, T., & Shirai, K. (2009). Clinical significance of serum 7-ketocholesterol concentrations in the progression of coronary atherosclerosis. J Atheroscler Thromb, 16(4), 363–370. https://doi.org/10.5551/jat.no703
Huang, J. D., Amaral, J., Lee, J. W., & Rodriguez, I. R. (2014). 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS ONE, 9(7), e100985. https://doi.org/10.1371/journal.pone.0100985
Hunt, N. J., Lockwood, G. P., Warren, A., Mao, H., McCourt, P. A. G., Le Couteur, D. G., et al. (2019). Manipulating fenestrations in young and old liver sinusoidal endothelial cells. American Journal of Physiology. Gastrointestinal and Liver Physiology, 316(1), G144–G154. https://doi.org/10.1152/ajpgi.00179.2018
Kato, Y., Kubo, Y., Iwata, D., Kato, S., Sudo, T., Sugiura, T., et al. (2010). Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharmaceutical Research, 27(5), 832–840. https://doi.org/10.1007/s11095-010-0076-z
Kim, J. H., Jittiwat, J., Ong, W. Y., Farooqui, A. A., & Jenner, A. M. (2010). Changes in cholesterol biosynthetic and transport pathways after excitotoxicity. Journal of Neurochemistry, 112(1), 34–41. https://doi.org/10.1111/j.1471-4159.2009.06449.x
Koka, S., Xia, M., Chen, Y., Bhat, O. M., Yuan, X., Boini, K. M., et al. (2017). Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol, 13, 336–344. https://doi.org/10.1016/j.redox.2017.06.004
Larrayoz, I. M., Huang, J. D., Lee, J. W., Pascual, I., & Rodriguez, I. R. (2010). 7-ketocholesterol-induced inflammation: involvement of multiple kinase signaling pathways via NFkappaB but independently of reactive oxygen species formation. Investigative Ophthalmology & Visual Science, 51(10), 4942–4955. https://doi.org/10.1167/iovs.09-4854
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol, 1(6), a001651. https://doi.org/10.1101/cshperspect.a001651
Lemaire, S., Lizard, G., Monier, S., Miguet, C., Gueldry, S., Volot, F., et al. (1998). Different patterns of IL-1beta secretion, adhesion molecule expression and apoptosis induction in human endothelial cells treated with 7alpha-, 7beta-hydroxycholesterol, or 7-ketocholesterol. FEBS Letters, 440(3), 434–439. https://doi.org/10.1016/s0014-5793(98)01496-3
Li, G., Shofer, J. B., Rhew, I. C., Kukull, W. A., Peskind, E. R., McCormick, W., et al. (2010). Age-varying association between statin use and incident Alzheimer’s disease. Journal of the American Geriatrics Society, 58(7), 1311–1317. https://doi.org/10.1111/j.1532-5415.2010.02906.x
Li, W., Ghosh, M., Eftekhari, S., & Yuan, X. M. (2011). Lipid accumulation and lysosomal pathways contribute to dysfunction and apoptosis of human endothelial cells caused by 7-oxysterols. Biochemical and Biophysical Research Communications, 409(4), 711–716. https://doi.org/10.1016/j.bbrc.2011.05.071
Li, X., Zhang, Y., Xia, M., Gulbins, E., Boini, K. M., & Li, P. L. (2014). Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS ONE, 9(1), e87552. https://doi.org/10.1371/journal.pone.0087552
Lyons, M. A., Samman, S., Gatto, L., & Brown, A. J. (1999). Rapid hepatic metabolism of 7-ketocholesterol in vivo: implications for dietary oxysterols. Journal of Lipid Research, 40(10), 1846–1857.
Marks, F., & Furstenberger, G. (2000). Cancer chemoprevention through interruption of multistage carcinogenesis. The lessons learnt by comparing mouse skin carcinogenesis and human large bowel cancer. European Journal of Cancer, 36(3), 314–329. https://doi.org/10.1016/s0959-8049(99)00318-4
Murakami, H., Tamasawa, N., Matsui, J., Yasujima, M., & Suda, T. (2000). Plasma oxysterols and tocopherol in patients with diabetes mellitus and hyperlipidemia. Lipids, 35(3), 333–338. https://doi.org/10.1007/s11745-000-0530-1
Ohtsuka, M., Miyashita, Y., & Shirai, K. (2006). Lipids deposited in human atheromatous lesions induce apoptosis of human vascular smooth muscle cells. J Atheroscler Thromb, 13(5), 256–262. https://doi.org/10.5551/jat.13.256
Ong, W. Y., Farooqui, T., Ho, C. F. Y., Ng, Y. K., & Farooqui, A. A. (2017). Use of Phytochemicals against Neuroinflammation. In Neuroprotective Effects of Phytochemicals in Neurological Disorders: Wiley.
Ong, W. Y., Go, M. L., Wang, D. Y., Cheah, I. K., & Halliwell, B. (2020). Effects of Antimalarial Drugs on Neuroinflammation-Potential Use for Treatment of COVID-19-Related Neurologic Complications. Molecular Neurobiology. https://doi.org/10.1007/s12035-020-02093-z
Ong, W. Y., Goh, E. W., Lu, X. R., Farooqui, A. A., Patel, S. C., & Halliwell, B. (2003). Increase in cholesterol and cholesterol oxidation products, and role of cholesterol oxidation products in kainate-induced neuronal injury. Brain Pathology, 13(3), 250–262. https://doi.org/10.1111/j.1750-3639.2003.tb00026.x
Paul, B. D., & Snyder, S. H. (2010). The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death and Differentiation, 17(7), 1134–1140. https://doi.org/10.1038/cdd.2009.163
Pedruzzi, E., Guichard, C., Ollivier, V., Driss, F., Fay, M., Prunet, C., et al. (2004). NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Molecular and Cellular Biology, 24(24), 10703–10717. https://doi.org/10.1128/MCB.24.24.10703-10717.2004
Qiu, J., Yuan, H., Chen, S., Zhou, Y., Song, D., & Chen, R. (2016). TNFalpha up-regulates COX-2 in chronic progressive nephropathy through nuclear accumulation of RelB and NF-kappaB2. Archives of Physiology and Biochemistry, 122(2), 88–93. https://doi.org/10.3109/13813455.2016.1141961
Romeo, G. R., & Kazlauskas, A. (2008). Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. Journal of Biological Chemistry, 283(15), 9595–9605. https://doi.org/10.1074/jbc.M710092200
Rosa-Fernandes, L., Maselli, L. M. F., Maeda, N. Y., Palmisano, G., & Bydlowski, S. P. (2017). Outside-in, inside-out: Proteomic analysis of endothelial stress mediated by 7-ketocholesterol. Chemistry and Physics of Lipids, 207(Pt B), 231–238. https://doi.org/10.1016/j.chemphyslip.2017.06.008
Samadi, A., Gurlek, A., Sendur, S. N., Karahan, S., Akbiyik, F., & Lay, I. (2019). Oxysterol species: reliable markers of oxidative stress in diabetes mellitus. Journal of Endocrinological Investigation, 42(1), 7–17. https://doi.org/10.1007/s40618-018-0873-5
Seibert, K., & Masferrer, J. L. (1994). Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor, 4(1), 17–23.
Song, J., Wang, D., Chen, H., Huang, X., Zhong, Y., Jiang, N., et al. (2017). Association of Plasma 7-Ketocholesterol With Cardiovascular Outcomes and Total Mortality in Patients With Coronary Artery Disease. Circulation Research, 120(10), 1622–1631. https://doi.org/10.1161/CIRCRESAHA.117.311049
Svistounov, D., Warren, A., McNerney, G. P., Owen, D. M., Zencak, D., Zykova, S. N., et al. (2012). The Relationship between fenestrations, sieve plates and rafts in liver sinusoidal endothelial cells. PLoS ONE, 7(9), e46134. https://doi.org/10.1371/journal.pone.0046134
Tang, R. M. Y., Cheah, I. K., Yew, T. S. K., & Halliwell, B. (2018). Distribution and accumulation of dietary ergothioneine and its metabolites in mouse tissues. Sci Rep, 8(1), 1601. https://doi.org/10.1038/s41598-018-20021-z
Tani, M., Kamata, Y., Deushi, M., Osaka, M., & Yoshida, M. (2018). 7-Ketocholesterol enhances leukocyte adhesion to endothelial cells via p38MAPK pathway. PLoS ONE, 13(7), e0200499. https://doi.org/10.1371/journal.pone.0200499
Testa, G., Staurenghi, E., Zerbinati, C., Gargiulo, S., Iuliano, L., Giaccone, G., et al. (2016). Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol, 10, 24–33. https://doi.org/10.1016/j.redox.2016.09.001
Tucker, R. A. J., Cheah, I. K., & Halliwell, B. (2019). Specificity of the ergothioneine transporter natively expressed in HeLa cells. Biochemical and Biophysical Research Communications, 513(1), 22–27. https://doi.org/10.1016/j.bbrc.2019.02.122
Vicen, M., Vitverova, B., Havelek, R., Blazickova, K., Machacek, M., Rathouska, J., et al. (2019). Regulation and role of endoglin in cholesterol-induced endothelial and vascular dysfunction in vivo and in vitro. The FASEB Journal, 33(5), 6099–6114. https://doi.org/10.1096/fj.201802245R
Wang, M., Long, W., Li, D., Wang, D., Zhong, Y., Mu, D., et al. (2017). Plasma 7-ketocholesterol levels and the risk of incident cardiovascular events. Heart, 103(22), 1788–1794. https://doi.org/10.1136/heartjnl-2016-310914
Weksler, B., Romero, I. A., & Couraud, P. O. (2013). The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS, 10(1), 16. https://doi.org/10.1186/2045-8118-10-16
Weksler, B. B., Subileau, E. A., Perriere, N., Charneau, P., Holloway, K., Leveque, M., et al. (2005). Blood-brain barrier-specific properties of a human adult brain endothelial cell line. The FASEB Journal, 19(13), 1872–1874. https://doi.org/10.1096/fj.04-3458fje
Yuan, X., Wang, L., Bhat, O. M., Lohner, H., & Li, P. L. (2018). Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: Antioxidant action of butyrate. Redox Biol, 16, 21–31. https://doi.org/10.1016/j.redox.2018.02.007
Zhou, L., Shi, M., Guo, Z., Brisbon, W., Hoover, R., & Yang, H. (2006). Different cytotoxic injuries induced by lysophosphatidylcholine and 7-ketocholesterol in mouse endothelial cells. Endothelium, 13(3), 213–226. https://doi.org/10.1080/10623320600780926
Acknowledgements
This work was supported by grants from the Ministry of Education, Singapore (R-181-000-183-114, WYO) and the National University Health System, Singapore (NUHSRO/2019/051/T1/Seed-Mar/04, WYO).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Koh, S.S., Ooi, S.CY., Lui, N.MY. et al. Effect of Ergothioneine on 7-Ketocholesterol-Induced Endothelial Injury. Neuromol Med 23, 184–198 (2021). https://doi.org/10.1007/s12017-020-08620-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-020-08620-4