
Abstract
From the clinical standpoint, systemic sclerosis (SSc) is characterized by skin and internal organ fibrosis, diffuse fibroproliferative vascular modifications, and autoimmunity. Clinical presentation and course are highly heterogenous and life expectancy variably affected mostly dependent on lung and heart involvement. SSc touches more women than men with differences in disease severity and environmental exposure. Pathogenetic events originate from altered homeostasis favored by genetic predisposition, environmental cues and a variety of endogenous and exogenous triggers. Epigenetic modifications modulate SSc pathogenesis which strikingly associate profound immune-inflammatory dysregulation, abnormal endothelial cell behavior, and cell trans-differentiation into myofibroblasts. SSc myofibroblasts show enhanced survival and enhanced extracellular matrix deposition presenting altered structure and altered physicochemical properties. Additional cell types of likely pathogenic importance are pericytes, platelets, and keratinocytes in conjunction with their relationship with vessel wall cells and fibroblasts. In SSc, the profibrotic milieu is favored by cell signaling initiated in the one hand by transforming growth factor-beta and related cytokines and in the other hand by innate and adaptive type 2 immune responses. Radical oxygen species and invariant receptors sensing danger participate to altered cell behavior. Conventional and SSc-specific T cell subsets modulate both fibroblasts as well as endothelial cell dysfunction. Beside autoantibodies directed against ubiquitous antigens important for enhanced clinical classification, antigen-specific agonistic autoantibodies may have a pathogenic role. Recent studies based on single-cell RNAseq and multi-omics approaches are revealing unforeseen heterogeneity in SSc cell differentiation and functional states. Advances in system biology applied to the wealth of data generated by unbiased screening are allowing to subgroup patients based on distinct pathogenic mechanisms. Deciphering heterogeneity in pathogenic mechanisms will pave the way to highly needed personalized therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic sclerosis (SSc) is clinically characterized in the one hand by fibrosis of skin and internal organs leading to altered organ structure and ultimately organ dysfunction and on the other hand by functional and structural vasculopathy resulting among others in Raynaud phenomenon, digital ulcers, pulmonary artery hypertension, and renal crisis [1]. In SSc, fibrosis and vasculopathy are intimately associated and lead to highly heterogeneous clinical manifestations with a widely variable prognosis. Main causes of death are lung and heart involvement which may occur early or late in the disease course [2]. Standardized mortality rates range from 2.82 to 3.64 in the most recent meta-analysis [3]. In addition, SSc imposes high burden in terms of quality of life and social cost.
Inflammation is the physiological response to altered tissue and organ homeostasis and is the common denominator to SSc pathogenesis. We believe that inflammatory processes are keys to initiation and progression toward both fibrosis and structural vasculopathy in response to events perturbing homeostasis. However, deciphering the multiple components of inflammation, which simultaneously act in many different, often opposing, directions remains an important aim to understand SSc pathophysiology.
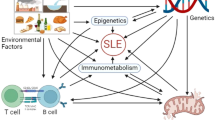
From the pathogenic point of view, the questions to be answered are many and should address the predisposing genetic background, the trigger(s) as well as the mechanisms involved in the initiation, and further development of both fibrosis and vasculopathy simultaneously taking into account clinical heterogeneity (Fig. 1). The term of “intermediate pathophenotypes” has been proposed by C. Feghali-Bostwick and J. Varga to accommodate the dynamic processes underlying heterogeneity in SSc and our understanding of the mechanisms involved in SSc pathogenesis at cellular and tissue levels.

Overview of conditions and events leading to systemic sclerosis. Schematic diagram highlighting the complex interplay thought to play a role in susceptibility and initiation of SSc in which genetic predisposition and environmental cues under the pressure of a variety of triggers lead to perturbed homeostasis with ensuing autoimmunity. Autoimmunity is represented as the common denominator of the three fundamental aspects of SSc: inflammation, vasculopathy, and fibrosis. Heterogeneous clinical manifestations would then develop according to variable amplification mechanisms resulting in recognized clinical subsets and organ damage
Excellent reviews have been recently published addressing various aspects of SSc pathogenesis [4,5,6,7,8,9,10,11,12,13]. Here, we will attempt to provide a synthetic view of the main aspects of SSc pathogenesis.
SSc Disease State
The most commonly postulated model of disease progression in SSc is sequential, with immune activation and subsequent vasculopathy leading to activation of fibroblasts and fibrosis as the end effect of these processes. However, substantial debate animates the SSc community on what order these events take place. According to the definition provided by Stern and Denton, the disease state is only tolerated if there is simultaneous dysregulation of the immune system, vascular endothelium, and connective tissue repair system [6]. Thus, SSc can be viewed as a three-leg pathology in which major dysfunctional cell types are immune cells, endothelial cells, and fibroblasts which intensely interact mostly via soluble mediators directly or indirectly leading to myo-fibroblast hyperactivation. This cell and soluble factor three-leg network establishes further interactions with many other cell types of which keratinocytes, pericytes, platelets, and adipocytes have attracted particular attention in recent years (Fig. 2).

Major cell types and their multiple interactions in SSc pathogenesis. SSc is here viewed as a three-leg pathology in which major dysfunctional cell types are immune cells, endothelial cells, and fibroblasts which directly or indirectly intensely interact leading to myofibroblast hyperactivation. This cell and soluble factor three-leg network establishes further interactions with many other cell types including adipocytes, keratinocytes, pericytes, and platelets. The concentric reddish shadow highlights the influence of the various cell types on the activation of myofibroblasts. Two-head red arrows indicate multiple, reciprocal interactions mainly ensured by soluble mediators of inflammation. Dashed arrow indicates increase in extracellular matrix (ECM) deposition by myofibroblasts
SSc Genetic Background
Family Studies
Compared to the general population, the risk of developing SSc is higher in first-degree relatives of persons suffering of SSc and strong family clustering, with an estimated risk of 1.6% versus a 0.026% [14]. However, the concordance rate for clinical disease in twins is relatively low (4.7% in one study) with higher frequency in concordance for the presence of autoantibodies and T cell responses irrespective of clinical expression [14,15,16]. This is strong evidence for the heritability of SSc, at the same time indicating a weak association with disease phenotype.
HLA
Systemic autoimmunity is favored by a genetic background in which genes and gene polymorphisms associated with the major histopathologic complex (MHC) or human leucocyte antigens (HLAs) are of major importance. This is the case also for SSc, but most interestingly, the associations between HLA haplotypes and SSc vary according to ethnicity and autoantibody (autoAb) status. Thus, risk alleles may be different in Fareast Asia compared to Europe or America and within USA according to ethnic origin. For example, in European Americans (EA) and Latino Americans (LA), the DRB1*1104, DQA1*0501, DQB1*0301 haplotype, and DQB1 alleles encoding a non-leucine residue at position 26 (DQB1 26 epi) showed the strongest associations with SSc, while the strongest association for African Americans (AA) was with DRB1*0804 and HLA-DRB1*1102. DRB1*0804, DQA1*0501, DQB1*0301, and DPB1*1301 alleles showed the highest odds ratio for anti-topoisomerase autoAb (ATA) (OR = 14) and HLA-DRB1*0804 for antifibrillarin autoAb (AFA) (odds ratio = 7.4) in AA. The anti-centromere autoAb (ACA) were best explained by DQB1*0501 and DQB1*26 epi alleles and anti-RNA polymerase autoAb (ARA) by DRB1*0404, DRB1*11, and DQB1*03 alleles in EA and LA subjects. Nonetheless, HLA-DPB1*1301 allele was associated with the ATA+ in both AA and EA patients demonstrating a transancestry effect [17, 18].
GWAS
In addition to the impact of HLA genes, candidate gene approaches and more substantially genome-wide association studies (GWAS) by assessing SNP (single nucleotide polymorphism) associations provided evidence on the contribution of chromosomal locations to the risk of developing SSc. Interestingly, most of the identified genetic regions which polymorphisms are associated with SSc involve intronic or intergenic regions. Recent evidence suggests that these regions may have regulatory function by interacting with gene promoters or enhancers. A recent collaborative effort, by applying a meta-analysis on 14 independent European cohorts comprising a total of 26,679 individuals (9095 SSc patients and 17,584 healthy controls) has identified 23 genomic regions significantly associated with SSc of which 12 most likely causal [19]. Interestingly, these authors identified 43 robust target genes of these regions, thus showing that the expression of more than one gene is influenced by these polymorphisms. Remarkably, the majority of recognized polymorphisms are relevant for the immune response particularly associated with five main molecular pathways identified by in silico analysis: (a) IFN-I signaling pathway, (b) T cell activation, (c) B cell activation, (d) NFkB pathway, and (e) immune system process. However, additional susceptibility genes are relevant for endothelial cells therefore potentially associated with vasculopathy, and fibroblasts with fibrosis. Table 1 inspired from [20] reports enriched SSc risk genes, their function, and cells likely involved.
Epigenetic Regulation
Substantial differences have been demonstrated in the epigenetic tags when SSc were compared to healthy fibroblasts. In one study, hypomethylated genes included ITGA9, encoding an α integrin and other relevant genes such as ADAM12, COL23A1, COL4A2, and MYO1E, and transcription factors genes RUNX1, RUNX2, and RUNX3 were hypomethylated in both dSSc and lSSc. Pathway analysis of differentially methylated genes in both dSSc and lSSc revealed enrichment of genes involved in extracellular matrix-receptor interaction and focal adhesion [21]. Another study focusing on Wnt signaling in mononuclear cells and fibroblasts found that the promoters of DKK1 (Dickkopf WNT signaling pathway inhibitor 1) and SFRP1 (secreted frizzled-related protein 1) were hypermethylated in SSc. Promoter hypermethylation resulted in impaired transcription and decreased expression of DKK1 and SFRP1 in SSc [22]. Since DKK1 is an inhibitor of the Wnt/β-catenin signaling cascade which deeply involved in fibrosis development, decreased DKK1 expression may account for greater pro-fibrotic signaling.
Gene transcription is also regulated by miRNA, of which some have been associated with SSc; miR-21 and miR-155 appear to have profibrotic properties, while let-7 and miR-29 are rather profibrotic. In addition, a significant decrease in the levels of miR-29 has been found in lesional SSc skin [23]. Increased expression of miR-92a was reported in SSc fibroblasts resulting in reduced MMP-1 expression [24].
SSc Triggers
Within genetic susceptibility, many triggers may be involved in disease initiation. They may operate sequentially and manifest gender preferences.
Chemicals
The association between environmental risk factors and SSc has been extensively analyzed, but the role of the environment is not yet fully understood [25, 26]. Environmental factors can be classified as occupational (silica, organic solvents) and non-occupational/non-infectious (drugs, pesticides, silicones, heavy metals) [25, 27]. According to a recent meta-analysis, the strongest evidence indicates that silica and organic solvents are risk factors for SSc. Exposure to vinyl chloride, white spirit, solvents, crystalline silica among others, and use of tryptophane have been associated with SSc or SSc-like disorders [28]. While there is substantial evidence that exposure to silicones is not a risk factor for SSc, the meta-analysis of breast implants exposure highlighted a slight over-risk [overall OR 1.68 (95%CI 1.65–1.71; p < 0.001)]. The risk of SSc following exposure to silica is higher in males compared with females with more frequent diffuse cutaneous SSc and lower survival rates [26, 29].
Infectious Agents
Infectious agents may participate in breaking T and B cell tolerance by molecular mimicry and by the simultaneous activation of innate responses when pathogen-associated molecular patterns (PAMPs) activate pattern recognition receptors (PRRs), thus tuning the immune system to enhanced responses. Immune effector mechanisms may then participate to cell damage. Parvovirus B19, cytomegalovirus (CMV), Epstein–Barr virus (EBV), and retroviruses have all been proposed as initiating triggers of SSc [30, 31]. Particular attention has been attracted by CMV which genetic material has been found in endothelial cells and suspected to elicit IgG that specifically recognized the CMV late protein UL94 and the endothelial cell surface integrin–NAG-2 protein complex, thereby inducing endothelial cell apoptosis [32]. These IgG may also activate fibroblasts and enhance collagen production [33]. The presence of Parvovirus B19 DNA in the bone marrow and/or skin biopsies has been reported. By in situ RT-PCR, the presence of Parvovirus B19 DNA and TNF was demonstrated in endothelium and fibroblasts [34]. While not replicating in fibroblasts, Parvovirus B19 can activate many genes involved in inflammation and fibrosis [35]. Similarly, EBV infection was shown to induce aberrant toll-like receptor (TLR) activation pathway and fibroblast-myofibroblast conversion in scleroderma [36]. From a different angle, when bioinformatically predicting the T cell immunodominant peptides of topoisomerase 1, fibrillarin, and centromere protein A in association with selected HLA α/β allelic heterodimers, it was reported that these autoantigens are homologous to viral protein sequences from the Mimiviridae and Phycodnaviridae families. These data suggest a possible link between HLA alleles, autoantibodies, and infectious triggers in the pathogenesis of SSc [18].
Neoplastic Diseases
SSc has complex relationships with many different types of cancer [9]. A close temporal association between the onset of SSc and the detection of cancer has been described in a subset of patients positive for anti-RNA polymerase III (RNApol III) antibodies [37]. This observation led to the discovery that mutated autoantigens (RNApol3) are present in the tumors obtained from these patients and result in mutant-specific T cell immune responses as well as in the generation cross-reactive autoantibodies [38]. These findings support the possibility that, at least in some patients, an abnormal (mutated) cancer antigen may be the initial trigger for an autoimmune T cell activation in SSc and autoAb recognizing the mutated RNApol III, which then cross-react with the wild-type autoantigen.
Microchimerism
Feto-maternal microchimerism, which is the transplacental passage of semi-allogenic fetal cells to the mother or vice versa the passage of semi-allogenic maternal cells to the fetus, may trigger autoimmunity in SSc [39]. It is supposed that microchimeric cells may provide chronic stimulation due to MHC-mismatch with enhanced expansion of alloreactive, profibrotic Th2 cells [40]. Exposure to vinyl chloride may enhance the pathogenic role of microchimeric cells in murine models [41], an interesting example of the combined effect of multiple triggers operating in conjunction or sequentially to favor SSc.
Sex Bias in SSc
As many other systemic autoimmune disorders, SSc preferentially affects women with a female to male ratio exceeding 4 to 1 [42,43,44]. Substantial differences in the clinical presentation and environmental exposure underlie gender differences in SSc. Thus, at diagnosis men preferentially present an active and diffuse form of the disease with more frequent heart and lung involvement which may impact on survival [45 46]. Exposure to chemicals is more frequent in males [28, 43], suggesting that perturbed homeostasis by environmental cues substantially adds to pathogenetic mechanisms which are enhanced in females.
Sex Hormones
Sex hormones and their cyclic variation during the fertile years have profound impact on the immune response and likely they play a role on female preponderance in SSc. Broadly speaking, estrogens tend to enhance the adaptive immune responses and in particular the production of (auto)-antibodies, while progesterone and androgens may exert inhibitory functions [47,48,49]. A recent systematic review of the literature conducted on the role of sex hormones in SSc reported that estrogens may be simultaneously fibrogenic and vasodilatory. Within the limitation of the small numbers of individuals studied, compared to healthy controls women with SSc tended to have lower levels of androgens, non-significantly higher levels of estradiol, while men had increased levels of estradiol [50].
X-chromosome
The large excess in genes present in the X-chromosome compared to the Y-chromosome is compensated by the inactivation of one X-chromosome (XCI) copy of the two present in females. This is a random and active process implicating the long non-coding RNA named XIST, which silences by epigenetic modifications almost all genes present in X-chromosome [51]. In females, escape from XCI may thus allow the expression of two copies of the genes encoded in the X-chromosomes, of which many are relevant for the immune response and for which the escape from inactivation has been demonstrated [52]. For instance, the expression of two copies of TLR7 in B cells of healthy females was shown to result in higher production of antibodies [53]. The relevance in SSc of such a mechanism is currently being explored. Enhanced X monosomy in SSc women [54] and specific patterns of X chromosome gene methylation in peripheral lymphocytes from monozygotic twins discordant for scleroderma [55] have been demonstrated. Intriguingly, extreme bias in XCI has been shown in SSc and correlated to a decreased expression of FOXP3 and reduced Treg function [56]. Further, single nucleotide polymorphisms (SNPs) enriched in SSc have been identified in X chromosome genes involved in the immune response such as IL13RA2, IRAK1, and FOXP3, and while this has not formally being proven, these SNPs may contribute to SSc development in females [57,58,59,60]. About 10% of miRNAs are located on X-chromosome and may escape inactivation or be subjected to skewed X inactivation; therefore, they may also participate in gender-related differences in SSc pathogenic mechanisms [61].
SSc Initial Events
The question of what is first in SSc pathogenesis has no definitive answer and spurs substantial debate. From the clinical stand point, Raynaud phenomenon in the large majority of cases initiate months to years before other clinical manifestations become apparent, including skin and organ fibrosis. Based on this chronological clinical order, many authors suspect that vasculopathy is the initiating event. In this perspective repeated vasospastic episodes triggered by cold exposure occurring in the appropriate genetic background may result in altered homeostasis in relationship to ischemia/reperfusion processes and may provide the substrate for inflammatory responses and structural vasculopathy. Endothelial cell injury is proposed as a crucial initiating event leading to vascular remodeling with intimal proliferation of arterioles and capillary breakdown and finally, blood vessel occlusion [7, 62, 63]. Of note, recognized mechanisms leading to endothelial cell injury are mostly immunologic in nature. Anti-endothelial cell autoantibodies through ADCC, anti-endothelin, and anti-angiotensin agonistic antibodies, cytolytic CD4+ T cells, γ/δ T cells, and NK cells have been described as effector of endothelial cell (EC) activation and/or damage [7, 64,65,66,67] If this is true, then vasculopathy follows innate and adaptive immune responses (Fig. 1). Consistently with this kinetic, the presence of serum anti-nuclear antibodies—evidence for adaptive immune responses—detected at first evaluation of Raynaud’s is considered an important, independent predictive element to classify Raynaud as secondary to SSc [68, 69]. Taken from a different perspective, it is known that monocyte/macrophage and T cell inflammatory perivascular infiltrates are detectable early in SSc [70] and ultrastructural EC damage appears to follow the appearance of inflammatory mononuclear infiltrates [71]. Thus, intricate mechanisms are at play in early events leading to SSc in which components of the immune response in relationship with EC and vessel function and integrity play a role, well before fibrosis initiate developing.
SSc Vasculopathy
Fibroproliferative modifications of vessel walls and rarefaction of capillaries underpin vasculopathy in SSc which affects mainly the micro-circulation, but also the macro-circulation. Endothelial cell (EC) dysfunction and damage are considered cornerstones of SSc vasculopathy (Fig. 3). Indeed, structural damage and inappropriate repair events distinguish primary form secondary Raynaud. Initial mechanisms may involve selective increased expression of alpha 2 adrenergic receptors on vascular smooth muscle cells (vSMC) with increased response to catecholamines [72]. Imbalance between vasodilating and vasoconstricting agents with reduced production of nitric oxide (NO) and enhanced production of endothelin-1 (ET-1) may lead to ischemia / reperfusion and subsequent increased oxidative stress which impact on EC [73]. Platelet activation may participate by releasing potent vasoconstrictors such as thromboxane and serotonin [74]. Transition to inflammatory events then occurs with opening of tight EC junctions, fluid leakage in the extravascular space, and enhanced expression of adhesion molecules, all favoring the recruitment of mononuclear cells. EC injury may lead to EC apoptosis [75]. Infectious agents, autoantibodies, toxic compounds, and cytolytic T and NK cells may be causes of EC apoptosis. Significant intimal proliferation and accumulation of proteoglycans in the arterioles and small sized arteries are common in SSc [76]. Moreover, abnormality of the vessel wall is likely to result from increased synthesis of extracellular matrix (ECM) by intimal and adventitial fibroblasts. Transdifferentiation of EC via the process of endothelial-mesenchymal transition (EndoMT) and more likely of pericytes into profibrotic myofibroblasts may contribute further to vascular wall fibrosis [77]. On the other hand, vSMC, under the influence of hypoxia, cytokines and growth factors may migrate into the intima, differentiate, and then synthesize the matrix of the fibrotic vascular lesions.

Vasculopathy in SSc. In SSc, under the influence of a variety of stimuli here depicted as a bicolor arrowhead, EC become dysfunctional and undergo damage. Excess in vasoconstricting over vasodilating agents, as well as enhanced fibroproliferative events of the vessel wall associated with reduced angiogenesis and vasculogenesis are characteristic. Vasculopathic alterations contribute to the developments of fibrosis. ET-1, endothelin-1; IL, interleukin; NO, nitric oxide; ROS, radical oxygen species
Both defective angiogenesis (growth of new vessels from existing vessels) and vasculogenesis (de novo formation of new vessels) likely contribute to capillary rarefactions. Angiogenesis is disturbed through expression of inefficient pro-angiogenic mediators, upregulation of inhibitors of angiogenesis and by alteration of transcripts involved in signal transduction pathways [78]. Hypoxia enhances the production of VEGF which is detectable in high amount in SSc sera. However, the relative abundance of a non-signaling variant (VEGF165b) and alterations at the receptors level may contribute to altered angiogenesis [79]. Imbalance between other pro-angiogenic factors and their receptors are also at play [80]. Endothelial progenitor cells (EPCs) originating from the bone marrow are fundamental for vasculogenesis. Although discrepancies between various reports exist, possibly related to differences in the markers used for the identification of these progenitors, the number of circulating EPC appears to be reduced in SSc, which may contribute to defective vasculogenesis. Alternatively, their recruitment at lesional sites could be impaired as suggested by the relative lack of the recruitment factor cellular communication network factor-1(CCN1) reported in SSc digital ulcers [81].
EC may respond to and produce a variety of cytokines and other soluble products of inflammation. Thus, they may influence the behavior of resident or recently recruited cell types in the skin and other organs. Among many others, interleukin-1 (IL-1), thymic stromal lymphopoietin (TSLP) [82], and IL-33 appear to play important roles in the interaction with macrophages, other innate immune cells, fibroblasts, and adipocytes. IL-33, which levels are increased early in the SSc disease course, might mediate very early pathogenic events of SSc through recruitment and stimulation of cells expressing the appropriate receptor [83,84,85,86].
Fibrosis and Fibroblasts in SSc
Fibrosis
Fibrosis is the default inflammatory response to chronic tissue injury of whatever cause aiming at containing and circumscribing tissue damage. Fibrosis itself consist in the enhanced deposition over resorption of ECM. In fibrotic tissues, the ECM appears to be structurally altered. Fibrosis in SSc can be seen as a process resembling wound healing in which the resolution phase is not efficacious or even does not occur.
The Pro-fibrotic Milieu
When examined in animal models, wound healing processes and fibrotic responses are characterized by type 2-like environment governed by the presence of IL-4, IL-13, ILC2, Th2-like T cells, and M2 macrophages—also named alternatively activated macrophages—all discussed in following paragraphs [87,88,89]. Very likely, type 2 environment plays an important role in SSc, particularly in skin fibrosis [90]. Specificities related to organs and tissues undergoing fibrotic changes are being unraveled by “omics” studies at single-cell level and are revealing the presence of rare cell types with specific phenotypic and functional characteristics [91]. Within this framework, the response to the master pro-fibrotic cytokine TGF-β is thought to be dysregulated in SSc. TGF-β is considered to be, at least partly, responsible for the fibrotic disease component. TGF-β induces fibroblast migration, proliferation, and differentiation and enhances ECM production components including various collagens [92, 93]. TGF-β has pleiotropic functions, is produced by many cell types in association with latency-associated peptide (LAP), interacts with the ECM, and requires processing to become biologically active. It binds to a heterodimeric receptor which intracellular signal is mediated by canonical SMAD signaling and complex, additional non-canonical pathways. The activity of TGF-β is tightly regulated at several levels including the availability of the biological active form, receptor binding, and most importantly the intracellular signaling pathway level which offers potential targets of treatment [94]. Connective tissue growth factor (CTGF) also known as CCN2 appears to be a necessary cofactor for TGF-β to activate or sustain extracellular matrix (ECM) production in both healthy and disease states [95]. Platelet-derived growth factor (PDGF), IL-6, Wnt/β-catenin (Wnt: Wingless and Int), and hedgehog signalling are some of the other important components of the profibrotic milieu [96]. As a word of caution, our understanding of the main forces involved in fibrosis, namely in SSc skin fibrosis, remains imprecisely defined. When submitting skin biopsies from the involved SSc skin to unbiased gene expression studies, heterogeneous results were obtained across skin samples. Patient samples were grouped according to the main gene expressed into an “inflammatory,” “fibroproliferative,” “limited,” or “normal-like” gene-signature [97,98,99,100]. These results point to heterogeneous mechanisms leading to skin fibrosis which do not match, or match only partially, to clinical classifications and histories.
Myofibroblast
Large agreement identifies in myofibroblasts the professional cells involved in the enhanced ECM deposition occurring during fibrosis development. At variance of what occurs during wound healing, in fibrotic processes myofibroblasts after having been activated or transdifferentiated do not stop producing ECM, possibly because they become resistant to apoptosis-inducing signals [101] (Fig. 4). Most recently, it has been proposed that myofibroblasts are characterized by increased levels of pro-apoptotic intracellular mediators, compensated by even higher levels of anti-apoptotic intracellular mediators. Among the most likely mechanisms responsible for such altered balance set-point, stiffness of tissue undergoing fibrosis transduced by mechano-sensors to myo-fibroblasts appears to play an important role [102]. It is unlikely that fibroblasts autonomously initiate the fibrotic response, however with time they may become independent from initiating stimuli. As an example, ECM stiffness enhances the release of latency associated peptide (LAP), followed by activation of transforming growth factor-β (TGF-β) by αv-integrins which then favors further ECM deposition [103]. Similarly, fibronectin extracellular domain A (FNEDA), expressed in high amounts in involved SSc skin, was shown to bind TLR4 and enhance collagen production in an in vivo murine model of scleroderma. FNEDA production is induced by TGF-β and simultaneously enhances TGF-β production by fibroblasts thus providing a positive feedback loop potentially able to maintain in an autonomous manner sustained fibroblast activation [104]. It is also likely that many stimuli of different origin may converge on fibroblasts which response is then monomorphic [96]. It is however important to stress that several subpopulations of fibroblasts have been documented which may play distinct and dynamic roles in tissue homeostasis and fibrosis [105,106,107]. In this respect, myofibroblasts are capable of contractile properties and are considered professional ECM producers [88, 108, 109]. Their origin is debated and has been ascribed variably at resident fibroblasts, at circulating fibrocytes (cells of hematopoietic origin with mesenchymal properties including the capacity to produce collagen), at smooth muscle cells, at epithelial cells undergoing mesenchymal transition, or similarly at endothelial cells undergoing mesenchymal transition (Fig. 4) [110]. Cell fate tracing in vivo experiments has however pointed to a larger contribution of pericyte transdifferentiation for the generation of myofibroblasts. Pericytes are naturally endowed with contractile properties and acquire the capacity to produce ECM components upon migration into tissues undergoing fibrosis [88]. Thus, migration, proliferation, differentiation of fibroblasts and the relationship they establish with ECM and tissue physical properties via mechanosensors are key to fibrosis development and persistence [91] (Fig. 4).

Myofibroblasts and their centrality in the development of fibrosis in SSc. Depicted are the cells potentially giving origin to myofibroblasts, as well as the main signals involved in their activation and survival. αSMA, alpha smooth muscle actin; BCL, B-cell lymphoma; ECM, extracellular matrix; FAK, focal adhesion kinase; MRTF, myocardin-related transcription factors; ROCK, Rho-associated creatinine kinase; SMAD, small mothers against decapentaplegic; TAZ, transcriptional co-activator with PDZ-binding motif; TGF-β, transforming growth factor-beta; YAP, Yes kinase-associated protein
Keratinocytes in SSc
The epidermis and in particular keratinocytes participate to dermal homeostasis by releasing factors that target dermal fibroblast. Reciprocally, keratinocytes respond to soluble mediators released by dermal fibroblasts [111]. Thus, it is not surprising that SSc epidermis presents a variety of abnormalities including altered differentiation, active TGF-β signaling, increased production of antimicrobial peptides, with DAMP properties, enhanced capacity to stimulate lattice contraction and inflammatory responses in dermal fibroblasts [112,113,114,115,116,117]. Furthermore, epithelial deficiency of the transcription factor Fli1 in mice is sufficient to induce a SSc-like phenotype, including fibrosis and systemic autoimmunity [118]. Within this framework, SSc keratinocytes, engineered epidermal equivalents, or organotypic full skin cultures were shown to respond to cytokines which levels are increased in SSc by further modulating dermal fibroblast responses. IL-17A and IL-22 in conjunction with TNF were shown to enhance inflammatory dermal responses and in particular IL-17A was shown to counteract, at least partially, the profibrotic activity of TGF-β by modulating the Wnt/β-catenin signals [119, 120]. These are examples of intercellular circuitries potentially aiming at reducing fibrosis still participating to inflammation in SSc altered homeostatic conditions (Fig. 5).

Altered cross-talk between keratinocytes and dermal fibroblasts in SSc. The homeostatic relationship between epidermis and dermis includes reciprocal signaling here represented by IL-1 produced by keratinocytes and KGF by fibroblasts. Cytokines dysregulated in SSc alter this cross-talk and variably affect the inflammatory and ECM deposition properties of dermal fibroblasts. ECM, extracellular matrix; KGF, keratinocyte growth factor; IL, interleukin; TGF-β, transforming growth factor beta; TNF, tumor necrosis factor
Autoimmunity and Inflammation in SSc
Immunological Tolerance Defects in SSc
SSc is considered a systemic autoimmune disorder, characterized by the presence of autoantibodies directed against ubiquitous (mostly nuclear auto-antigens) as well as cell-specific autoantigens. Similarly, autoreactive T cells have been demonstrated recognizing epitopes of the ubiquitous autoantigens topoisomerase-I (topo-I) and RNA polymerase III (RNApol-III) [121, 122]. Indirect proof of autoimmunity from the T cell point of view is the oligoclonal distribution of TcRs of T cells retrieved from SSc blood, skin, and lung, suggestive of an (auto)antigen-driven clonal expansion [67, 123]. In addition, the strong association of SSc with specific HLA alleles supports an immune component in the pathogenesis of SSc. Consistent with this view the survival advantage provided by profound pharmacological immunosuppression rescued by autologous hematopoietic stem cell transplantation in severe SSc [124,125,126]. However, standard immunosuppression has limited efficacy in SSc when compared to other autoimmune systemic disorders. Thus, while defective immune tolerance has a role in SSc pathogenesis, possibly in very initial events, important additional singularities characterize SSc leading to sustained vasculopathy and fibrosis. Interestingly, the presence of autoAb directed against distinct ubiquitous autoantigens is usually mutually exclusive and clinical manifestations segregate with the type of autoAb, which supports a pathogenic link between autoAb specificities and clinical manifestations. However, there is no experimental proof of such a link and for the moment being autoAb directed against ubiquitous antigens are considered only as epiphenomena, tough clinically useful as biomarkers.
Autoimmunity in SSc requires both innate and adaptive immune responses at humoral and cellular levels which participate to disease initiation under the influence of some of the triggers previously mentioned. While no animal model faithfully reproduces all the clinical and biological features of SSc, it is worth to stress that repeated mice immunization with T and B cell autoantigen Topo-I and concomitant stimulation of the innate immune response by complete Freund adjuvant results in a disease characterized by skin and lung fibrosis and autoimmunity in C57Bl/6 mice [127]. No such results were obtained in autoimmune prone mice when immunized with Topo-I in the absence of solid innate immunity activation [128]. The evidence thus generated strongly support the need of multiple, sustained hits to break tolerance and initiate processes leading to SSc. In humans, molecular mimicry is potentially implicated in tolerance breakdown. For instance, experimental evidence suggestive for SSc having a paraneoplastic origin has been provided in association with mutated RNA pol III antigen [38]. Similarly, cross-reactive antibodies between CMV UL94 antigen and endothelial cells have been documented [32].
Innate Immune Cells, Soluble Products, and Receptors in SSc
All innate immune cells and their capacity to be activated in the one hand by pathogen (PAMP) or danger due to tissue damage (DAMP) molecular patterns via PRRs and on the other hand by soluble mediators of inflammation (cytokines, chemokines, lipidic mediators, NO, etc.) participate to SSc pathogenesis. Of interest, PRR are not only expressed by innate immune cells but also by stromal cells including fibroblasts and endothelial cells, where they are thought to play a substantial role. Similarly, stromal cells can produce and respond to soluble mediators of inflammation. Thus, an intricate web of signals to cells and responses by cells constitute the inflammatory network that in SSc extends well beyond the “classical” components of the immune system. Deciphering this network will potentially provide hierarchically important nodes as target for therapeutic interventions. Broadly speaking, by sensing altered homeostasis and tissue damage, cells of the innate immune system may contribute in many ways to initiation and amplification of inflammatory events leading to fibrosis [91]. On the one hand, the release by cells submitted to stressful signals of pro-inflammatory cytokines, including IL-1, tumor necrosis factor (TNF), and IL-6 may turn on macrophages which may initiate TGF-β release and activation. In the other hand alarmins release, including IL-33, IL-25 (also known as IL-17E), and thymic stroma lymphopoietin (TSLP), may activate type 2 innate lymphoid cells (ILC2), which participate to Th2-like T cell responses and enhance the production of IL-4 and IL-13 which directly and indirectly participate to enhanced ECM deposition. Furthermore, myeloid dendritic cells (mDCs) and plasmacytoid DC (pDC) contribute to generate the fibroproliferative milieu by releasing type I interferon (Fig. 6).

Contribution of cells and soluble products of the innate immune system to enhanced ECM deposition. Parallel, not mutually exclusive pathways involving cells of the innate immune system and their soluble products, converge on fibroblasts enhancing their ECM synthetic capacity. CXCL, chemokine containing the CXC motif; DAMP, danger associated molecular patterns; IFN-I, type I interferon; IL, interleukin; ILC, innate lymphoid cell; pDC, plasmacytoid dendritic cell; TGF-β, transforming growth factor-beta; Th2, type 2 T helper cell; TNF, tumor necrosis factor; TSLP, thymic stromal lympopoietin
PRR and SSc
Given their central role in sensing danger, whether due to infectious agents or tissue damage, PRR undoubtedly plays a major role in SSc [129]. The contribution of PRR to SSc pathogenesis has received increasing attention in two distinct directions: the role of PRR in the production of type I interferons (IFN-I) or other pro-inflammatory cytokines and the contribution of TLRs in activating mesenchymal cells, in particular fibroblasts. Here follow a few examples. TLR4, which expression is increased on SSc fibroblasts [130], mediates chronic fibroblast activation by sensing FNEDA [104]. Consistently with a role in fibroblast activation, an amelioration of tissue fibrosis was observed in TLR4 knockout in murine models of systemic sclerosis [131]. TLR8, expressed in monocytes, may mediate their transdifferentiation in fibroblasts, potentially responding to lytic EBV infection [132]. SSc monocytes upon TLR8 activation by ssRNA (and to a lesser extent by LPS/TLR4) produce enhanced levels of tissue inhibitor of matrix metalloproteinase (TIMP)-1 [133]. TLR8, paradoxically expressed in SSc pDC, plays a role in IFN-I production [134]. Always in pDC, TLR7 and TLR9 play a role in sensing DNA or RNA shuttled by autoantibodies via Fc-gamma receptors or by CXCL4 into the endosomal compartment thus also participating to enhanced levels of IFN-I in SSc [135, 136]. In addition to TLR9, cytosol-located GAS-STING activation by mitochondrial DNA—which concentration is increased in SSc plasma—was shown to be positively associated with IFN-I and IL-6 expression and SSc-ILD progression [137].
In SSc, circulating monocytes and tissue-resident macrophages, potentially under the influence of type 2 cytokines (IL-4, IL-13), appear to preferentially express CD163 and CD204 and promote fibrogenesis by increasing the production of TGF-β. They are involved also in the production of a large variety of other inflammatory mediators including chemokines, cytokines, matrix metalloproteinases (MMPs) and their inhibitors (TIMPs), which composition and role may depend on timing and localization. They are likely involved both in vasculopathy as well as in fibrosis and they may play a role in the perpetuation of the disease having pro-reparative properties inefficiently terminated [138]. In an experimental murine model of SSc, it was shown that epigenetic modifications of macrophages (trained immunity) induced by activation in the one hand with low-dose lipopolysaccharide (LPS), on the other hand by BCG (Bacillus Calmette Guérin) could deeply influence the fibrotic response with reduced or enhanced fibrosis, respectively [139]. Thus, macrophages sensing pro-fibrotic cues may propagate or amplify tissue fibrosis. mDCs, beside their role as antigen-presenting cells (APCs) may play relevant inflammatory functions in SSc [138]. Tissue-resident plasmacytoid DC (pDC) also may play a substantial role. pDC were shown to respond to CXCL4 (CXC chemokine ligand 4, also known as platelet factor 4, PF4) which levels are highly increased in SSc sera [140] and forms complexes with DNA [136]. These complexes are shuttled into the endosomal compartment where by interacting with TLR8 or TLR9 favor the production of IFN-I, highly increased in about 50% of SSc individuals [134, 136]. The relevance of these findings was highlighted by the prevention of skin inflammation and fibrosis in xenotransplant human-mouse model of scleroderma by targeting human pDC [141]. The most recently described in the innate cell family are the innate lymphoid cells (ILCs). They are endowed with the capacity to rapidly produce polarized subsets of cytokines under the control of differentially expressed master transcription factors. They are activated by PRR ligation and are fast producers of cytokines. Relatively little is known yet about ILCs in SSc; however, evidence points to an expansion of ILC2 (producing IL-4/IL-13) in the blood and in the skin. Thus, they may contribute to a dysregulated environment favoring fibrosis [142]. ILC2, in particular the KLRG1neg ILC2 subset numbers appear to be increased in SSc skin correlating with the extent of skin fibrosis. Of note, TGFβ favors the expansion of the KLRG1neg ILC2 subset and simultaneously decreases their production of IL10, which regulates negatively collagen production by dermal fibroblasts [143]. This example highlights the intricate relationship in the cytokine network that portends enhanced deposition of ECM.
ROS in SSc
An imbalance between oxidant and anti-oxidant states is observed in SSc, with increase in the blood of oxidative stress biomarkers such as malondialdehyde (MDA—a marker of lipid peroxydation), nitric oxide and endogenous nitric oxide inhibitor asymmetric dimethylarginine (ADMA) and decreased anti-oxidative biomarkers, such as superoxide dismutase and vitamin C [144]. ROS may impact on monocyte/macrophages polarization favoring M2-like differentiation [145]. ROS participate to fibroblasts activation triggering the production of pro-inflammatory cytokines such as IL-1β and fibroblasts from SSc are a potent source of ROS through an up-regulation of NOX-2 and NOX-4 [146]. Further, inflammasome, in particular NLR family pyrin domain containing 3 (NLRP3) inflammasome is thought to be involved in fibroblasts [147, 148], endothelial cells, and macrophages activation in SSc [149]. NLRP3 expression is increased in SSc skin and NLRP3-deficient mice are resistant to bleomycin-induced fibrosis. It is possible that oxidative stress could participate to NLRP3 activation [10].
Adaptive Immunity in SSc
T Cells in SSc Skin
Compared to healthy skin, T cells are abundant in involved SSc more so early in disease course and active collagen synthesis is more pronounced around inflammatory infiltrates [150, 151]. T cell effector functions are highly heterogenous and differentially affect fibrosis and vasculopathy. Within the adagio that type 2 responses favor repair and fibrosis, Th2 cells (CD4+T cells producing IL-4 and IL-13) as well as Tc2 (CD8 T cells producing IL-13), in conjunction with the previously mentioned ILC2 may be actively involved in enhancing ECM deposition, since both IL-4 and IL-13 can directly enhance collagen production by fibroblasts [90, 152]. Further, IL-13 may enhance the production of TGF-β by macrophages thus indirectly enhancing ECM deposition [153]. In addition, SSc-skin infiltrating Treg cells, under the influence of IL-33, may become Th2-like effectors and release profibrotic cytokines contributing to enhanced ECM deposition (Fig. 7). However, Th1, Th17, and Th22 cells may also be enriched in SSc skin where they potentially participate to inflammation simultaneously, and most importantly, opposing fibrosis [119, 120, 154, 155]. From another angle, CD4+ and CD8+ T cells with cytolytic potential present in SSc skin may participate to vasculopathy by enhancing endothelial cell apoptosis [67]. The presence of high-affinity, isotype switched, autoantibodies characteristic of SSc is further strong evidence for the role and contribution of T helper cells, in particular of follicular T cells (TFH) in SSc. Indeed, TFH cells are increased in SSc peripheral blood and in the skin, they present an activated phenotype, increased capacity to produce IL-21, and higher capacity to stimulate the differentiation of CD19+CD27+CD38hi B cells and their secretion of IgG and IgM through the IL-21 pathway than healthy controls. In experimental SSc, the blockade of IL-21 or of inducible T cell co-stimulator ICOS (expressed by TFH) resulted in decreased skin fibrosis establishing a link between TFH cells and an immune-mediated fibrotic reaction [156, 157]. Finally, a study based on single-cell RNAseq has identified eight distinct T cell clusters, of which one uniquely present in SSc skin https://doi.org/10.1136/annrheumdis-2021-220209. This CD4+ T cell subset is characterized by the expression of CXCL13 and IL-21 in addition to an TFH-like gene expression signature and that appears to be poised to promote B-cell responses within the inflamed skin of patients. Thus, the composite picture provided by studies focusing on T cells in SSc is highly complex which may depend on the timing along disease course in which the study is made, with a relative predominance of type 2 responses early and of type 1 (IFN-γ) and 17 later in disease course, when fibrosis tends to decrease, at least in the skin (Fig. 7).

Adaptive immune responses and their roles in SSc. T cells, B cells, and their products contribute to both enhanced ECM deposition and vasculopathy. This schematic representation highlights the characteristics of conventionally defined as well as of SSc-restricted T cell subsets. They may have enhancing or inhibitory functions (blunted heads: inhibitory function; arrowhead: enhancing function). T-B cell interactions are important for both the generation of agonist/antagonist autoAb and tissue damage. CD, cluster of differentiation; CXCL, chemokine containing the CXC motif; IFN-γ, interferon-gamma; IL, interleukin; TFH, T follicular helper cell; Th, T helper cell. Autoantibody specificities: AFA, anti-fibroblast; AT1R, angiotensin-II receptor; ECA, endothelial cell; ETAR, endothein-1 receptor A; MMP, matrix metalloproteinase; NAG2, also known as transmembrane 4 superfamily member 7; PDGF-Rα, platelet-derived growth factor receptor-alpha; UL-94, gene coding for the cytomegalovirus (CMV) cytoplasmic envelopment protein 2
B Cells and Humoral Immunity in SSc
B cells participate deeply to SSc pathogenic events both as precursors of autoAb producing cells and as inflammatory cells infiltrating tissues undergoing fibrosis, namely the skin and the lung [97, 158] where they release cytokines and may influence the behavior of fibroblasts and other mesenchymal cells [159] (Fig. 7). Reduced numbers of the Breg subset with decreased production of IL-10 have also been documented and may participate to the dysregulated regulatory network in SSc [160].
Beyond autoAb directed against nuclear antigens characteristic of SSc, cell-specific autoAb may very well be pathologically relevant. Examples are autoAb directed against the receptor A of endothelin-1 (ETAR) or the angiotensin-II type 1 receptor (AT1R), which were shown to affect several processes ranging from production of collagen by skin fibroblasts to angiogenesis modulation [7]. Antagonist autoAb directed against MMP1 and MMP3 were described to block the enzymatic activity of these proteins, thus reducing the digestion of matrix [161, 162]. In the other hand, autoAb with relevant agonist properties are those directed against the PDGFRα with induction of the ROS-ERK1/2-Ha-Ras loop and increased collagen gene transcription in human fibroblasts in vitro and in vivo in a humanized mouse model of skin fibrosis [163, 164]. These autoAb can be considered pathogenic and participate to disease progression in conjunction with those directed against endothelial cells [64].
Microbiome and SSc
No doubt that microbiome influences deeply the immune response and this in two main ways [165] first, representing an antigenic challenge with whom the immune system needs to cope, mostly by establishing tolerance via different mechanisms but also generating specific innate and adaptive responses; second, by enforcing nutritional and metabolic cues that influence the immune response, beside the behavior of other host systems. Dysbiosis is a modification of microbiota with relevant immunological and metabolic consequences. Of interest, main organs affected in SSc are the skin, the lung and the gut, which are barrier organs in which the microbiota resides. SSc-associated dysbiosis has been documented in the skin with decreased lipophilic taxa and a marked increase in a wide range of gram-negative taxa [166]. In the gut, a distinct microbial signature in SSc patients compared with healthy controls has been documented [167, 168] with indications for a reduction in protective butyrate-producing bacteria and by an increase in proinflammatory noxious genera, especially Desulfovibrio [169]. Similar findings were reported in gut microbiomes of IgG4-related disease and SSc patients showing increase in opportunistic pathogenic Clostridium and Streptococcus species, while butyrate-producing species were depleted. Interestingly, the gut microbiomes of IgG4-RD and SSc showed signatures similar to those found in multiple sclerosis and rheumatoid arthritis, but not those found in inflammatory bowel diseases where the most differentially abundant taxa are facultative anaerobes [170]. Thus, it is likely that the dysbiosis may influence disease initiation and disease evolution. At this time point, however, whether the microbiome alterations documented in SSc are primary or secondary to organ pathology and/or medication use is not yet established.
System Biology Approaches to Decipher SSc
Within the last decade or so, we have witnessed the increased application of techniques based on the unbiased identification of gene expressed in SSc affected organs, particularly but not exclusively the skin and the peripheral blood, and more recently single cell RNAseq, that exponentially increase the amount of information on cellular and tissue alterations characterizing SSc. Additionally, multi-“omics” approaches exploring metabolism, epigenetic modifications, phenotypes, etc. further contribute novel information. The wealth of data is then submitted to sophisticated analysis based on complex algorithms aiming at reducing the catalogued data to integrated dimensions that are comprehensible, simultaneously providing new understanding or novel perspectives for old knowledge. This type of studies should provide a wider conceptual framework to better understand SSc physiopathology.
Historically, the Whitfield group published the first gene array study on skin biopsies. Expressed genes differentiated SSc from healthy controls and were similarly expressed in involved and not involved skin [97]. Further analysis based on genes expressed in skin provided evidence for the existence of intrinsic SSc subsets named “inflammatory”, “fibroproliferative,” “limited,” or “normal-like” [171]. Active immune and defense responses were associated with the inflammatory subset; proliferation and cell cycle programs with the fibroproliferative subset; and the normal-like subset was associated with a distinct lack of inflammatory signature coupled with fatty-acid metabolism. The limited subset showed deregulation of pathways associated with cell adhesion, cardiovascular system development, ECM, and immune and inflammatory responses [172]. According to these authors, the SSc intrinsic subsets were relatively stable throughout disease course and unlikely to change over time [98]. To identify genes co-expressed across various cohorts, consensus clustering analysis led to the identification of conserved genes and networks common to distinct subsets [173]. The connected gene-gene networks included the terms: “adaptive immunity,” “interferon,” “M2 macrophages,” “ECM,” and “proliferation.” A meta-analysis of genes expressed in multiple end-target organs including the skin, the lung, the esophagus and the peripheral blood provided evidence for the occurrence across organs of the intrinsic subsets, pointing to the existence of pro-fibrotic macrophages in multiple tissues [174].
Similar, but not identical, gene signatures were found by other authors with the identification of two prominent transcriptomes in SSc skin: named the “keratin” and “fibroinflammatory” signatures. The first associated with shorter disease duration the second with diffuse cutaneous involvement and a higher modified Rodnan skin score (mRSS). A subgroup of patients with significantly longer disease duration had a normal-like transcript pattern [175]. Further data from the same group reinforced the concept that gene expressed in early disease had higher adaptive immune cell signatures than in later disease, while fibroblast and macrophage cell type signatures were associated with higher mRSS. Of further interest, the immune cell signatures correlated with the rate of skin thickness progression prior to, but not after, biopsy [151]. Overall, these results support the concept that the pathological processes characterizing SSc may be different during the disease evolution and enrich our understanding by subgrouping patients on the basis of preferential gene expression in target organs.
In another study, by generating a normalized catalog of differentially expressed genes (DEGs) from 344 skin samples of 173 patients and submitting DEG to pathway analysis, patients with SSc were grouped into four distinct clusters that differed in activation levels of SSc-relevant signaling pathways. In this analysis, the phosphoinositide-3-kinase protein kinase B (PI3K-Akt) signaling pathway showed the closest correlation and temporal association to mRSS. Interestingly, the inflammatory subtype was related to significant improvement in skin fibrosis at follow-up in the absence of specific treatment [176].
The identification of 415 DEG in skin differentiating SSc from HC allowed the generation of a score, named 4S, correlating with mRSS and potentially useful to predict response to treatment [99].
The identification of genes defining intrinsic SSc subsets provides ground for applying precision medicine in therapeutic approaches. Retrospective analysis of data generated during therapeutic trials has indeed offered elements supportive for responses to therapeutic agents depending on the intrinsic subset of the treated individual. Thus, agents like mycophenolate mofetil or abatacept targeting the immune response may be efficacious in the inflammatory subset [177,178,179], while “anti-fibrotic” agents may be more efficacious for the fibroproliferative subset [180, 181]. Potentially surprising, in a post hoc analysis, individuals belonging to the fibroproliferative subset presented a significant advantage in event free survival when undergoing hematopoietic stem cell transplantation (HSCT) compared to individuals treated with cyclophosphamide in the SCOT trial [182]. HSCT tended to confer an advantage over cyclophosphamide to individuals of the inflammatory subset, with no differences between treatment arms for individuals belonging to the normal-like subset. Limiting factor in the interpretation of these data was the low number of samples and individual trajectories available for these analyses; that however could provide novel dimensions in the selection of patients included in clinical trial beside classic clinical classification.
To summarize pathophysiological information gathered until now by using big data and unbiased methods to identify SSc specificities, it appears that SSc heterogeneity extends beyond and does not overlap with classical clinical and serological parameters, that predominant gene signatures—intrinsic subsets—differ among SSc individuals and tend to persist during disease evolution, with however enrichment for immune response genes earlier, and macrophage—fibroblast gene later in disease course in severe cases. Not yet confirmed in prospective studies, responses to therapeutic approaches may differ among patient subsets according to the mechanism of the therapeutic agent assessed.
Conclusions and Perspectives
SSc represents a major challenge for our understating of physio-pathological processes leading to disease state and disease progression. The heterogeneity in SSc clinical manifestations influences disease identification and classification and, to a certain extent, our approach to medical management. However, the subtle mechanisms underpinning clinical heterogeneity are, by far, poorly understood. Major progress in our understanding, based on increasingly more precise identification of cell types and intercellular signaling as well as of intracellular molecular cues at play in SSc physio-pathology has spurred enthusiasm in the scientific community and has led to the recent approval of therapeutic agents that may alter the disease course. We should admit, however, that the pace of improvement is slow and a major gap still exist between scientific advancement and clinical application. We believe that the integration of “omics” approaches to sophisticated system biology analyses will contribute to the refinement of our understanding and should be intensively applied, particularly in controlled clinical trials. By employing these methods, the comparisons in tissue responses between placebo and active arms in well characterized patient populations should provide interesting new information on mechanisms at play in subsets of patients and their deviation under the pressure of therapeutic agents. In view of SSc clinical heterogeneity, possibly linked to heterogeneity in pathogenic mechanisms, it is unlikely that the responses to a given agent will be homogeneous. We believe that differences in responses within supposedly homogenous subsets of patients will be extremely informative from the pathogenic point of view and will provide substantial advancement. In this perspective, we propose that small rather than large trials, in which deep “omics” will be applied to extremely selected group of patients will provide relevant information. To overcome the constrains linked to the rarity of the disease, these trials should be conducted across multiple, well-coordinated, integrated, and equipped centers. Trials in which mechanisms will be primary outcomes will provide solid ground for solid therapeutic secondary outcomes.
References
Denton CP, Khanna D (2017) Systemic sclerosis. Lancet 390:1685–1699. https://doi.org/10.1016/s0140-6736(17)30933-9
Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, Riemekasten G, Airo P, Joven B, Vettori S, Cozzi F, Ullman S, Czirjak L, Tikly M, Muller-Ladner U, Caramaschi P, Distler O, Iannone F, Ananieva LP, Hesselstrand R, Becvar R, Gabrielli A, Damjanov N, Salvador MJ, Riccieri V, Mihai C, Szucs G, Walker UA, Hunzelmann N, Martinovic D, Smith V, Muller CS, Montecucco CM, Opris D, Ingegnoli F, Vlachoyiannopoulos PG, Stamenkovic B, Rosato E, Heitmann S, Distler JHW, Zenone T, Seidel M, Vacca A, Langhe E, Novak S, Cutolo M, Mouthon L, Henes J, Chizzolini C, Muhlen CAV, Solanki K, Rednic S, Stamp L, Anic B, Santamaria VO, De Santis M, Yavuz S, Sifuentes-Giraldo WA, Chatelus E, Stork J, Laar JV, Loyo E, de la Pena Garcia, Lefebvre P, Eyerich K, Cosentino V, Alegre-Sancho JJ, Kowal-Bielecka O, Rey G, Matucci-Cerinic M, Allanore Y (2017) Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis 76:1897–1905. https://doi.org/10.1136/annrheumdis-2017-211448
Moore DF, Steen VD (2020) Racial disparities in systemic sclerosis. Rheum Dis Clin North Am 46:705–712. https://doi.org/10.1016/j.rdc.2020.07.009
Abraham DJ, Varga J (2005) Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol 26:587–595
Katsumoto TR, Whitfield ML, Connolly MK (2011) The pathogenesis of systemic sclerosis. Ann Rev Pathol 6:509–537. https://doi.org/10.1146/annurev-pathol-011110-130312
Stern EP, Denton CP (2015) The pathogenesis of systemic sclerosis. Rheum Dis Clin North Am 41:367–382. https://doi.org/10.1016/j.rdc.2015.04.002
Cabral-Marques O, Riemekasten G (2016) Vascular hypothesis revisited: role of stimulating antibodies against angiotensin and endothelin receptors in the pathogenesis of systemic sclerosis. Autoimmun Rev 15:690–694. https://doi.org/10.1016/j.autrev.2016.03.005
Bergmann C, Distler JH (2017) Epigenetic factors as drivers of fibrosis in systemic sclerosis. Epigenomics 9:463–477. https://doi.org/10.2217/epi-2016-0150
Maria ATJ, Partouche L, Goulabchand R, Rivière S, Rozier P, Bourgier C, Le Quellec A, Morel J, Noël D, Guilpain P (2018) Intriguing relationships between cancer and systemic sclerosis: role of the immune system and other contributors. Front Immunol 9:3112. https://doi.org/10.3389/fimmu.2018.03112
Doridot L, Jeljeli M, Chêne C, Batteux F (2019) Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol 25:101122. https://doi.org/10.1016/j.redox.2019.101122
Zhu H, Chen W, Liu D, Luo H (2019) The role of metabolism in the pathogenesis of systemic sclerosis. Metabolism 93:44–51. https://doi.org/10.1016/j.metabol.2018.12.004
Kania G, Rudnik M, Distler O (2019) Involvement of the myeloid cell compartment in fibrogenesis and systemic sclerosis. Nat Rev Rheumatol 15:288–302. https://doi.org/10.1038/s41584-019-0212-z
Herrick AL, Wigley FM (2020) Raynaud’s phenomenon. Best Pract Res Clin Rheumatol 34:101474. https://doi.org/10.1016/j.berh.2019.101474
Agarwal SK, Tan FK, Arnett FC (2008) Genetics and genomic studies in scleroderma (systemic sclerosis). Rheum Dis Clin N Am 34:17–40. https://doi.org/10.1016/j.rdc.2007.10.001
Kuwana M, Feghali CA, Medsger TA Jr, Wright TM (2001) Autoreactive T cells to topoisomerase I in monozygotic twins discordant for systemic sclerosis. Arthritis Rheum 44:1654–1659
Feghali-Bostwick C, Medsger TA Jr, Wright TM (2003) Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum 48:1956–1963. https://doi.org/10.1002/art.11173
Arnett FC, Gourh P, Shete S, Ahn CW, Honey RE, Agarwal SK, Tan FK, McNearney T, Fischbach M, Fritzler MJ, Mayes MD, Reveille JD (2010) Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis 69:822–827. https://doi.org/10.1136/ard.2009.111906
Gourh P, Safran SA, Alexander T, Boyden SE, Morgan ND, Shah AA, Mayes MD, Doumatey A, Bentley AR, Shriner D, Domsic RT, Medsger TA Jr, Ramos PS, Silver RM, Steen VD, Varga J, Hsu V, Saketkoo LA, Schiopu E, Khanna D, Gordon JK, Kron B, Criswell LA, Gladue H, Derk CT, Bernstein EJ, Bridges SL Jr, Shanmugam VK, Kolstad KD, Chung L, Kafaja S, Jan R, Trojanowski M, Goldberg A, Korman BD, Steinbach PJ, Chandrasekharappa SC, Mullikin JC, Adeyemo A, Rotimi C, Wigley FM, Kastner DL, Boin F, Remmers EF (2020) HLA and autoantibodies define scleroderma subtypes and risk in African and European Americans and suggest a role for molecular mimicry. Proc Natl Acad Sci U S A 117:552–562. https://doi.org/10.1073/pnas.1906593116
López-Isac E, Acosta-Herrera M, Kerick M, Assassi S, Satpathy AT, Granja J, Mumbach MR, Beretta L, Simeón CP, Carreira P, Ortego-Centeno N, Castellvi I, Bossini-Castillo L, Carmona FD, Orozco G, Hunzelmann N, Distler JHW, Franke A, Lunardi C, Moroncini G, Gabrielli A, de Vries-Bouwstra J, Wijmenga C, Koeleman BPC, Nordin A, Padyukov L, Hoffmann-Vold AM, Lie B, Proudman S, Stevens W, Nikpour M, Vyse T, Herrick AL, Worthington J, Denton CP, Allanore Y, Brown MA, Radstake T, Fonseca C, Chang HY, Mayes MD, Martin J (2019) GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat Commun 10:4955. https://doi.org/10.1038/s41467-019-12760-y
Orvain C, Assassi S, Avouac J, Allanore Y (2020) Systemic sclerosis pathogenesis: contribution of recent advances in genetics. Curr Opin Rheumatol 32:505–514. https://doi.org/10.1097/bor.0000000000000735
Altorok N, Tsou PS, Coit P, Khanna D, Sawalha AH (2015) Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann Rheum Dis 74:1612–1620. https://doi.org/10.1136/annrheumdis-2014-205303
Dees C, Schlottmann I, Funke R, Distler A, Palumbo-Zerr K, Zerr P, Lin NY, Beyer C, Distler O, Schett G, Distler JH (2014) The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis 73:1232–1239. https://doi.org/10.1136/annrheumdis-2012-203194
Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH, Gay S, Distler O (2010) MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 62:1733–1743. https://doi.org/10.1002/art.27443
Sing T, Jinnin M, Yamane K, Honda N, Makino K, Kajihara I, Makino T, Sakai K, Masuguchi S, Fukushima S, Ihn H (2012) microRNA-92a expression in the sera and dermal fibroblasts increases in patients with scleroderma. Rheumatology (Oxford) 51:1550–1556. https://doi.org/10.1093/rheumatology/kes120
Mora GF (2009) Systemic sclerosis: environmental factors. J Rheumatol 36:2383–2396. https://doi.org/10.3899/jrheum.090207
Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, De Roos AJ (2012) Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun 39:259–271. https://doi.org/10.1016/j.jaut.2012.05.002
Marie I (2019) Systemic sclerosis and exposure to heavy metals. Autoimmun Rev 18:62–72. https://doi.org/10.1016/j.autrev.2018.11.001
Rubio-Rivas M, Moreno R, Corbella X (2017) Occupational and environmental scleroderma. Systematic review and meta-analysis. Clin Rheumatol 36:569–582. https://doi.org/10.1007/s10067-016-3533-1
Freire M, Alonso M, Rivera A, Sousa A, Soto A, Gomez-Sousa JM, Baroja A, Vazquez-Trinanes C, Sopena B (2015) Clinical peculiarities of patients with scleroderma exposed to silica: A systematic review of the literature. Semin Arthritis Rheum 45:294–300. https://doi.org/10.1016/j.semarthrit.2015.06.004
Randone SB, Guiducci S, Cerinic MM (2008) Systemic sclerosis and infections. Autoimmun Rev 8:36–40. https://doi.org/10.1016/j.autrev.2008.07.022
Moroncini G, Mori S, Tonnini C, Gabrielli A (2013) Role of viral infections in the etiopathogenesis of systemic sclerosis. Clin Exp Rheumatol 31:3–7
Lunardi C, Bason C, Navone R, Millo E, Damonte G, Corrocher R, Puccetti A (2000) Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med 6:1183–1186
Lunardi C, Dolcino M, Peterlana D, Bason C, Navone R, Tamassia N, Beri R, Corrocher R, Puccetti A (2006) Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: a gene array approach. PLoS Med 3:e2
Ferri C, Zakrzewska K, Longombardo G, Giuggioli D, Storino FA, Pasero G, Azzi A (1999) Parvovirus B19 infection of bone marrow in systemic sclerosis patients. Clin Exp Rheumatol 17:718–720
Arvia R, Margheri F, Stincarelli MA, Laurenzana A, Fibbi G, Gallinella G, Ferri C, Del Rosso M, Zakrzewska K (2020) Parvovirus B19 activates in vitro normal human dermal fibroblasts: a possible implication in skin fibrosis and systemic sclerosis. Rheumatology (Oxford) 59:3526–3532. https://doi.org/10.1093/rheumatology/keaa230
Farina A, Cirone M, York M, Lenna S, Padilla C, McLaughlin S, Faggioni A, Lafyatis R, Trojanowska M, Farina GA (2014) Epstein-Barr virus infection induces aberrant TLR activation pathway and fibroblast-myofibroblast conversion in scleroderma. J Invest Dermatol 134:954–964. https://doi.org/10.1038/jid.2013.423
Shah AA, Rosen A, Hummers L, Wigley F, Casciola-Rosen L (2010) Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum 62:2787–2795. https://doi.org/10.1002/art.27549
Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, Boin F, Fava A, Thoburn C, Kinde I, Jiao Y, Papadopoulos N, Kinzler KW, Vogelstein B, Rosen A (2014) Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 343:152–157. https://doi.org/10.1126/science.1246886
Nelson JL, Furst DE, Maloney S, Gooley T, Evans PC, Smith A, Bean MA, Ober C, Bianchi DW (1998) Microchimerism and HLA-compatible relationships of pregnancy in scleroderma. Lancet 351:559–562
Scaletti C, Vultaggio A, Bonifacio S, Emmi L, Torricelli F, Maggi E, Romagnani S, Piccinni MP (2002) Th2-oriented profile of male offspring T cells present in women with systemic sclerosis and reactive with maternal major histocompatibility complex antigens. Arthritis Rheum 46:445–450
Christner PJ, Artlett CM, Conway RF, Jimenez SA (2000) Increased numbers of microchimeric cells of fetal origin are associated with dermal fibrosis in mice following injection of vinyl chloride. Arthritis Rheum 43:2598–2605
Invernizzi P, Pasini S, Selmi C, Gershwin ME, Podda M (2009) Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun 33:12–16. https://doi.org/10.1016/j.jaut.2009.03.005
Peoples C, Medsger TA Jr, Lucas M, Rosario BL, Feghali-Bostwick CA (2016) Gender differences in systemic sclerosis: relationship to clinical features, serologic status and outcomes. J Scleroderma Relat Disord 1:177–240. https://doi.org/10.5301/jsrd.5000209
Coi A, Barsotti S, Santoro M, Almerigogna F, Bargagli E, Caproni M, Emmi G, Frediani B, Guiducci S, Matucci Cerinic M, Mosca M, Parronchi P, Prediletto R, Selvi E, Simonini G, Tavoni AG, Bianchi F, Pierini A (2021) Epidemiology of systemic sclerosis: a multi-database population-based study in Tuscany (Italy). Orphanet J Rare Dis 16:90. https://doi.org/10.1186/s13023-021-01733-4
Elhai M, Avouac J, Walker UA, Matucci-Cerinic M, Riemekasten G, Airò P, Hachulla E, Valentini G, Carreira PE, Cozzi F, Balbir Gurman A, Braun-Moscovici Y, Damjanov N, Ananieva LP, Scorza R, Jimenez S, Busquets J, Li M, Müller-Ladner U, Kahan A, Distler O, Allanore Y (2016) A gender gap in primary and secondary heart dysfunctions in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis 75:163–169. https://doi.org/10.1136/annrheumdis-2014-206386
Carreira PE, Carmona L, Joven BE, Loza E, Andreu JL, Riemekasten G, Vettori S, Balbir-Gurman A, Airò P, Walker UA, Damjanov N, Matucci-Cerinic M, Ananieva LP, Rednic S, Czirják L, Distler O, Farge D, Hesselstrand R, Corrado A, Caramaschi P, Tikly M, Allanore Y (2018) Gender differences in early systemic sclerosis patients: a report from the EULAR scleroderma trials and research group (EUSTAR) database. Clin Exp Rheumatol. 36(Suppl 113):68–75
Gubbels Bupp MR, Jorgensen TN (2018) Androgen-induced immunosuppression. Front Immunol 9:794. https://doi.org/10.3389/fimmu.2018.00794
Bereshchenko O, Bruscoli S, Riccardi C (2018) Glucocorticoids, sex hormones, and immunity. Front Immunol 9:1332. https://doi.org/10.3389/fimmu.2018.01332
Cutolo M, Straub RH (2020) Sex steroids and autoimmune rheumatic diseases: state of the art. Nat Rev Rheumatol 16:628–644. https://doi.org/10.1038/s41584-020-0503-4
Ciaffi J, van Leeuwen NM, Schoones JW, Huizinga TWJ, de Vries-Bouwstra JK (2020) Sex hormones and sex hormone-targeting therapies in systemic sclerosis: A systematic literature review. Semin Arthritis Rheum 50:140–148. https://doi.org/10.1016/j.semarthrit.2019.07.007
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N (1996) Requirement for Xist in X chromosome inactivation. Nature 379:131–137. https://doi.org/10.1038/379131a0
Youness A, Miquel CH, Guéry JC (2021) Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. Int J Mol Sci 22. https://doi.org/10.3390/ijms22031114
Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, Pienkowski C, Chaumeil J, Mejía JE, Guéry JC (2018) TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol 3. https://doi.org/10.1126/sciimmunol.aap8855
Invernizzi P, Miozzo M, Selmi C, Persani L, Battezzati PM, Zuin M, Lucchi S, Meroni PL, Marasini B, Zeni S, Watnik M, Grati FR, Simoni G, Gershwin ME, Podda M (2005) X chromosome monosomy: a common mechanism for autoimmune diseases. J Immunol 175:575–578. https://doi.org/10.4049/jimmunol.175.1.575
Selmi C, Feghali-Bostwick CA, Lleo A, Lombardi SA, De Santis M, Cavaciocchi F, Zammataro L, Mitchell MM, Lasalle JM, Medsger T Jr, Gershwin ME (2012) X chromosome gene methylation in peripheral lymphocytes from monozygotic twins discordant for scleroderma. Clin Exp Immunol 169:253–262. https://doi.org/10.1111/j.1365-2249.2012.04621.x
Broen JC, Wolvers-Tettero IL, Geurts-van Bon L, Vonk MC, Coenen MJ, Lafyatis R, Radstake TR, Langerak AW (2010) Skewed X chromosomal inactivation impacts T regulatory cell function in systemic sclerosis. Ann Rheum Dis 69:2213–2216. https://doi.org/10.1136/ard.2010.129999
Granel B, Allanore Y, Chevillard C, Arnaud V, Marquet S, Weiller PJ, Durand JM, Harlé JR, Grange C, Frances Y, Berbis P, Gaudart J, de Micco P, Kahan A, Dessein A (2006) IL13RA2 gene polymorphisms are associated with systemic sclerosis. J Rheumatol 33:2015–2019
Dieudé P, Bouaziz M, Guedj M, Riemekasten G, Airò P, Müller M, Cusi D, Matucci-Cerinic M, Melchers I, Koenig W, Salvi E, Wichmann HE, Cuomo G, Hachulla E, Diot E, Hunzelmann N, Caramaschi P, Mouthon L, Riccieri V, Distler J, Tarner I, Avouac J, Meyer O, Kahan A, Chiocchia G, Boileau C, Allanore Y (2011) Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum 63:3979–3987. https://doi.org/10.1002/art.30640
Carmona FD, Cénit MC, Diaz-Gallo LM, Broen JC, Simeón CP, Carreira PE, Callejas-Rubio JL, Fonollosa V, López-Longo FJ, González-Gay MA, Hunzelmann N, Riemekasten G, Witte T, Kreuter A, Distler JH, Madhok R, Shiels P, van Laar JM, Schuerwegh AJ, Vonk MC, Voskuyl AE, Fonseca C, Denton CP, Herrick A, Worthington J, Arnett FC, Tan FK, Assassi S, Radstake TR, Mayes MD, Martín J (2013) New insight on the Xq28 association with systemic sclerosis. Ann Rheum Dis 72:2032–2038. https://doi.org/10.1136/annrheumdis-2012-202742
D’Amico F, Fiorito G, Skarmoutsou E, Granata M, Rossi GA, Trovato C, Bellocchi C, Marchini M, Beretta L, Mazzarino MC (2018) FOXP3, ICOS and ICOSL gene polymorphisms in systemic sclerosis: FOXP3 rs2294020 is associated with disease progression in a female Italian population. Immunobiology 223:112–117. https://doi.org/10.1016/j.imbio.2017.10.004
Fioretto BS, Rosa I, Romano E, Wang Y, Guiducci S, Zhang G, Manetti M, Matucci-Cerinic M (2020) The contribution of epigenetics to the pathogenesis and gender dimorphism of systemic sclerosis: a comprehensive overview. Ther Adv Musculoskelet Dis. 12: 1759720x20918456. https://doi.org/10.1177/1759720x20918456
Matucci-Cerinic M, Kahaleh B, Wigley FM (2013) Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum 65:1953–1962. https://doi.org/10.1002/art.37988
Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G (1996) Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest 98:785–792. https://doi.org/10.1172/jci118851
Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G (2000) Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum 43:2550–2562
Giacomelli R, Matucci-Cerinic M, Cipriani P, Ghersetich I, Lattanzio R, Pavan A, Pignone A, Cagnoni ML, Lotti T, Tonietti G (1998) Circulating Vdelta1+ T cells are activated and accumulate in the skin of systemic sclerosis patients. Arthritis Rheum 41:327–334
Benyamine A, Magalon J, Sabatier F, Lyonnet L, Robert S, Dumoulin C, Morange S, Mazodier K, Kaplanski G, Reynaud-Gaubert M, Rossi P, Dignat-George F, Granel B, Paul P (2018) Natural Killer Cells Exhibit a Peculiar Phenotypic Profile in Systemic Sclerosis and Are Potent Inducers of Endothelial Microparticles Release. Front Immunol 9:1665. https://doi.org/10.3389/fimmu.2018.01665
Maehara T, Kaneko N, Perugino CA, Mattoo H, Kers J, Allard-Chamard H, Mahajan VS, Liu H, Murphy SJ, Ghebremichael M, Fox DA, Payne AS, Lafyatis R, Stone JH, Khanna D, Pillai S (2020) Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J Clin Invest. https://doi.org/10.1172/JCI131700
Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, Goulet JR, Rich E, Grodzicky T, Raymond Y, Senecal JL (2008) Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum 58:3902–3912. https://doi.org/10.1002/art.24038
Ingegnoli F, Ughi N, Crotti C, Mosca M, Tani C (2017) Outcomes, rates and predictors of transition of isolated Raynaud’s phenomenon: a systematic review and meta-analysis. Swiss Med Wkly 147:w14506. https://doi.org/10.4414/smw.2017.14506
Roumm AD, Whiteside TL, Medsger TA Jr, Rodnan GP (1984) Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum 27:645–653
Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P (1992) Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol 166:255–263
Freedman RR, Baer RP, Mayes MD (1995) Blockade of vasospastic attacks by alpha 2-adrenergic but not alpha 1-adrenergic antagonists in idiopathic Raynaud’s disease. Circulation 92:1448–1451. https://doi.org/10.1161/01.cir.92.6.1448
Vona R, Giovannetti A, Gambardella L, Malorni W, Pietraforte D, Straface E (2018) Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J Cell Mol Med 22:3308–3314. https://doi.org/10.1111/jcmm.13630
Ntelis K, Solomou EE, Sakkas L, Liossis SN, Daoussis D (2017) The role of platelets in autoimmunity, vasculopathy, and fibrosis: Implications for systemic sclerosis. Semin Arthritis Rheum 47:409–417. https://doi.org/10.1016/j.semarthrit.2017.05.004
Fleischmajer R, Perlish JS (1980) Capillary alterations in scleroderma. J Am Acad Dermatol 2:161–170. https://doi.org/10.1016/s0190-9622(80)80396-3
Rodnan GP, Myerowitz RL, Justh GO (1980) Morphologic changes in the digital arteries of patients with progressive systemic sclerosis (scleroderma) and Raynaud phenomenon. Medicine (Baltimore) 59:393–408. https://doi.org/10.1097/00005792-198011000-00001
Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, Ibba-Manneschi L, Matucci-Cerinic M (2017) Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis 76:924–934. https://doi.org/10.1136/annrheumdis-2016-210229
Giusti B, Fibbi G, Margheri F, Serratì S, Rossi L, Poggi F, Lapini I, Magi A, Del Rosso A, Cinelli M, Guiducci S, Kahaleh B, Bazzichi L, Bombardieri S, Matucci-Cerinic M, Gensini GF, Del Rosso M, Abbate R (2006) A model of anti-angiogenesis: differential transcriptosome profiling of microvascular endothelial cells from diffuse systemic sclerosis patients. Arthritis Res Ther 8:R115. https://doi.org/10.1186/ar2002
Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M (2011) Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res 109:e14-26. https://doi.org/10.1161/circresaha.111.242057
Mostmans Y, Cutolo M, Giddelo C, Decuman S, Melsens K, Declercq H, Vandecasteele E, De Keyser F, Distler O, Gutermuth J, Smith V (2017) The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun Rev 16:774–786. https://doi.org/10.1016/j.autrev.2017.05.024
Saigusa R, Asano Y, Taniguchi T, Yamashita T, Takahashi T, Ichimura Y, Toyama T, Tamaki Z, Tada Y, Sugaya M, Kadono T, Sato S (2015) A possible contribution of endothelial CCN1 downregulation due to Fli1 deficiency to the development of digital ulcers in systemic sclerosis. Exp Dermatol 24:127–132. https://doi.org/10.1111/exd.12602
Truchetet ME, Demoures B, Eduardo Guimaraes J, Bertrand A, Laurent P, Jolivel V, Douchet I, Jacquemin C, Khoryati L, Duffau P, Lazaro E, Richez C, Seneschal J, Doutre MS, Pellegrin JL, Constans J, Schaeverbeke T, Blanco P, Contin-Bordes C (2016) Platelets Induce Thymic Stromal Lymphopoietin Production by Endothelial Cells:Contribution to Fibrosis in Human Systemic Sclerosis. Arthritis & rheumatology. 68:2784–2794. https://doi.org/10.1002/art.39817
Choi Y-S, Choi H-J, Min J-K, Pyun B-J, Maeng Y-S, Park H, Kim J, Kim Y-M, Kwon Y-G (2009) Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 114:3117–3126. https://doi.org/10.1182/blood-2009-02-203372
Manetti M, Ibba-Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, Marrelli A, Conforti ML, Romano E, Giacomelli R, Matucci-Cerinic M, Cipriani P (2010) The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis 69:598–605. https://doi.org/10.1136/ard.2009.119321
Vettori S, Cuomo G, Iudici M, D’Abrosca V, Giacco V, Barra G, De Palma R, Valentini G (2014) Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol 34:663–668. https://doi.org/10.1007/s10875-014-0037-0
Zhang YJ, Zhang Q, Yang GJ, Tao JH, Wu GC, Huang XL, Duan Y, Li XP, Ye DQ, Wang J (2018) Elevated serum levels of interleukin-1β and interleukin-33 in patients with systemic sclerosis in Chinese population. Z Rheumatol 77:151–159. https://doi.org/10.1007/s00393-016-0202-3
Wynn TA (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4:583–594
Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2013) Host responses in tissue repair and fibrosis. Ann Rev Pathol 8:241–276. https://doi.org/10.1146/annurev-pathol-020712-163930
Gieseck RL 3rd, Wilson MS, Wynn TA (2018) Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18:62–76. https://doi.org/10.1038/nri.2017.90
Chizzolini C, Boin F (2015) The role of the acquired immune response in systemic sclerosis. Semin Immunopathol 37:519–528. https://doi.org/10.1007/s00281-015-0509-1
Henderson NC, Rieder F, Wynn TA (2020) Fibrosis: from mechanisms to medicines. Nature 587:555–566. https://doi.org/10.1038/s41586-020-2938-9
Varga J, Pasche B (2009) Transforming growth factor β as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol 5:200–206. https://doi.org/10.1038/nrrheum.2009.26
Frangogiannis N (2020) Transforming growth factor-β in tissue fibrosis. J Exp Med 217:e20190103. https://doi.org/10.1084/jem.20190103
Györfi AH, Matei AE, Distler JHW (2018) Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol 68–69:8–27. https://doi.org/10.1016/j.matbio.2017.12.016
Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL, Renzoni EA, Carter DE, Bou-Gharios G, Stratton RJ, Pearson JD, Beier F, Lyons KM, Black CM, Abraham DJ, Leask A (2006) CCN2 is necessary for adhesive responses to transforming growth factor-β1 in embryonic fibroblasts*. J Biol Chem 281:10715–10726. https://doi.org/10.1074/jbc.M511343200
Distler JHW, Gyorfi AH, Ramanujam M, Whitfield ML, Konigshoff M, Lafyatis R (2019) Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 15:705–730. https://doi.org/10.1038/s41584-019-0322-7
Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, McCalmont TH, Brown PO, Botstein D, Connolly MK (2003) Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A 100:12319–12324. https://doi.org/10.1073/pnas.1635114100
Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML (2012) Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol 132:1363–1373. https://doi.org/10.1038/jid.2011.472
Lofgren S, Hinchcliff M, Carns M, Wood T, Aren K, Arroyo E, Cheung P, Kuo A, Valenzuela A, Haemel A, Wolters PJ, Gordon J, Spiera R, Assassi S, Boin F, Chung L, Fiorentino D, Utz PJ, Whitfield ML, Khatri P (2016) Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI Insight 1:e89073. https://doi.org/10.1172/jci.insight.89073
Franks JM, Martyanov V, Cai G, Wang Y, Li Z, Wood TA, Whitfield ML (2019) A machine learning classifier for assigning individual patients with systemic sclerosis to intrinsic molecular subsets. Arthritis Rheumatol 71:1701–1710. https://doi.org/10.1002/art.40898
Mutsaers SE, Bishop JE, McGrouther G, Laurent GJ (1997) Mechanisms of tissue repair: from wound healing to fibrosis. Int J Biochem Cell Biol 29:5–17
Hinz B, Lagares D (2020) Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol 16:11–31. https://doi.org/10.1038/s41584-019-0324-5
Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G (2001) Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol 159:1009–1020. https://doi.org/10.1016/S0002-9440(10)61776-2
Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, White ES, Varga J (2014) FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 6:232ra250. https://doi.org/10.1126/scitranslmed.3008264
Lynch MD, Watt FM (2018) Fibroblast heterogeneity: implications for human disease. J Clin Invest 128:26–35. https://doi.org/10.1172/jci93555
Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, Liu N, Kulur V, Yao C, Chen P, Liu Z, Stripp B, Tang J, Liang J, Noble PW, Jiang D (2018) Single-Cell Deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep 22:3625–3640. https://doi.org/10.1016/j.celrep.2018.03.010
Griffin MF, desJardins-Park HE, Mascharak S, Borrelli MR, Longaker MT (2020) Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis Models Mech 13 https://doi.org/10.1242/dmm.044164
Gabbiani G, Majno G (1972) Dupuytren’s contracture: fibroblast contraction? An ultrastructural study. Am J Pathol 66:131–146
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816. https://doi.org/10.2353/ajpath.2007.070112
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180:1340–1355. https://doi.org/10.1016/j.ajpath.2012.02.004
Russo B, Brembilla NC, Chizzolini C (2020) Interplay between keratinocytes and fibroblasts: a systematic review providing a new angle for understanding skin fibrotic disorders. Front Immunol 11:648. https://doi.org/10.3389/fimmu.2020.00648
Aden N, Shiwen X, Aden D, Black C, Nuttall A, Denton CP, Leask A, Abraham D, Stratton R (2008) Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology (Oxford) 47:1754–1760. https://doi.org/10.1093/rheumatology/ken370
Aden N, Nuttall A, Shiwen X, de Winter P, Leask A, Black CM, Denton CP, Abraham DJ, Stratton RJ (2010) Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. J Invest Dermatol 130:2191–2200. https://doi.org/10.1038/jid.2010.120
Canady J, Arndt S, Karrer S, Bosserhoff AK (2013) Increased KGF expression promotes fibroblast activation in a double paracrine manner resulting in cutaneous fibrosis. J Invest Dermatol 133:647–657. https://doi.org/10.1038/jid.2012.389
Nikitorowicz-Buniak J, Shiwen X, Denton CP, Abraham D, Stratton R (2014) Abnormally differentiating keratinocytes in the epidermis of systemic sclerosis patients show enhanced secretion of CCN2 and S100A9. J Invest Dermatol 134:2693–2702. https://doi.org/10.1038/jid.2014.253
McCoy SS, Reed TJ, Berthier CC, Tsou PS, Liu J, Gudjonsson JE, Khanna D, Kahlenberg JM (2017) Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology (Oxford) 56:1970–1981. https://doi.org/10.1093/rheumatology/kex280
Russo B, Borowczyk J, Boehncke WH, Truchetet ME, Modarressi A, Brembilla NC, Chizzolini C (2021) Dysfunctional keratinocytes increase dermal inflammation in systemic sclerosis. Results from tissue-engineered scleroderma epidermis. Arthritis Rheumatol. https://doi.org/10.1002/art.41659
Takahashi T, Asano Y, Sugawara K, Yamashita T, Nakamura K, Saigusa R, Ichimura Y, Toyama T, Taniguchi T, Akamata K, Noda S, Yoshizaki A, Tsuruta D, Trojanowska M, Sato S (2017) Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: Possible roles in scleroderma. J Exp Med 214:1129–1151. https://doi.org/10.1084/jem.20160247
Brembilla NC, Dufour AM, Alvarez M, Hugues S, Montanari E, Truchetet ME, Lonati P, Fontao L, Gabrielli A, Vettori S, Valentini G, Boehncke WH, Meroni P, Chizzolini C (2016) IL-22 capacitates dermal fibroblast responses to TNF in scleroderma. Ann Rheum Dis 75:1697–1705. https://doi.org/10.1136/annrheumdis-2015-207477
Dufour AM, Borowczyk-Michalowska J, Alvarez M, Truchetet ME, Modarressi A, Brembilla NC, Chizzolini C (2020) IL-17A dissociates inflammation from fibrogenesis in systemic sclerosis (scleroderma). J Invest Dermatol 140:103–112. https://doi.org/10.1016/j.jid.2019.05.026
Kuwana M, Medsger TA Jr, Wright TM (1995) T cell proliferative response induced by DNA topoisomerase I in patients with systemic sclerosis and healthy donors. J Clin Invest 96:586–596
Fava A, Cimbro R, Wigley FM, Liu QR, Rosen A, Boin F (2016) Frequency of circulating topoisomerase-I-specific CD4 T cells predicts presence and progression of interstitial lung disease in scleroderma. Arthritis Res Ther 18:99. https://doi.org/10.1186/s13075-016-0993-2
Sakkas LI, Xu B, Artlett CM, Lu S, Jimenez SA, Platsoucas CD (2002) Oligoclonal T cell expansion in the skin of patients with systemic sclerosis. J Immunol 168:3649–3659
Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, Craig R, Hirano I, Marshall K, Ruderman E, Jovanovic B, Milanetti F, Jain S, Boyce K, Morgan A, Carr J, Barr W (2011) Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet 378:498–506. https://doi.org/10.1016/S0140-6736(11)60982-3
van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, Schuerwegh AJ, Marijt EW, Vonk MC, Schattenberg AV, Matucci-Cerinic M, Voskuyl AE, van de Loosdrecht AA, Daikeler T, Kotter I, Schmalzing M, Martin T, Lioure B, Weiner SM, Kreuter A, Deligny C, Durand JM, Emery P, Machold KP, Sarrot-Reynauld F, Warnatz K, Adoue DF, Constans J, Tony HP, Del Papa N, Fassas A, Himsel A, Launay D, Lo Monaco A, Philippe P, Quere I, Rich E, Westhovens R, Griffiths B, Saccardi R, van den Hoogen FH, Fibbe WE, Socie G, Gratwohl A, Tyndall A (2014) Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA J Am Med Assoc 311:2490–2498. https://doi.org/10.1001/jama.2014.6368
Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, Mayes MD, Nash RA, Crofford LJ, Eggleston B, Castina S, Griffith LM, Goldstein JS, Wallace D, Craciunescu O, Khanna D, Folz RJ, Goldin J, St Clair EW, Seibold JR, Phillips K, Mineishi S, Simms RW, Ballen K, Wener MH, Georges GE, Heimfeld S, Hosing C, Forman S, Kafaja S, Silver RM, Griffing L, Storek J, LeClercq S, Brasington R, Csuka ME, Bredeson C, Keever-Taylor C, Domsic RT, Kahaleh MB, Medsger T, Furst DE (2018) Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med 378:35–47
Yoshizaki A, Yanaba K, Ogawa A, Asano Y, Kadono T, Sato S (2011) Immunization with DNA topoisomerase I and Freund’s complete adjuvant induces skin and lung fibrosis and autoimmunity via interleukin-6 signaling. Arthritis Rheum 63:3575–3585. https://doi.org/10.1002/art.30539
Hu PQ, Hurwitz AA, Oppenheim JJ (2007) Immunization with DNA topoisomerase I induces autoimmune responses but not scleroderma-like pathologies in mice. J Rheumatol 34:2243–2252
Henderson J, Bhattacharyya S, Varga J, O’Reilly S (2018) Targeting TLRs and the inflammasome in systemic sclerosis. Pharmacol Ther 192:163–169. https://doi.org/10.1016/j.pharmthera.2018.08.003
Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, Lee J, Christmann RB, Lafyatis R (2014) Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther 16:R136. https://doi.org/10.1186/ar4598
Takahashi T, Asano Y, Ichimura Y, Toyama T, Taniguchi T, Noda S, Akamata K, Tada Y, Sugaya M, Kadono T, Sato S (2015) Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol 67:254–265. https://doi.org/10.1002/art.38901
Farina A, Peruzzi G, Lacconi V, Lenna S, Quarta S, Rosato E, Vestri AR, York M, Dreyfus DH, Faggioni A, Morrone S, Trojanowska M, Farina GA (2017) Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res Ther 19:39. https://doi.org/10.1186/s13075-017-1237-9
Ciechomska M, Huigens CA, Hugle T, Stanly T, Gessner A, Griffiths B, Radstake TR, Hambleton S, O’Reilly S, van Laar JM (2013) Toll-like receptor-mediated, enhanced production of profibrotic TIMP-1 in monocytes from patients with systemic sclerosis: role of serum factors. Ann Rheum Dis 72:1382–1389. https://doi.org/10.1136/annrheumdis-2012-201958
Kioon MDA, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, Gordon JK, Barrat FJ (2018) Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 10. https://doi.org/10.1126/scitranslmed.aam8458
Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, Alm GV, Ronnblom L (2010) Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 69:1396–1402. https://doi.org/10.1136/ard.2009.121400
Lande R, Lee EY, Palazzo R, Marinari B, Pietraforte I, Santos GS, Mattenberger Y, Spadaro F, Stefanantoni K, Iannace N, Dufour AM, Falchi M, Bianco M, Botti E, Bianchi L, Alvarez M, Riccieri V, Truchetet ME, G CLW, Chizzolini C, Frasca L, (2019) CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-alpha production in systemic sclerosis. Nat Commun 10:1731. https://doi.org/10.1038/s41467-019-09683-z
Ryu C, Walia A, Ortiz V, Perry C, Woo S, Reeves BC, Sun H, Winkler J, Kanyo JE, Wang W, Vukmirovic M, Ristic N, Stratton EA, Meena SR, Minasyan M, Kurbanov D, Liu X, Lam TT, Farina G, Gomez JL, Gulati M, Herzog EL (2020) Bioactive plasma mitochondrial DNA is associated with disease progression in scleroderma-associated interstitial lung disease. Arthritis Rheumatol 72:1905–1915. https://doi.org/10.1002/art.41418
Kania G, Rudnik M, Distler O (2019) Involvement of the myeloid cell compartment in fibrogenesis and systemic sclerosis. Nat Rev Rheumatol 15:288–302. https://doi.org/10.1038/s41584-019-0212-z
Jeljeli M, Riccio LGC, Doridot L, Chene C, Nicco C, Chouzenoux S, Deletang Q, Allanore Y, Kavian N, Batteux F (2019) Trained immunity modulates inflammation-induced fibrosis. Nat Commun 10:5670. https://doi.org/10.1038/s41467-019-13636-x
van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, Farina GA, Stifano G, Mathes AL, Cossu M, York M, Collins C, Wenink M, Huijbens R, Hesselstrand R, Saxne T, DiMarzio M, Wuttge D, Agarwal SK, Reveille JD, Assassi S, Mayes M, Deng Y, Drenth JP, de Graaf J, den Heijer M, Kallenberg CG, Bijl M, Loof A, van den Berg WB, Joosten LA, Smith V, de Keyser F, Scorza R, Lunardi C, van Riel PL, Vonk M, van Heerde W, Meller S, Homey B, Beretta L, Roest M, Trojanowska M, Lafyatis R, Radstake TR (2014) Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med 370:433–443. https://doi.org/10.1056/NEJMoa1114576
Ross RL, Corinaldesi C, Migneco G, Carr IM, Antanaviciute A, Wasson CW, Carriero A, Distler JHW, Holmes S, El-Sherbiny YM, McKimmie CS, Del Galdo F (2021) Targeting human plasmacytoid dendritic cells through BDCA2 prevents skin inflammation and fibrosis in a novel xenotransplant mouse model of scleroderma. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2020-218439
Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, Gelse K, Distler O, Schett G, Distler JH, Ramming A (2015) Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2015-207388
Laurent P, Allard B, Manicki P, Jolivel V, Levionnois E, Jeljeli M, Henrot P, Izotte J, Leleu D, Groppi A, Seneschal J, Constans J, Chizzolini C, Richez C, Duffau P, Lazaro E, Forcade E, Schaeverbeke T, Pradeu T, Batteux F, Blanco P, Contin-Bordes C, Truchetet ME (2021) TGFβ promotes low IL10-producing ILC2 with profibrotic ability involved in skin fibrosis in systemic sclerosis. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2020-219748
Luo JY, Liu X, Jiang M, Zhao HP, Zhao JJ (2017) Oxidative stress markers in blood in systemic sclerosis: A meta-analysis. Mod Rheumatol 27:306–314. https://doi.org/10.1080/14397595.2016.1206510
Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG (2013) ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res 23:898–914. https://doi.org/10.1038/cr.2013.75
Spadoni T, Svegliati Baroni S, Amico D, Albani L, Moroncini G, Avvedimento EV, Gabrielli A (2015) A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol 67:1611–1622. https://doi.org/10.1002/art.39084
Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD (2017) Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther 19:144. https://doi.org/10.1186/s13075-017-1331-z
Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD (2011) The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum 63:3563–3574. https://doi.org/10.1002/art.30568
Henderson J, O’Reilly S (2017) Inflammasome lights up in systemic sclerosis. Arthritis Res Ther 19:205. https://doi.org/10.1186/s13075-017-1420-z
Scharffetter K, Lankat-Buttgereit B, Krieg T (1988) Localization of collagen mRNA in normal and scleroderma skin by in-situ hybridization. Eur J Clin Invest 18:9–17
Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, Hant FN, Gordon JK, Shah AA, Zhu L, Zheng WJ, Browning JL, Barron AMS, Wu M, Visvanathan S, Baum P, Franks JM, Whitfield ML, Shanmugam VK, Domsic RT, Castelino FV, Bernstein EJ, Wareing N, Lyons MA, Ying J, Charles J, Mayes MD, Assassi S (2020) Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 79:379–386. https://doi.org/10.1136/annrheumdis-2019-215894
Fuschiotti P (2020) T cells in SSc skin lesions: knowing your enemy. Nat Rev Rheumatol 16:253–254. https://doi.org/10.1038/s41584-020-0404-6
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, Senior RM, Elias JA (2001) Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194:809–821
Chizzolini C, Rezzonico R, Ribbens C, Burger D, Wollheim FA, Dayer JM (1998) Inhibition of type I collagen production by dermal fibroblasts upon contact with activated T cells: different sensitivity to inhibition between systemic sclerosis and control fibroblasts. Arthritis Rheum 41:2039–2047
Truchetet ME, Brembilla NC, Montanari E, Lonati P, Raschi E, Zeni S, Fontao L, Meroni PL, Chizzolini C (2013) Interleukin-17A+ cell counts are increased in systemic sclerosis skin and their number is inversely correlated with the extent of skin involvement. Arthritis Rheum 65:1347–1356. https://doi.org/10.1002/art.37860
Taylor DK, Mittereder N, Kuta E, Delaney T, Burwell T, Dacosta K, Zhao W, Cheng LI, Brown C, Boutrin A, Guo X, White WI, Zhu J, Dong H, Bowen MA, Lin J, Gao C, Yu L, Ramaswamy M, Gaudreau MC, Woods R, Herbst R, Carlesso G (2018) T follicular helper-like cells contribute to skin fibrosis. Sci Transl Med 10. https://doi.org/10.1126/scitranslmed.aaf5307
Ricard L, Jachiet V, Malard F, Ye Y, Stocker N, Riviere S, Senet P, Monfort JB, Fain O, Mohty M, Gaugler B, Mekinian A (2019) Circulating follicular helper T cells are increased in systemic sclerosis and promote plasmablast differentiation through the IL-21 pathway which can be inhibited by ruxolitinib. Ann Rheum Dis 78:539–550. https://doi.org/10.1136/annrheumdis-2018-214382
Lafyatis R, O’Hara C, Feghali-Bostwick CA, Matteson E (2007) B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum 56:3167–3168
Francois A, Chatelus E, Wachsmann D, Sibilia J, Bahram S, Alsaleh G, Gottenberg JE (2013) B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthritis Res Ther 15:R168. https://doi.org/10.1186/ar4352
Mavropoulos A, Simopoulou T, Varna A, Liaskos C, Katsiari C, Bogdanos DP, Sakkas LI (2015) B regulatory cells are decreased and functionally impaired in patients with systemic sclerosis. Arthritis Rheumatol https://doi.org/10.1002/art.39437
Sato S, Hayakawa I, Hasegawa M, Fujimoto M, Takehara K (2003) Function blocking autoantibodies against matrix metalloproteinase-1 in patients with systemic sclerosis. J Invest Dermatol 120:542–547
Nishijima C, Hayakawa I, Matsushita T, Komura K, Hasegawa M, Takehara K, Sato S (2004) Autoantibody against matrix metalloproteinase-3 in patients with systemic sclerosis. Clin Exp Immunol 138:357–363
Luchetti MM, Moroncini G, Jose Escamez M, Svegliati Baroni S, Spadoni T, Grieco A, Paolini C, Funaro A, Avvedimento EV, Larcher F, Del Rio M, Gabrielli A (2016) Induction of scleroderma fibrosis in skin-humanized mice by administration of anti-platelet-derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol 68:2263–2273. https://doi.org/10.1002/art.39728
Moroncini G, Svegliati Baroni S, Gabrielli A (2018) Agonistic antibodies in systemic sclerosis. Immunol Lett 195:83–87. https://doi.org/10.1016/j.imlet.2017.10.007
Palm NW, de Zoete MR, Flavell RA (2015) Immune-microbiota interactions in health and disease. Clin Immunol 159:122–127. https://doi.org/10.1016/j.clim.2015.05.014
Johnson ME, Franks JM, Cai G, Mehta BK, Wood TA, Archambault K, Pioli PA, Simms RW, Orzechowski N, Arron S, Whitfield ML (2019) Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression in systemic sclerosis skin. Arthritis Res Ther 21:49. https://doi.org/10.1186/s13075-019-1816-z
Volkmann ER, Chang YL, Barroso N, Furst DE, Clements PJ, Gorn AH, Roth BE, Conklin JL, Getzug T, Borneman J, McGovern DP, Tong M, Jacobs JP, Braun J (2016) Association of systemic sclerosis with a unique colonic microbial consortium. Arthritis Rheumatol 68:1483–1492. https://doi.org/10.1002/art.39572
Patrone V, Puglisi E, Cardinali M, Schnitzler TS, Svegliati S, Festa A, Gabrielli A, Morelli L (2017) Gut microbiota profile in systemic sclerosis patients with and without clinical evidence of gastrointestinal involvement. Sci Rep 7:14874. https://doi.org/10.1038/s41598-017-14889-6
Bellocchi C, Fernández-Ochoa Á, Montanelli G, Vigone B, Santaniello A, Milani C, Quirantes-Piné R, Borrás-Linares I, Ventura M, Segura-Carrettero A, Alarcón-Riquelme ME, Beretta L (2018) Microbial and metabolic multi-omic correlations in systemic sclerosis patients. Ann N Y Acad Sci 1421:97–109. https://doi.org/10.1111/nyas.13736
Plichta DR, Somani J, Pichaud M, Wallace ZS, Fernandes AD, Perugino CA, Lähdesmäki H, Stone JH, Vlamakis H, Chung DC, Khanna D, Pillai S, Xavier RJ (2021) Congruent microbiome signatures in fibrosis-prone autoimmune diseases: IgG4-related disease and systemic sclerosis. Genome Med 13:35. https://doi.org/10.1186/s13073-021-00853-7
Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, Whitfield ML (2008) Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One 3:e2696. https://doi.org/10.1371/journal.pone.0002696
Derrett-Smith EC, Martyanov V, Chighizola CB, Moinzadeh P, Campochiaro C, Khan K, Wood TA, Meroni PL, Abraham DJ, Ong VH, Lafyatis R, Whitfield ML, Denton CP (2017) Limited cutaneous systemic sclerosis skin demonstrates distinct molecular subsets separated by a cardiovascular development gene expression signature. Arthritis Res Ther 19:156. https://doi.org/10.1186/s13075-017-1360-7
Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, Hinchcliff ME, Whitfield ML (2015) Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol 11:e1004005. https://doi.org/10.1371/journal.pcbi.1004005
Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, Lafyatis RA, Denton CP, Hinchcliff ME, Pioli PA, Mahoney JM, Whitfield ML (2017) A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med 9:27. https://doi.org/10.1186/s13073-017-0417-1
Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, Tashkin DP, Jahan-Tigh RR, Mayes MD, Gudjonsson JE, Chang JT (2015) Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 67:3016–3026. https://doi.org/10.1002/art.39289
Moon SJ, Bae JM, Park KS, Tagkopoulos I, Kim KJ (2019) Compendium of skin molecular signatures identifies key pathological features associated with fibrosis in systemic sclerosis. Ann Rheum Dis 78:817–825. https://doi.org/10.1136/annrheumdis-2018-214778
Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, Tamaki Z, Lee J, Carns M, Podlusky S, Sirajuddin A, Shah SJ, Chang RW, Lafyatis R, Varga J, Whitfield ML (2013) Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 133:1979–1989. https://doi.org/10.1038/jid.2013.130
Chakravarty EF, Martyanov V, Fiorentino D, Wood TA, Haddon DJ, Jarrell JA, Utz PJ, Genovese MC, Whitfield ML, Chung L (2015) Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res Ther 17:159. https://doi.org/10.1186/s13075-015-0669-3
Khanna D, Spino C, Johnson S, Chung L, Whitfield ML, Denton CP, Berrocal V, Franks J, Mehta B, Molitor J, Steen VD, Lafyatis R, Simms RW, Gill A, Kafaja S, Frech TM, Hsu V, Domsic RT, Pope JE, Gordon JK, Mayes MD, Schiopu E, Young A, Sandorfi N, Park J, Hant FN, Bernstein EJ, Chatterjee S, Castelino FV, Ajam A, Wang Y, Wood T, Allanore Y, Matucci-Cerinic M, Distler O, Singer O, Bush E, Fox DA, Furst DE (2020) Abatacept in early diffuse cutaneous systemic sclerosis: results of a Phase II investigator-initiated, multicenter, double-blind, randomized. placebo-controlled trial. Arthritis Rheumatol 72:125–136. https://doi.org/10.1002/art.41055
Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, Milano A, Connolly MK, Ratiner BD, Wiskocil RL, Whitfield ML, Chang HY, Robinson WH (2009) Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum 60:584–591. https://doi.org/10.1002/art.24221
Martyanov V, Kim GJ, Hayes W, Du S, Ganguly BJ, Sy O, Lee SK, Bogatkevich GS, Schieven GL, Schiopu E, Marangoni RG, Goldin J, Whitfield ML, Varga J (2017) Novel lung imaging biomarkers and skin gene expression subsetting in dasatinib treatment of systemic sclerosis-associated interstitial lung disease. PLoS One 12:e0187580. https://doi.org/10.1371/journal.pone.0187580
Franks JM, Martyanov V, Wang Y, Wood TA, Pinckney A, Crofford LJ, Keyes-Elstein L, Furst DE, Goldmuntz E, Mayes MD, McSweeney P, Nash RA, Sullivan KM, Whitfield ML (2020) Machine learning predicts stem cell transplant response in severe scleroderma. Ann Rheum Dis 79:1608–1615. https://doi.org/10.1136/annrheumdis-2020-217033
Acknowledgements
We thank Dr Paolo Airò (Rheumatology, Brescia, Italy) for his critical reading of our manuscript.
Funding
Open Access funding provided by Université de Genève. Work supported in part by grant 310030-159999 from the Swiss National Science Foundation and by a grant from sclerodermie.ch (Swiss Scleroderma Patient organization) to CC and by the French Rheumatology Society Rhumato-network to MET.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Truchetet, M.E., Brembilla, N.C. & Chizzolini, C. Current Concepts on the Pathogenesis of Systemic Sclerosis. Clinic Rev Allerg Immunol 64, 262–283 (2023). https://doi.org/10.1007/s12016-021-08889-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08889-8