
Abstract
Purpose of Review
Ischemic cardiomyopathy refers to systolic left ventricular dysfunction in the setting of obstructive coronary artery disease and represents the most common cause of heart failure worldwide. It is often the combination of an irreversible loss of viable mass following an acute myocardial infarction (AMI) with a dysfunctional, but still viable, myocardium in the context of a chronically reduced myocardial blood flow and reduced coronary reserve. Medical treatments aiming at modulating neurohumoral response and restoring blood flow to the ischemic cardiomyocytes were shown to dramatically abate the occurrence of ventricular dysfunction and adverse remodeling in ischemic cardiomyopathy.
Recent Findings
Novel therapeutic approaches, such as mechanical unloading and modulation of the inflammatory response, appear to be promising. Furthermore, the understanding of the mechanisms by which, despite optimal treatment, heart failure ensues after AMI, with or without adverse remodeling and systolic dysfunction, is a critical step in the search for novel ways to tackle heart failure risk beyond preservation of left ventricular volumes and systolic function.
Summary
In this review article, we explore the principal pathophysiological mechanisms and pathways of heart failure in ischemic cardiomyopathy, therapeutic opportunities, and knowledge gaps in this area.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic cardiomyopathy refers to systolic left ventricular (LV) dysfunction in the setting of obstructive coronary artery disease (CAD), and represents the most common cause of heart failure (HF) worldwide [1]. Left ventricular dysfunction in patients with CAD is often the consequence of an irreversible loss of viable mass following an acute myocardial infarction (AMI), occasionally in combination with loss of contractility in ischemic, but still viable, myocardium (hibernating myocardium) [1, 2]. This has relevant therapeutic and prognostic implications since loss of contractility in hibernating myocardium is potentially reversible upon resolution of ischemia [3]. Irrespective of the underlying mechanism, patients with ischemic cardiomyopathy may have some degree of HF symptoms. The improvements in the acute treatment of myocardial infarction, revascularization strategies, and guideline-directed medical therapies led at reducing the incidence and degree of systolic dysfunction and adverse remodeling following AMI, yet the risk of HF remains substantial, suggesting that other mechanisms beyond LV systolic dysfunction and remodeling may also be implicated in HF symptoms following AMI [4,5,6, 7•, 8•, 9, 10].
In this review article, we explore the principal pathophysiological mechanisms and pathways of HF in ischemic cardiomyopathy, therapeutic opportunities, and knowledge gaps in this area.
Determinants of Left Ventricular Dysfunction in Ischemic Cardiomyopathy
Infarct Size
The infarct size following AMI is the most important predictor of LV dysfunction and remodeling after AMI [11, 12]. Larger infarct size due to late presentation or late reperfusion, no or minimal collateral flow, or anterior location is associated with greater LV dysfunction, adverse cardiac remodeling, and HF over time [13]. In a patient-level meta-analysis of 10 randomized trials including patients undergoing primary percutaneous coronary intervention (PCI), infarct size was strongly associated with subsequent mortality and hospitalization for HF [14].
Myocardial Stunning
The degree of LV dysfunction during the acute phase of AMI is not only the result of the irreversibly myocardial loss but may be greater due to the contribution of myocardial “stunning,” regions of depressed cardiac function following the reperfusion of an occluded artery that gradually recover over the course of a days following a successful reperfusion [15]. The underlying mechanisms involve oxygen radical damage following reperfusion and altered calcium flux with calcium overload that then desensitizes the myofilaments [16]. This phenomenon, initially described following reperfusion using thrombolytic therapy, has also been shown to play a role in the current era of primary PCI, and contributes to the overall LV dysfunction [15].
Coronary Microvascular Injury
Coronary microvascular injury following the recanalization of an occluded epicardial coronary artery is another important determinant of the final infarct size, and a predictor of future adverse remodeling and outcomes [17, 18]. This phenomenon has now been better characterized using CMR as a typical pattern with contrast-enhanced infarct area and contrast-void infarct core, known as “microvascular obstruction” (MVO) [17]. MVO is associated with further cardiomyocyte injury and inflammation through the development of a severe microvascular damage, which can cause the occurrence of intramyocardial hemorrhage with red blood cell extravasation and residual iron deposits triggering intramyocardial inflammation [17]. The mechanisms involved in the pathogenesis of coronary microvascular injury include ischemia- and reperfusion-related injury, distal embolization, and individual susceptibility.
All these mechanisms contribute to a worse prognosis by causing a larger infarct size, myocardial dysfunction, and adverse remodeling [19].
Changes in Hemodynamics and Neurohormonal Activation
After AMI, the loss of contractile myocytes and reduced stroke volume leads to LV dilatation. This phenomenon can be acutely beneficial by maintaining stroke volume through the Frank-Starling mechanism. However, the LV wall stress is proportional to the radius of the ventricular cavity and inversely correlated to LV wall thickness, as explained by Laplace law [20]. Hence, LV dilatation increases wall stress and therefore oxygen consumption, leading to subendocardial myocardial ischemia via impaired coronary perfusion pressure, which fosters impaired LV contractility, reduced cardiac output, and the activation of adverse neurohormonal pathways over time. As part of the adverse ventricular remodeling post-AMI, cardiac geometry changes from elliptic shape to a spherical one, which is mainly driven by hypertrophic myocyte elongation in the non-infarcted zone, resulting in increase in wall mass and LV enlargement [20]. Furthermore, remote myocardial dysfunction could be seen as secondary to morphological changes in the infarct region, leading to an increased systolic longitudinal wall stress and loss in global ventricular function [21]. A relative increase in LV end-diastolic volume (LVEDV) of at least 20% using echocardiography has been traditionally used to define LV adverse remodeling. Compared with patients without adverse remodeling, those experiencing adverse remodeling had a greater risk of the composite of cardiovascular death, AMI, HF, stroke, or resuscitated cardiac arrest. This global adverse remodeling response leads to increases in both LV end-diastolic and end-systolic volumes and reduced LV ejection fraction (LVEF) [22,23,24]. Myocardial structural and functional changes in patients with ischemic cardiomyopathy are summarized in Table 1.
Reduced cardiac output promotes the activation of renin–angiotensin–aldosterone system (RAAS). Angiotensin II alters gene expression contributing to myocyte growth, affects cardiac metabolism and energetics, and increases protein synthesis in both fibroblasts and myocytes, thus increasing extracellular matrix deposition and fibrosis [25].
Inflammation
AMI is a trigger for the inflammatory response aimed at clearing necrotic debris, which is then followed by an anti-inflammatory reparative phase leading to myocardial scar formation. Perturbation of this balance may acutely worsen the infarct size and chronically contributes to adverse remodeling and post-AMI [26]. While some degree of response is necessary to recruit leukocytes and clear the tissue debris, inflammation becomes itself a mechanism of disease. A sustained and dysregulated inflammation promotes cardiomyocyte death (through apoptosis or pyroptosis), impairs contractile function of the surviving cardiomyocytes through the cardiodepressant effects of some cytokines (i.e., interleukin 1), and promotes the disruption of the interstitial tissue hampering the formation of a structured infarct scar [26]. Elevated levels of C-reactive protein (a marker of systemic inflammatory response) have been associated with worse prognosis in patients with AMI and HF [27,28,29]. The inflammatory response is proportional to the degree of myocardial injury, so that larger AMI are associated with a greater inflammatory response.
Hibernating Myocardium
LV dysfunction in the setting of CAD may also occur in patients without history of AMI. Repeated episodes of stunning as well as chronic low flow may induce metabolic and cellular adaptation leading to development of dedifferentiation of cardiomyocytes (hibernating myocardium) to prevent myocardial necrosis. Hibernating myocardium is viable myocardium, and therefore retains the ability to recover function upon revascularization [3]. The most common cause for the development of this condition is that a region of the myocardium is supplied by a stenotic coronary artery in which enough blood supply is available to maintain viability but not enough to maintain normal contractility of the region [2].
Other Factors
Besides scar and stunning/hibernation, LV dysfunction in patients with CAD may also be the result of other contributing, pre-existent, or superimposed factors, such as ventricular interdependency, electromechanical desynchrony (i.e., left bundle brunch block), prior scar, or other causes of cardiomyopathies (genetic, metabolic, or toxic) that may coexist and meaningfully contribute to LV remodeling, dysfunction, and HF syndrome.
Therapeutic Options
Several treatment options are currently available, and supported by strong evidence, in order to prevent or treat HF in ischemic cardiomyopathy. Prompt reperfusion of the infarct-related artery remains the mainstay treatment for AMI. As cardiomyocyte death per unit of time appears to be curvilinear with time from arterial occlusion, achievement of arterial patency in the first few hours after AMI allows for myocardial salvage and reduced infarct size, and subsequently might reduce the incidence of heart failure and other adverse outcomes [14, 30]. Thrombolysis was the first reperfusion strategy for AMI. In addition to be effective in abating in-hospital mortality [31], thrombolytic therapy was shown to significantly reduce infarct size by approximately 30% compared to placebo, with the largest benefit observed among patients who received thrombolysis within 1 h from the onset of symptoms [32]. Primary PCI was subsequently shown to allow for higher rates of effective revascularization compared to thrombolysis, which, combined with lower incidence of bleeding and reinfarction, led to a further improvement in outcomes for AMI patients [33]. In addition, primary PCI was shown to further reduce infarct size [34]. In patients with hibernating myocardium, revascularization of the viable myocardium is associated with improvement in cardiac systolic function, reverse remodeling, patient functional status, and HF symptoms. A comprehensive review of contemporary viability imaging techniques and their applications in clinical practice goes beyond the scope of this review article and can be found elsewhere [2].
Subsequent management strategies focus on modulating and interrupting the mal-adaptive process that led to infarct expansion and replacement fibrosis deposition within the cardiac muscle. Contemporary state-of-the-art pharmacological treatment focuses on modulation of the renin–angiotensin–aldosterone and of beta-adrenergic receptors. Of note, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARB) were shown to reduce the development of myocardial fibrosis and hypertrophy, as well as LV adverse remodeling, hence reducing the onset of HF [35]. Table 2 summarizes the current indications to neurohormonal blockade in patients with ST elevation AMI.
Sacubitril-valsartan was recently studied in patients with AMI and high-risk features (LVEF ≤ 40% and/or pulmonary congestion at admission; STEMI 76%). At 2-year follow-up, sacubitril-valsartan failed to reduce the incidence of the combined primary of cardiovascular death, first HF hospitalization, or development of outpatient HF, which occurred in 11.9% of the sacubitril-valsartan group vs 13.2% of the ramipril group (HR 0.90; 95% CI 0.78–1.04, p = 0.17), despite the trends toward lower number of events for the individual endpoints with sacubitril-valsartan [36, 37]. Mineralocorticoid receptor antagonists have also been associated with better outcomes in patients with ST elevation AMI and reduced EF when initiated early after AMI [38, 39]. Beta-blockers indirectly modulate the renin–angiotensin–aldosterone system and may also have a direct antifibrotic effect at a cellular level, and thus were shown to reduce adverse remodeling and HF development after AMI [40]. Newer drugs, such as sodium-glucose cotransporter 2 (SGLT2) inhibitors, exert beneficial effects across of a wide spectrum of cardiovascular diseases, and are indicated among patients with HF regardless of the cause [41]. Ivabradine, a potent anti-anginal drug targeting heart rate hence reducing myocardial energy expenditure, was shown to have selective prognostic benefits in HF [42].
Considering the potential for functional recovery of the hibernating myocardium after perfusion restoration, revascularization has always been considered an attractive option for ischemic cardiomyopathy [43]. In fact, initial non-randomized studies showed a significant mortality benefit of coronary artery bypass graft (CABG) versus medical therapy among individuals with ischemic cardiomyopathy and a reduced LVEF [44]. The subsequent Surgical Treatment for Ischemic Heart Failure (STICH) enrolled 2112 patients with CAD and an LVEF ≤ 35% to assess the role of surgery in improving ventricular function and prognosis [45]. Among these, 1212 subjects with a coronary anatomy amenable to CABG were randomized to optimal medical treatment (OMT) or OMT plus CABG [45]. At 5-year follow-up, CABG resulted in significant reduction of death and hospitalization for cardiovascular causes (hazard ratio with CABG, 0.74; 95% CI, 0.64 to 0.85; P < 0.001) [45]. Long-term follow-up data showed a sustained benefit of CABG over OMT, resulting in a number needed to treat of 14 patients to prevent one death [46]. A significant interaction between CAD extent and outcomes was detected, with more extensive coronary artery involvement associated with greater benefit from surgery [46, 47]. In patients with ischemic cardiomyopathy, multivessel or left main CAD, and coronary anatomy not amenable to CABG, PCI should be considered even if there are not high quality data to support this approach. Notably, sub-analyses of randomized controlled trial showed that achievement of complete revascularization was associated to improved prognosis in ischemic cardiomyopathy [47, 48]. Based on this data, both American and European guidelines support revascularization when ischemia is believed to be a contributor to HFrEF [49, 50]. Whether the prognostic advantage is mediated by an improvement in myocardial contractility, electrical stabilization with the prevention of malignant ventricular arrhythmias, a reduction in ischemic events and cardiac death, or a combination of these factors is not currently known. In addition, extensive revascularization is associated with an improvement of LVEF [51]. A recently-published, real-world retrospective study including 10,071 patients from the Veteran Administrations showed that an increase in LVEF of more than 5% after revascularization was associated with significantly lower rates of mortality and HF hospitalization [52]. Similarly, a sub-analysis of the STICH trial including 618 patients showed that an increase in LVEF ≥ 10% after 24 months from enrolment was associated with better overall prognosis [53]. Interestingly, the occurrence of LVEF improvement was not different between patients who did or did not undergo revascularization, and CABG remained significantly associated to better outcomes even after adjusting for LVEF changes [53].
In patients with ischemic cardiomyopathy, an implantable cardioverter-defibrillator (ICD) and/or cardiac resynchronization therapy (CRT) pacemaker may be indicated. The current indications to ICD and/or CRT are found in detail elsewhere. CRT is indicated in particular in patients symptomatic for HF, in sinus rhythm, with a reduced LVEF (≤ 35%) and a QRS duration ≥ 130 ms [50]. QRS morphology (i.e., left bundle branch block), QRS width, sex (i.e., female), and low myocardial scar burden all predict a favorable response to CRT in terms of reverse remodeling and improved morbidity and mortality. Finally, for patients with severe chronic secondary (functional) mitral regurgitation, LVEF ≤ 50%, and New York Heart Association (NYHA) functional class II–IVa (ambulatory) HF despite optimum evidence-based management (pharmacologic therapy plus CRT, as indicated), transcatheter edge-to-edge repair can be considered if anatomically amenable [50].
New Approaches to Ischemic Cardiomyopathy
Recent translational evidence has shown that unloading the LV through the use of a mechanical circulatory support device prior to revascularization during AMI has the potential to reduce the infarct size [54]. Proposed mechanisms for that include reduced LV work expenditure and oxygen consumption, activation of intracellular cardioprotective system, and increased collateral coronary circulation and myocardial perfusion [55]. The clinical implementation of a mechanical unloading strategy using intra-aortic balloon pump (IABP) was tested in the Counterpulsation and Infarct Size in Patients With Acute Anterior Myocardial Infarction (CRISP-AMI) trial [56]. The CRISP-AMI study recruited 337 patients with AMI, which were randomized 1:1 to receive IABP or not prior to PCI; the use of IABP was not associated to reduction in infarct size [56]. Pre-clinical data on transaortic unloading with microaxial pumps (i.e., Impella, Abiomed) appear to be more promising [57]. More recently, initial experience with LV unloading using Impella in humans has been published, showing excellent safety for this approach [58•]. Other percutaneous mechanical circulatory supports, such as transeptal centrifugal assist device (TandemHeart, Livanova) system, share similar hemodynamic effects with transaortic pump, including the unloading of the left ventricle by preload reduction, and may be have similar biological effect in terms of infarct size reduction, although data are currently lacking [59].
Similarly, the use of pressure-controlled intermittent coronary sinus occlusion (PiCSO), a mechanical catheter-based device placed into the coronary sinus after initial primary PCI, has shown to be effective in small, initial experiences in humans [60]. The system consists of a balloon-tipped catheter and a driving console, placed in the coronary sinus and provoking intermittent coronary sinus occlusion, hence redistributing blood flow to the border zone of deprived myocardium, enhancing the washout of deleterious agents, and increasing expression of vascular endothelial growth factor in the myocardium.
After revascularization, modulation of the inflammatory response appears to be an interesting target to improve infarct healing processes. Interleukin-1β (IL-1β) is a master cytokine in the cascade, released following activation of the inflammasome, which is able to induce local and systemic inflammation [61]. Notably, the inflammasome is activated in the setting of AMI, and the local activation of IL-1β was shown to foster ischemia reperfusion injury, favor infarction expansion, and overall increase the incidence of HF [61]. In the Virginia Commonwealth University Anakinra Remodeling Trial 3 (VCUART3), the administration of anakinra, recombinant IL-1 receptor antagonist, in the first 2 weeks after AMI significantly improved the incidence of HF or death [7•]. In particular, 26% of patients in placebo group either died or had new onset of HF, compared with 9% in the anakinra group, and 11% were hospitalized for HF as compared to 0% in the anakinra group [7•]. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial randomized 10,061 patients with prior AMI and residual inflammation; canakinumab, a humanized IL-1β antibody or placebo given once every 3 months, subcutaneously showed to significantly reduce HF hospitalizations and HF-related mortality [62••]. Finally, a recent small trial Assessing the Effect of Anti-IL-6 Treatment in Myocardial Infarction (ASSAIL-MI) (n = 199) showed that tocilizumab, an inhibitor of the inflammatory cytokine interleukin-6 (IL-6) pathway, which is a downstream effector of IL-1β, increased the extent of myocardial recovery after reperfusion when compared with matching placebo [63••].
New Paradigm of HF Without LV Dysfunction and Remodeling After AMI
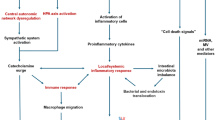
The advances in technology, reperfusion strategies, and the implementation of cardioprotective medications have led to a significant mitigation of LV systolic dysfunction and adverse cardiac remodeling after AMI. Strategies aimed at preventing LV dysfunction and remodeling may in fact also prevent the incidence of HF. HF after AMI is, however, described also in the absence of systolic dysfunction and adverse remodeling (Fig. 1) [64]. In a study including 374 patients undergoing CMR during hospitalization for STEMI and after 6 months, there was no significant change in LVEDV and LVEF at follow-up, but up to 7.5% of patients experienced a HF hospitalization at long-term follow-up [8•]. In a trial of colchicine versus placebo in STEMI, HF occurred in 13% of placebo patients despite minimal changes in relative remodeling at follow-up [9]. In the trial of cyclosporine versus placebo, 10% of STEMI patients experienced a HF hospitalization at 1 year, independent of changes in cardiac dimensions and systolic function (LVEDV and LVEF) [4]. In the aforementioned VCUART3 trial of anakinra versus placebo in patients with STEMI, the combined endpoint of new-onset HF and HF hospitalization or death occurred in 26% of patients in the placebo arm despite lack of changes in LVEDV and LVEF [7•]. In the pooled data analysis of three VCUART clinical including 139 patients with STEMI, all-cause death or new-onset HF occurred in 29.1% of placebo group at 1-year follow-up. Of these, only half of the patients had a reduced LVEF at follow-up (LVEF < 40%) [65].

Heart failure and ischemic cardiomyopathy. Left ventricular dysfunction in patients with coronary artery disease (i.e., ischemic cardiomyopathy) is often the consequence of an irreversible loss of viable myocardium following a large AMI occasionally in combination with loss of contractility in ischemic, but still viable, myocardium (hibernating myocardium). However, even smaller infarcts, not necessarily associated with cardiac dilatation and dysfunction at rest, may put that patient at risk of developing HF with preserved EF as result of the combination of the post-infarction inflammatory, hemodynamic and neurohormonal response, and predisposing individual risk factors (i.e., obesity, older age, hypertension, chronic kidney disease, diabetes)
Pathophysiologic mechanisms leading to HF in patients with CAD may be in fact multifactorial and only partially explained by measures of LV systolic dysfunction at rest or changes in LV volumes. The acute myocardial damage is only one of the factors implicated in the determination of cardiac contractility, LV filling pressure, and cardiac output reserve. Post-infarction inflammatory, hemodynamic and neurohormonal response, chamber compliance, myocardial stiffness, cardiac pre-existing individual risk factors, and subclinical comorbidities all contribute at vary degree to determine cardiac function and decline in cardiorespiratory fitness [66, 67]. Resting systolic function may be preserved but patients may still experience symptoms during exercise due to the loss of cardiac systolic and diastolic reserve, as it happens in HF with preserved LVEF [68,69,70]. Myocardial stiffness due to persistent chronic ischemia or previous subclinical functional structural changes may limit diastolic reserve and contribute to the reduced cardiorespiratory fitness [71].
Conclusions
Ischemic cardiomyopathy is the most common cause of HF in the general population. Treatments aiming at modulating the neurohumoral response and, in selected cases, restoring blood flow to the ischemic cardiomyocytes were shown to dramatically abate the occurrence of HFrEF in ischemic cardiomyopathy. Novel therapeutic approaches, such as mechanical unloading and modulation of the inflammatory response, appear to be promising. Furthermore, the understanding of the mechanisms by which, despite optimal treatment, HF ensues after AMI, with or without adverse remodeling and systolic dysfunction, is a critical step in the search for novel ways to measure HF risk beyond preservation of LV volumes and LVEF, and for novel interventions to reduce the incidence of HF after AMI with subsequent consequences on morbidity and mortality. The scarcity of data exploring cardiac reserve and cardiorespiratory fitness following an AMI paves the way for more systematic investigations to provide novel pathophysiological insights and a better understanding of the problem.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Felker GM, Shaw LK, O’Connor CM. A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol. 2002;39:210–8. https://doi.org/10.1016/s0735-1097(01)01738-7.
Almeida AG, Carpenter JP, Cameli M, Donal E, Dweck MR, Flachskampf FA, et al. Multimodality imaging of myocardial viability: an expert consensus document from the European Association of Cardiovascular Imaging (EACVI). Eur Heart J Cardiovasc Imaging. 2021;22:e97–125. https://doi.org/10.1093/ehjci/jeab053.
Page BJ, Banas MD, Suzuki G, Weil BR, Young RF, Fallavollita JA, et al. Revascularization of chronic hibernating myocardium stimulates myocyte proliferation and partially reverses chronic adaptations to ischemia. J Am Coll Cardiol. 2015;65:684–97. https://doi.org/10.1016/j.jacc.2014.11.040.
Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–31. https://doi.org/10.1056/NEJMoa1505489.
Mathur A, Fernández-Avilés F, Bartunek J, Belmans A, Crea F, Dowlut S, et al. The effect of intracoronary infusion of bone marrow-derived mononuclear cells on all-cause mortality in acute myocardial infarction: the BAMI trial. Eur Heart J. 2020;41:3702–10. https://doi.org/10.1093/eurheartj/ehaa651.
Bulluck H, Carberry J, Carrick D, McEntegart M, Petrie MC, Eteiba H, et al. Redefining adverse and reverse left ventricular remodeling by cardiovascular magnetic resonance following ST-segment-elevation myocardial infarction and their implications on long-term prognosis. Circ Cardiovasc Imaging. 2020;13:e009937. https://doi.org/10.1161/circimaging.119.009937.
• Abbate A, Trankle CR, Buckley LF, Lipinski MJ, Appleton D, Kadariya D, et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc. 2020;9:e014941. https://doi.org/10.1161/jaha.119.014941. In this small trial of anakinra, recombinant IL-1 receptor antagonist, versus placebo in patients with STEMI, anakinra reduced the incidence of heart failure or death in patients with STEMI independent of changes in left ventricular remodeling.
• Rodriguez-Palomares JF, Gavara J, Ferreira-González I, Valente F, Rios C, Rodríguez-García J, et al. Prognostic value of initial left ventricular remodeling in patients with reperfused STEMI. JACC Cardiovasc Imaging. 2019;12:2445–56. https://doi.org/10.1016/j.jcmg.2019.02.025. In this study including 374 patients undergoing cardiac magnetic resonance during hospitalization for STEMI and after 6 months, there was no significant change in left ventricular diastolic volume and ejection fraction at follow-up, but up to 7.5% of patients experienced a heart failure hospitalization at long-term follow-up.
Mewton N, Roubille F, Bresson D, Prieur C, Bouleti C, Bochaton T, et al. Effect of colchicine on myocardial injury in acute myocardial infarction. Circulation. 2021;144:859–69. https://doi.org/10.1161/circulationaha.121.056177.
Cavender MA, O’Donoghue ML, Abbate A, Aylward P, Fox KA, Glaser RX, et al. Inhibition of p38 MAP kinase in patients with ST-elevation myocardial infarction - findings from the LATITUDE-TIMI 60 trial. Am Heart J. 2022;243:147–57. https://doi.org/10.1016/j.ahj.2021.08.022.
Lund GK, Stork A, Muellerleile K, Barmeyer AA, Bansmann MP, Knefel M, et al. Prediction of left ventricular remodeling and analysis of infarct resorption in patients with reperfused myocardial infarcts by using contrast-enhanced MR imaging. Radiology. 2007;245:95–102. https://doi.org/10.1148/radiol.2451061219.
Del Buono MG, Montone RA, Rinaldi R, Gurgoglione FL, Meucci MC, Camilli M, et al. Clinical predictors and prognostic role of high Killip class in patients with a first episode of anterior ST-segment elevation acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2021;22:530–8. https://doi.org/10.2459/jcm.0000000000001168.
Chareonthaitawee P, Christian TF, Hirose K, Gibbons RJ, Rumberger JA. Relation of initial infarct size to extent of left ventricular remodeling in the year after acute myocardial infarction. J Am Coll Cardiol. 1995;25:567–73. https://doi.org/10.1016/0735-1097(94)00431-o.
Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, et al. Relationship between infarct size and outcomes following primary PCI: patient-level analysis from 10 randomized trials. J Am Coll Cardiol. 2016;67:1674–83. https://doi.org/10.1016/j.jacc.2016.01.069.
Sharif D, Matanis W, Sharif-Rasslan A, Rosenschein U. Doppler echocardiographic myocardial stunning index predicts recovery of left ventricular systolic function after primary percutaneous coronary intervention. Echocardiography. 2016;33:1465–71. https://doi.org/10.1111/echo.13305.
Kloner RA. Stunned and hibernating myocardium: where are we nearly 4 decades later? J Am Heart Assoc. 2020;9:e015502. https://doi.org/10.1161/jaha.119.015502.
Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G, et al. Optimized treatment of ST-elevation myocardial infarction. Circ Res. 2019;125:245–58. https://doi.org/10.1161/circresaha.119.315344.
Del Buono MG, Montone RA, Camilli M, Carbone S, Narula J, Lavie CJ, et al. Coronary microvascular dysfunction across the spectrum of cardiovascular diseases: JACC state-of-the-art review. J Am Coll Cardiol. 2021;78:1352–71. https://doi.org/10.1016/j.jacc.2021.07.042.
Liu T, Howarth AG, Chen Y, Nair AR, Yang HJ, Ren D, et al. Intramyocardial hemorrhage and the “wave front” of reperfusion injury compromising myocardial salvage. J Am Coll Cardiol. 2022;79:35–48. https://doi.org/10.1016/j.jacc.2021.10.034.
Mitchell GF, Lamas GA, Vaughan DE, Pfeffer MA. Left ventricular remodeling in the year after first anterior myocardial infarction: a quantitative analysis of contractile segment lengths and ventricular shape. J Am Coll Cardiol. 1992;19:1136–44. https://doi.org/10.1016/0735-1097(92)90314-d.
Bogaert J, Bosmans H, Maes A, Suetens P, Marchal G, Rademakers FE. Remote myocardial dysfunction after acute anterior myocardial infarction: impact of left ventricular shape on regional function: a magnetic resonance myocardial tagging study. J Am Coll Cardiol. 2000;35:1525–34. https://doi.org/10.1016/s0735-1097(00)00601-x.
Sutton MSJ, Pfeffer MA, Moye L, Plappert T, Rouleau JL, Lamas G, et al. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 1997;96:3294–9. https://doi.org/10.1161/01.cir.96.10.3294.
van der Bijl P, Abou R, Goedemans L, Gersh BJ, Holmes DR Jr, Ajmone Marsan N, et al. Left ventricular post-infarct remodeling: implications for systolic function improvement and outcomes in the Modern Era. JACC Heart Fail. 2020;8:131–40. https://doi.org/10.1016/j.jchf.2019.08.014.
Masci PG, Ganame J, Francone M, Desmet W, Lorenzoni V, Iacucci I, et al. Relationship between location and size of myocardial infarction and their reciprocal influences on post-infarction left ventricular remodelling. Eur Heart J. 2011;32:1640–8. https://doi.org/10.1093/eurheartj/ehr064.
Isnard R, Pousset F, Trochu J, Chafirovskaïa O, Carayon A, Golmard J, et al. Prognostic value of neurohormonal activation and cardiopulmonary exercise testing in patients with chronic heart failure. Am J Cardiol. 2000;86:417–21. https://doi.org/10.1016/s0002-9149(00)00957-7.
Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, et al. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2016;67:2050–60. https://doi.org/10.1016/j.jacc.2016.01.073.
Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group Fragmin during Instability in Coronary Artery Disease. N Engl J Med. 2000;343:1139–47. https://doi.org/10.1056/nejm200010193431602.
van Wezenbeek J, Canada JM, Ravindra K, Carbone S, Trankle CR, Kadariya D, et al. C-reactive protein and N-terminal pro-brain natriuretic peptide levels correlate with impaired cardiorespiratory fitness in patients with heart failure across a wide range of ejection fraction. Front Cardiovasc Med. 2018;5:178. https://doi.org/10.3389/fcvm.2018.00178.
Del Buono MG, Damonte JI, Trankle CR, Kadariya D, Carbone S, Thomas G, et al. Effect of interleukin-1 blockade with anakinra on leukocyte count in patients with ST-segment elevation acute myocardial infarction. Sci Rep. 2022;12:1254. https://doi.org/10.1038/s41598-022-05374-w.
Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA. 2005;293:979–86. https://doi.org/10.1001/jama.293.8.979.
Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet. 1996;348:771–5. https://doi.org/10.1016/s0140-6736(96)02514-7.
Simoons ML, Serruys PW, van den Brand M, Res J, Verheugt FW, Krauss XH, et al. Early thrombolysis in acute myocardial infarction: limitation of infarct size and improved survival. J Am Coll Cardiol. 1986;7:717–28. https://doi.org/10.1016/s0735-1097(86)80329-1.
Fazel R, Joseph TI, Sankardas MA, Pinto DS, Yeh RW, Kumbhani DJ, et al. Comparison of reperfusion strategies for ST-segment-elevation myocardial infarction: a multivariate network meta-analysis. J Am Heart Assoc. 2020;9:e015186. https://doi.org/10.1161/jaha.119.015186.
Schömig A, Kastrati A, Dirschinger J, Mehilli J, Schricke U, Pache J, et al. Coronary stenting plus platelet glycoprotein IIb/IIIa blockade compared with tissue plasminogen activator in acute myocardial infarction. Stent versus thrombolysis for occluded coronary arteries in patients with acute myocardial infarction study investigators. N Engl J Med. 2000;343:385–91. https://doi.org/10.1056/nejm200008103430602.
Ishii H, Amano T, Matsubara T, Murohara T. Pharmacological intervention for prevention of left ventricular remodeling and improving prognosis in myocardial infarction. Circulation. 2008;118:2710–8. https://doi.org/10.1161/circulationaha.107.748772.
Pfeffer MA, Claggett B, Lewis EF, Granger CB, Køber L, Maggioni AP, et al. Angiotensin receptor-neprilysin inhibition in acute myocardial infarction. N Engl J Med. 2021;385:1845–55. https://doi.org/10.1056/NEJMoa2104508.
Del Buono MG, Damonte JI, Trankle CR, Bhardwaj H, Markley R, Turlington J, et al. Sacubitril/valsartan for the prevention and treatment of postinfarction heart failure: ready to use? J Cardiovasc Pharmacol. 2021;78:331–3. https://doi.org/10.1097/fjc.0000000000001103.
Torrado J, Cain C, Mauro AG, Romeo F, Ockaili R, Chau VQ, et al. Sacubitril/valsartan averts adverse post-infarction ventricular remodeling and preserves systolic function in rabbits. J Am Coll Cardiol. 2018;72:2342–56. https://doi.org/10.1016/j.jacc.2018.07.102.
Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. https://doi.org/10.1161/circulationaha.113.001878.
Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. beta-adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation. 2000;101:2103–9. https://doi.org/10.1161/01.cir.101.17.2103.
Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: a review. JAMA. 2020;324:488–504. https://doi.org/10.1001/jama.2020.10262.
Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–85. https://doi.org/10.1016/s0140-6736(10)61198-1.
Pitt M, Lewis ME, Bonser RS. Coronary artery surgery for ischemic heart failure: risks, benefits, and the importance of assessment of myocardial viability. Prog Cardiovasc Dis. 2001;43:373–86. https://doi.org/10.1053/pcad.2001.20672.
O’Connor CM, Velazquez EJ, Gardner LH, Smith PK, Newman MF, Landolfo KP, et al. Comparison of coronary artery bypass grafting versus medical therapy on long-term outcome in patients with ischemic cardiomyopathy (a 25-year experience from the Duke Cardiovascular Disease Databank). Am J Cardiol. 2002;90:101–7. https://doi.org/10.1016/s0002-9149(02)02429-3.
Velazquez EJ, Lee KL, Deja MA, Jain A, Sopko G, Marchenko A, et al. Coronary-artery bypass surgery in patients with left ventricular dysfunction. N Engl J Med. 2011;364:1607–16. https://doi.org/10.1056/NEJMoa1100356.
Velazquez EJ, Lee KL, Jones RH, Al-Khalidi HR, Hill JA, Panza JA, et al. Coronary-artery bypass surgery in patients with ischemic cardiomyopathy. N Engl J Med. 2016;374:1511–20. https://doi.org/10.1056/NEJMoa1602001.
Burke DA, Kundi H, Almonacid A, O’Neill W, Moses J, Kleiman N, et al. The value of left ventricular support in patients with reduced left ventricular function undergoing extensive revascularization: an analysis from the PROTECT-II randomized trial. JACC Cardiovasc Interv. 2019;12:1985–7. https://doi.org/10.1016/j.jcin.2019.07.050.
Généreux P, Palmerini T, Caixeta A, Rosner G, Green P, Dressler O, et al. Quantification and impact of untreated coronary artery disease after percutaneous coronary intervention: the residual SYNTAX (Synergy Between PCI with Taxus and Cardiac Surgery) score. J Am Coll Cardiol. 2012;59:2165–74. https://doi.org/10.1016/j.jacc.2012.03.010.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147-239. https://doi.org/10.1016/j.jacc.2013.05.019.
McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2022;24:4–131. https://doi.org/10.1002/ejhf.2333.
Russo JJ, Prasad M, Doshi D, Karmpaliotis D, Parikh MA, Ali ZA, et al. Improvement in left ventricular function following higher-risk percutaneous coronary intervention in patients with ischemic cardiomyopathy. Catheter Cardiovasc Interv. 2020;96:764–70. https://doi.org/10.1002/ccd.28557.
Velagaleti RS, Vetter J, Parker R, Kurgansky KE, Sun YV, Djousse L, et al. Change in left ventricular ejection fraction with coronary artery revascularization and subsequent risk for adverse cardiovascular outcomes. Circ Cardiovasc Interv. 2022. https://doi.org/10.1161/circinterventions.121.011284.
Perry AS, Mann DL, Brown DL. Improvement of ejection fraction and mortality in ischaemic heart failure. Heart. 2020. https://doi.org/10.1136/heartjnl-2020-316975.
Kapur NK, Qiao X, Paruchuri V, Morine KJ, Syed W, Dow S, et al. Mechanical pre-conditioning with acute circulatory support before reperfusion limits infarct size in acute myocardial infarction. JACC Heart Fail. 2015;3:873–82. https://doi.org/10.1016/j.jchf.2015.06.010.
Esposito ML, Zhang Y, Qiao X, Reyelt L, Paruchuri V, Schnitzler GR, et al. Left Ventricular unloading before reperfusion promotes functional recovery after acute myocardial infarction. J Am Coll Cardiol. 2018;72:501–14. https://doi.org/10.1016/j.jacc.2018.05.034.
Patel MR, Smalling RW, Thiele H, Barnhart HX, Zhou Y, Chandra P, et al. Intra-aortic balloon counterpulsation and infarct size in patients with acute anterior myocardial infarction without shock: the CRISP AMI randomized trial. JAMA. 2011;306:1329–37. https://doi.org/10.1001/jama.2011.1280.
Swain L, Reyelt L, Bhave S, Qiao X, Thomas CJ, Zweck E, et al. Transvalvular ventricular unloading before reperfusion in acute myocardial infarction. J Am Coll Cardiol. 2020;76:684–99. https://doi.org/10.1016/j.jacc.2020.06.031.
• Kapur NK, Alkhouli MA, DeMartini TJ, Faraz H, George ZH, Goodwin MJ, et al. Unloading the left ventricle before reperfusion in patients with anterior ST-segment-elevation myocardial infarction. Circulation. 2019;139:337–46. https://doi.org/10.1161/circulationaha.118.038269. In this exploratory safety and feasibility trial, left ventricular unloading using the Impella CP device with a 30-min delay before reperfusion was feasible within a relatively short time period in anterior STEMI.
Burkhoff D, Sayer G, Doshi D, Uriel N. Hemodynamics of mechanical circulatory support. J Am Coll Cardiol. 2015;66:2663–74. https://doi.org/10.1016/j.jacc.2015.10.017.
Egred M, Bagnall A, Spyridopoulos I, Purcell IF, Das R, Palmer N, et al. Effect of Pressure-controlled intermittent coronary sinus occlusion (PiCSO) on infarct size in anterior STEMI: PiCSO in ACS study. Int J Cardiol Heart Vasc. 2020;28:100526. https://doi.org/10.1016/j.ijcha.2020.100526.
Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020;126:1260–80. https://doi.org/10.1161/circresaha.120.315937.
•• Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–99. https://doi.org/10.1161/circulationaha.118.038010. In the CANTOS trial including 10,061 patients with prior myocardial infarction and residual inflammation, canakinumab, a humanized IL-1beta antibody, showed to significantly reduce heart failure hospitalizations and heart failure-related mortality.
•• Broch K, Anstensrud AK, Woxholt S, Sharma K, Tøllefsen IM, Bendz B, et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2021;77:1845–55. https://doi.org/10.1016/j.jacc.2021.02.049. In a trial of tocilizumab (an anti-interleukin-6 (IL-6) receptor blockade), or placebo in patients with STEMI, tocilizumab increased the adjusted myocardial salvage index when compared with matching placebo.
Buono MGD, Garmendia CM, Seropian IM, Gonzalez G, Berrocal DH, Biondi-Zoccai G, et al. Heart failure after ST-elevation myocardial infarction: beyond left ventricular adverse remodeling. Curr Probl Cardiol. 2022. https://doi.org/10.1016/j.cpcardiol.2022.101215.
Abbate A, Wohlford GF, Del Buono MG, Chiabrando JG, Markley R, Turlington J, et al. Interleukin-1 blockade with Anakinra and heart failure following ST-segment elevation myocardial infarction: results from a pooled analysis of the VCUART clinical trials. Eur Heart J Cardiovasc Pharmacother. 2021. https://doi.org/10.1093/ehjcvp/pvab075.
Del Buono MG, Arena R, Borlaug BA, Carbone S, Canada JM, Kirkman DL, et al. Exercise intolerance in patients with heart failure: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2209–25. https://doi.org/10.1016/j.jacc.2019.01.072.
Potere N, Del Buono MG, Vecchié A, Porreca E, Abbate A, Dentali F, et al. Diabetes mellitus and heart failure: an update on pathophysiology and therapy. Minerva Cardiol Angiol. 2022. https://doi.org/10.23736/s2724-5683.22.05967-1.
Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD, et al. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010;56:845–54. https://doi.org/10.1016/j.jacc.2010.03.077.
Del Buono MG, Iannaccone G, Scacciavillani R, Carbone S, Camilli M, Niccoli G, et al. Heart failure with preserved ejection fraction diagnosis and treatment: an updated review of the evidence. Prog Cardiovasc Dis. 2020;63:570–84. https://doi.org/10.1016/j.pcad.2020.04.011.
Del Buono MG, Buckley L, Abbate A. Primary and secondary diastolic dysfunction in heart failure with preserved ejection fraction. Am J Cardiol. 2018;122:1578–87. https://doi.org/10.1016/j.amjcard.2018.07.012.
Obokata M, Reddy YNV, Melenovsky V, Kane GC, Olson TP, Jarolim P, et al. Myocardial injury and cardiac reserve in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2018;72:29–40. https://doi.org/10.1016/j.jacc.2018.04.039.
O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127:e362-425. https://doi.org/10.1161/CIR.0b013e3182742cf6.
Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018;39:119–77. https://doi.org/10.1093/eurheartj/ehx393.
Funding
Open access funding provided by Università Cattolica del Sacro Cuore within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All the authors have nothing to disclose regarding this article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Myocardial Disease
Marco Giuseppe Del Buono and Francesco Moroni contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Del Buono, M.G., Moroni, F., Montone, R.A. et al. Ischemic Cardiomyopathy and Heart Failure After Acute Myocardial Infarction. Curr Cardiol Rep 24, 1505–1515 (2022). https://doi.org/10.1007/s11886-022-01766-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-022-01766-6