
Abstract
Epidermal stem cells (ESCs) are critical for skin regeneration and repair. Previous studies have shown that ESCs are susceptible to oxidative stress, which in turn leads to lipid peroxidation and affects skin repair. Our study aims to explore how ESCs resist lipid peroxidation. By performing proteomics analysis, we found that the expression of Acyl-CoA thioesterase 7 (ACOT7) was positively correlated with the concentration of transferrin. Overexpression adenovirus vectors of ACOT7 were constructed and transfected into ESCs. Levels of lipid peroxidation by flow cytometry, cell viabilities, and MDA levels were measured. The results revealed that ACOT7 could inhibit lipid peroxidation, reduce the level of malondialdehyde (MDA), and improve the survival rate of ESCs induced by H2O2, Erastin, and RSL3. Our data suggest that ACOT7 has an effect on protecting ESCs against iron-dependent lipid peroxidation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The skin is the largest organ of the human body, which constantly renews itself to maintain its barrier function (Hennig et al. 2020). Being directly exposed to the environment, the skin is easily affected by ultraviolet radiation, pathogen invasion, and other external threats (Yin et al. 2019). These factors can cause oxidative stress, which in turn affects the function of skin cells. The epidermis, the outermost layer of the skin, is rich in lipids, proteins, and DNA, so it is susceptible to oxidative stress. Epidermal damage can lead to skin aging, poor wound healing, and even skin diseases (Klaunig et al. 2010; An et al. 2013). ESCs are a type of cells located in the basal part of the epidermis, which can differentiate into mature keratinocytes that have an indispensable role during skin healing after trauma (Liao et al. 2014). However, ESCs are easily affected by oxidative stress, which may affect their ability to proliferate and differentiate, thus delaying the wound healing process (Ud-Din et al. 2021).
Iron is a catalyst of oxidative stress. It can bind to different ligands and has the property of electron transfer (Wang et al. 2019). Excessive amounts of intracellular iron may destroy redox homeostasis through the Fenton reaction and catalyze reactive oxygen species (ROS) production, inducing the destruction of macromolecular substances, such as DNA, RNA, protein, and lipid (Fibach 2019). ROS then interacts with polyunsaturated fatty acids on the biomembrane, resulting in cellular structure and function changes, a process also known as lipid peroxidation, which then leads to cell death, i.e., ferroptosis (Tomita et al. 2019). Previous studies suggested that oxidative stress increases with overload iron, which may harm cell functions. This process may be ameliorated by chelating excess intracellular free iron through deferoxamine (Holden and Nair 2019). In addition, cells may upregulate the expression of some genes, such as ferritin, to relieve the effect. According to previous studies, in keratinocytes and fibroblasts, overexpressed ferritin caused by iron load may significantly inhibit the procedure of lipid peroxidation (Giordani et al. 2000; Zhang et al. 2021). However, the impact of iron load on ESCs needs to be further explored.
ACOT7 is an enzyme that catalyzes the hydrolysis procedure from acyl-CoA to free fatty acid and coenzyme A. In our study, we found that ACOT7 reduces the level of iron-dependent lipid peroxidation. The aim of the present study was to further explore the role of ACOT7 in inhibiting the procedure of lipid peroxidation in ESCs cultured in vitro.
Materials and methods
Animals
All animal studies (including the mouse euthanasia procedure) were done in compliance with Shandong University institutional animal care regulations and conducted according to the ARRIVE guidelines and the U.K. Animals (Scientific Procedures) Act 1986 guidelines. A total of 20 specific pathogen-free (SPF) wild-type male C57BL/6 J newborn mice were purchased from the experimental animal center of Shandong University (Permit number: SCXK Lu 20,190,001). All the animals were housed in an environment with a temperature of 22 ± 1°C, relative humidity of 50 ± 1%, and a light/dark cycle of 12/12 h.
Isolation, culture, and identification of ESCs
Processes of isolation, culture, and identification of ESCs were conducted according to previous studies (Zhang et al. 2018a, b, 2020). Briefly, 20 mice were used for this experiment. Skin tissues were collected and digested at + 4°C overnight in 0.5% Dispase II (Sigma, St. Louis, MO). Then, the epidermis was dissected from the dermis and digested in 0.25% trypsin (Gibco, Crand Island, NY) at + 37°C for 7 min; the process of digestion was terminated by adding RMPI 1640 (Thermo Fisher Scientific, Shanghai, China) and 10% FBS (Thermo Fisher Scientific). Samples were then filtrated and centrifuged to obtain cells, which were resuspended in a CnT-Prime culture medium (Celltec, Bern, Switzerland).
Cells were incubated in culture bottles containing type IV collagen (Sigma) at the density of 5 × 105–1 × 106/ml, and they were placed in normoxic incubators containing 5% CO2 at + 37°C for approximately 30 min. After cell adherence, the culture solution was discarded, and cells were gently rinsed with phosphate-buffered saline (PBS) three times. Cells were incubated in a fresh medium in normoxic incubators, and the culture solution was conducted every other day.
Flow cytometry, Western blot analysis, and immunofluorescence analysis were used to identify ESCs. Cytokeratin 14 (CK14), cytokeratin 15 (CK15), p63, and cytokeratin 10 (CK10) were used to differentiate ESCs from keratinocytes.
All experiments were performed with mycoplasma-free cells.
TMT quantitative proteomic analysis
ESCs were cultured in the medium containing 100 µg/ml holo-transferrin (Sigma) (experimental group) or without holo-transferrin (control group) for 2 d. Three samples from each group were used for TMT quantitative proteomic analysis. The TMT quantitative proteomic analysis was performed by Shanghai Applied Protein Technology Co., Ltd. (Project’s number: P20200801875).
Adenovirus transfection
After reaching a 70% confluency, the adenovirus carrying Acot7 (with Flag tag) and the vector adenovirus (without Flag tag) were transfected into ESCs. The multiplicity of infection (MOI) was determined to be 10. The ESCs were divided into the following three groups: (a) experimental group (ESCs were transfected by the adenovirus carrying Acot7); (b) control group (ESCs were transfected by the vector adenovirus); (c) the blank group (ESCs did not receive adenovirus transfection). The adenoviruses were provided by Shanghai Genechem Co., Ltd, Shanghai, China. The culture mediums were changed after the 10-h period of transfection. On the second day, Western blot analysis and immunofluorescence analysis were applied to investigate the transfection efficiency.
Detection of cell proliferation
Cell proliferation was analyzed using an IncuCyte S3 Living-Cell Analysis System (Sartorius, Gottingen, Germany). Briefly, ESCs were cultured in 96-well plates at the density of 5 × 103 cells/plate, and then incubated with CnT-Prime culture medium at 37°C and 5% CO2 for 48 h. By calculating the area confluence, which was normalized to 0 h, then the cell proliferation curves were drawn.
Detection of cell apoptosis
The Annexin V-PE/7-AAD apoptosis analysis kits (BD Biosciences, San Jose, CA) detected apoptosis (Meng et al. 2018). After centrifugation and washing with PBS, ESCs were resuspended using 1 × binding buffer at a density of 1 × 106 cells/ml, and then incubated with PE-conjugated Annexin V and 5 µl of 7-AAD for 15 min at room temperature (25°C) in the dark. Afterwards, samples were detected by CytoFLEX flow cytometer (Beckman Coulter, Indianapolis, IN). Early apoptotic, late apoptotic, and dead cells can be distinguished on the basis of a double-labeling for Annexin V-PE and 7-AAD.
Detection of cell viability
The impacts of H2O2, Erastin, and RSL3 on the cell viability of ESCs were measured by the Cell Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) method. Cells were inoculated in 96-well plates, and 500 µM H2O2 (Sigma), 20 µM Erastin (MedChemExpress, Shanghai, China), and 5 µM RSL3 (MedChemExpress) were added into plates. After 6 h, the culture solution was discarded. Cells were then incubated in 100 μl fresh culture solution and 10 μl CCK-8 at 37°C for 2 h. Next, the absorbance of the solution at 450 nm was measured by Spark (Tecan, Salzburg, Austria) to calculate the cell viability of ESCs.
Detection of the relative levels of lipid peroxidation and Fe.2+ by flow cytometry
The relative intracellular levels of lipid peroxidation and Fe2+ were measured by the C11-BODIPY lipid probe (Invitrogen, Carlsbad, CA) and FerroOrange (Dojindo). The cells were gently rinsed with PBS three times, and the adherent cells were digested with trypsin. After digestion, centrifugation was conducted to collect cell pellets, which were resuspended with 100-µl living cell image solution (Thermo Fisher Scientific), and incubated with 0.1 µl C11-BODIPY (Invitrogen, 1:1000) and 0.2 µl FerroOrange (Dojindo, 1:500) in the dark for 30 min. Next, to blow and mix the cells, a 400-µl living cell image solution was added. The wavelength of FerroOrange excitation light was 543 nm, and the wavelength of emission light was 580 nm. Oxidation of the polyunsaturated butadienyl portion of the C11-BODIPY resulted in a shift of the fluorescence emission peak from ~ 590 to ~ 510 nm. The relative levels of lipid peroxidation and Fe2+ were measured by flow cytometry, which was quantified by the FITC/PE value and the PE value, respectively (Cheloni and Slaveykova 2013; Mei et al. 2020).
Detection of MDA levels
The intracellular level of MDA was determined according to the instructions of lipid peroxidation (MDA) assay kit (Abcam, Cambridge, UK). In the lipid peroxidation assay protocol, the MDA in the sample was reacted with thiobarbituric acid (TBA) to generate an MDA-TBA adduct. The MDA-TBA adduct was easily quantified colorimetrically (OD = 532 nm) (Wang et al. 2021).
Protein extraction and Western blot
Western blot analysis was performed in order to measure the expression of proteins. The cell lysate was attained with RIPA lysate (Thermo Fisher Scientific) and the protein concentration of the samples was measured with BCA Protein Assay Kit (Thermo Fisher Scientific). Samples (approximately 40 µg) were separated by 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked in 5% non-fat milk for 2 h before being incubated with primary antibodies at 37°C overnight. The primary antibodies including anti-CK10 (1:1000, Abcam, ab76318), anti-CK14 (1:1000, Abcam, ab181595), anti-CK15 (1:1000, Abcam, ab52816), anti-p63 (1:1000, Abcam, ab124762), anti-GAPDH (1:1000, Cell Signaling Technology, #5174, Boston, MA), anti-ACOT7 (1:5000, Proteintech Group, 15,972–1-AP), anti-FLAG (1:500, Sigma, F7425), anti-FTH (1:1000, Invitrogen, 701,934), and anti-FTL (1:1000, Invitrogen, MA5-32,755). The membranes were then rinsed with TBST and incubated with the HRP-conjugated secondary antibody (1:5000, Cell Signaling Technology, #7074) at room temperature for 1 h. The iBright imaging system (Invitrogen) was applied to obtain images, and the ImageJ software was used to quantify the expression of proteins.
Immunofluorescence
Cell samples were fixed in 4% paraformaldehyde for 15 min at room temperature and washed three times in PBS, permeabilized 0.5% TritonX-100/DPBS (Invitrogen) for 10 min, and blocked with blocking solution for 1 h. After overnight incubation at 4℃ with primary antibodies, cell samples were incubated with secondary antibodies for 1 h. Finally, the cell nuclei were stained with DAPI. The antibody information has been described before. Cell immunofluorescence images were captured by an A1R confocal microscope (Nikon, Belmont, CA).
Statistical analysis
We used the GraphPad Prism version 8.0.1 for statistical analysis, and the quantitative data were expressed as mean ± standard deviation. All data were attained from at least three independent experiments. Group differences were explored by using the unpaired t-test, where P < 0.05 represented statistical significance. Asterisks represent significance between conditions for each group. P-values are labeled as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
ESCs showed high expression of CK14, CK15, and p63
After 2 days of culture, ESCs were observed under a light microscope (Fig. 1A). CK14, CK15, and p63 are markers of ESCs and CK10 is the marker of keratinocytes (Ghadially 2012). Next, Western blot analysis was performed to compare markers expressed on adherent and non-adherent cells. The expression of CK14, CK15, and p63 was significantly higher in the adherent cells than that in the non-adherent cells, and the expression of CK10 was significantly lower in the adherent cells than that in the non-adherent cells (Fig. 1B). Furthermore, flow cytometry suggested that the positive rates of obtained cells expressing CK14, CK15, and p63 were more than 80%, while the positive rate of CK10 was less than 10% (Fig. 1C). Similar results were observed by immunofluorescence, which suggested that these cells were ESCs (Fig. 1D).

ESCs showed high expression of CK14, CK15, and p63. (A) ESCs cultured for 2 d. Scale bar = 50 µm. (B) WB and quantification showed CK14, CK15, p63, and CK10 expression levels between adherent and non-adherent cells. (C) Flow cytometry and quantification of positive rates of obtained cells expressing CK14, CK15, p63, and CK10. (D) Immunofluorescence identification of obtained cells expressing CK14, CK15, p63, and CK10. Scale bar = 20 µm. Statistics: Data were obtained in triplicate experiments and are shown as the mean ± SD; unpaired t-test. *P < 0.05, **P < 0.01.
The expression of ACOT7 increases with the rise of iron load in ESCs
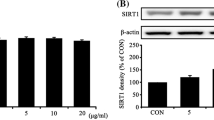
Next, ESCs were cultured in the medium with 0, 1, 10, 100, or 1000 µg/ml holo-transferrin. Flow cytometry suggested that with the increase of the concentration of holo-transferrin, there was a significant rise in the level of intracellular Fe2+ (Fig. 2A), while no differences were observed in the level of lipid peroxidation. After the ESCs were stimulated with 500 µM H2O2 for 6 h, no differences in the level of lipid peroxidation were observed (Fig. 2B). Because of the Fenton reaction, we suspected that some genes were upregulated to maintain a stable level of lipid peroxidation. Consequently, we found significant differences in the expression of some proteins, including ACOT7 and Ferritin Light Chain1 (FTL1), through TMT quantitative proteomic analysis (Fig. 2C), which was further confirmed by Western blot (WB). WB analysis suggested that ACOT7, Ferritin Heavy Chain (FTH), and Ferritin Light Chain (FTL) upregulation was positively correlated with the concentration of transferrin (Fig. 2D).

The expression of ACOT7 increases with the rise of iron load in ESCs. (A) Flow cytometry quantification of cellular Fe2+ level in the medium with 0, 1, 10, 100, or 1000 µg/ml holo-transferrin. (B) Flow cytometry quantification of cellular lipid peroxidation level with no irritants and 500 µM H2O2. (C) ESCs cultured in the medium with 100 μg/ml holo-transferrin and no holo-transferrin; ACOT7 and FTL1 showed differences through TMT quantitative proteomic analysis. (D) WB and quantification showed ACOT7, FTL, and FTH expression levels in the medium with 0, 1, 10, 100, or 1000 µg/ml holo-transferrin. Statistics: Data were obtained in triplicate experiments and are shown as the mean ± SD; unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
The overexpression of ACOT7 has no impact on the proliferation and apoptosis of ESCs
The experimental group (ESCs were transfected by the adenovirus carrying Acot7) and the control group (ESCs were transfected by the vector adenovirus) were differentiated using the Flag tag. Western blot analysis and immunofluorescence analysis suggested that ESCs in the experimental group expressed a high Flag level compared to the control group (Fig. 3A and B). The results indicated the efficiency of virus transfection in the present study. Moreover, the cell proliferation experiment and flow cytometry result showed no differences in the cell proliferation curve and the apoptosis ratio between the two groups (Fig. 3C and D).

The overexpression of ACOT7 has no impact on the proliferation and apoptosis of ESCs. (A, B) WB and immunofluorescence showed Flag expression levels between experimental group (ESCs were transfected by the adenovirus carrying Acot7) and the control group (ESCs were transfected by the vector adenovirus). Scale bar = 20 µm. (C) IncuCyte S3 Living-Cell Analysis System measured cell proliferation. (D) Flow cytometry detected cell apoptosis between experimental group and the control group. Statistics: Data were obtained in triplicate experiments and are shown as the mean ± SD; unpaired t-test. ns, not significant.
The overexpression of ACOT7 decreases the levels of MDA and lipid peroxidation
To verify the function of ACOT7, we divided the cells into the following three groups: the experimental group, the control group, and the blank group. There were no differences in the level of lipid peroxidation measured by flow cytometry without oxidative stress. After being incubated with H2O2, Erastin, and RSL3 for 6 h, the lipid peroxidation level was significantly lower in the experimental group than that in the control and blank groups (Fig. 4A). We also found that under the condition of the same oxidative stress, the MDA level was significantly lower in the experimental group than that in the control and blank groups (Fig. 4B). Besides, the result of the CCK-8 experiment showed that the cell viability of ESCs was significantly higher in the experimental group than that in the control and blank groups (Fig. 4C).

The overexpression of ACOT7 decreases the levels of MDA and lipid peroxidation. (A) Flow cytometry detected cellular lipid peroxidation levels with no irritants, H2O2, Erastin, and RSL3. (B) MDA assay showed MDA relative levels with no irritants, H2O2, Erastin, and RSL3. (C) CCK-8 experiment measured cell viability with H2O2, Erastin, and RSL3. Statistics: Data were obtained in triplicate experiments and are shown as the mean ± SD; unpaired t-test. *P < 0.05, **P < 0.01. ns, not significant.
Discussion
Iron plays a key role in oxidative stress and light-induced skin damage. It is essential for cells to regulate the balance of oxidative stress (Wang et al. 2014). Given continuous exposure to outside environment, how the skin cells resist oxidative stress is very important (Balic and Mokos 2019). Our study found that ACOT7, which may increase with the rising expression of iron load, has a protective effect on lipid peroxidation induced by H2O2, Erastin, and RSL3 in ESCs.
Previous studies showed that free iron induces the production of free radicals, contributing to an increase of the level of lipid peroxidation (Wuyun et al. 2019). The balance of intracellular iron homeostasis depends on the expression and activity of transferrin, ferritin, and other proteins, which regulate and store iron (Park and Chung 2019). When the amount of intracellular free iron is high, the expression of ferritin may store more excess iron (Shu et al. 2019). Autophagy also regulates intracellular free iron through ferritin degradation (Jacomin et al. 2019). Previous studies have shown that ACOT7 might regulate the metabolism of neuronal fatty acid and prevent neurotoxicity (Ellis et al. 2013). As ACOT7 is closely related to lipid metabolism, we discovered that ACOT7 might inhibit lipid peroxidation in ESCs. H2O2, Erastin, and RSL3 were applied to induce lipid peroxidation in ESCs because Erastin and RSL3 are classical inducers of ferroptosis (Shintoku et al. 2017). Erastin impairs cellular antioxidant defenses, facilitating toxic ROS accumulation by inhibiting system xc−, cysteine-dependent glutathione (GSH) synthesis, and inhibiting the trans-plasma membrane cysteine redox shuttle (Banjac et al. 2008; Dixon et al. 2012). RSL3 can directly decrease the expression of glutathione peroxidase 4 (GPX4), elevating the ROS level in cells (Sui et al. 2018).
We found that the overexpression of ACOT7 may decrease MDA and lipid peroxidation levels, resulting in a higher survival rate of ESCs. We also found that the overexpression of ACOT7 had no impact on cell proliferation and apoptosis, which suggests that ACOT7 may improve the cellular survival rate by inhibiting ferroptosis.
It is not clear how ACOT7 contributes to protecting ESCs against lipid peroxidation. Previous studies have shown that ACOT7 might interact with a series of long-chain acyl-CoA enzymes, especially arachidonoyl-CoA (AA-CoA), an important precursor of AA-phosphatidylethanolamine (AA-PE) that forms the cell membrane. Besides, AA-PE is likely to be attacked by oxidation, which is a signal of ferroptosis (Doll et al. 2017; Kagan et al. 2017). However, further research is needed to explore whether ACOT7 may regulate the level of AA-PE on the cell membrane to execute its function of inhibiting lipid peroxidation.
In conclusion, we found that ACOT7 may protect cells against lipid peroxidation and improve the cellular survival rate for the first time, which provides us with a new method to inhibit the damage of ESCs caused by lipid peroxidation under oxidative stress, a new idea to prevent ferroptosis and a new direction to explore how ACOT7 exercises its functions.
Data availability
Not applicable.
Code availability
Not applicable.
Abbreviations
- ESCs:
-
Epidermal stem cells
- ACOT7:
-
Acyl-CoA thioesterase 7
- MDA:
-
Malondialdehyde
- ROS:
-
Reactive oxygen species
- SPF:
-
Specific pathogen-free
- PBS:
-
Phosphate-buffered saline
- CK14:
-
Cytokeratin 14
- CK15:
-
Cytokeratin 15
- CK10:
-
Cytokeratin 10
- CCK-8:
-
Cell Counting Kit-8
- FTH:
-
Ferritin Heavy Chain
- FTL:
-
Ferritin Light Chain
- AA-CoA:
-
Arachidonoyl-CoA
- AA-PE:
-
AA-phosphatidylethanolamine
References
An JJ, Eum WS, Kwon HS, Koh JS, Lee SY, Baek JH, Cho YJ, Kim DW, Han KH, Park J, Jang SH, Choi SY (2013) Protective effects of skin permeable epidermal and fibroblast growth factor against ultraviolet-induced skin damage and human skin wrinkles. J Cosmet Dermatol 12:287–295
Balic A, Mokos M (2019) Do we utilize our knowledge of the skin protective effects of carotenoids enough? Antioxidants 8:19
Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kölle P, Tschoep K, Issels RD, Daniel PT, Conrad M, Bornkamm GW (2008) The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 27(11):1618–1628. https://doi.org/10.1038/sj.onc.1210796
Cheloni G, Slaveykova VI (2013) Optimization of the C11-BODIPY581/591 dye for the determination of lipid oxidation in Chlamydomonas reinhardtii by flow cytometry. Cytom Part A 83:952–961
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, IngoId I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao GW, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JPF, Conrad M (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13:91–98
Ellis JM, Wong W, Wolfgang MJ (2013) Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol 33:1869–1882
Fibach E (2019) Erythropoiesis in vitro-a research and therapeutic tool in thalassemia. J Clin Med 8:15
Ghadially R (2012) 25 years of epidermal stem cell research. J Invest Dermatol 132:797–810
Giordani A, Haigle J, Leflon P, Risler A, Salmon S, Aubailly M, Maziere JC, Santus R, Morliere P (2000) Contrasting effects of excess ferritin expression on the iron-mediated oxidative stress induced by tert-butyl hydroperoxide or ultraviolet-A in human fibroblasts and keratinocytes. J Photochem Photobiol B-Biol 54:43–54
Hennig P, Fenini G, Di Filippo M, Beer HD (2020) Electrophiles against (skin) diseases: more than Nrf2. Biomolecules 10:20
Holden P, Nair LS (2019) Deferoxamine: an angiogenic and antioxidant molecule for tissue regeneration. Tissue Eng Part B-Rev 25:461–470
Jacomin AC, Geraki K, Brooks J, Tjendana-Tjhin V, Collingwood JF, Nezis IP (2019) Impact of autophagy and aging on iron load and ferritin in Drosophila brain. Front Cell Dev Biol 7:12
Kagan VE, Mao GW, Qu F, Angeli JPF, Doll S, St Croix C, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang JF, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar H, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13:81–90
Klaunig JE, Kamendulis LM, Hocevar BA (2010) Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 38:96–109
Liao X, Xie GH, Liu HW, Cheng B, Li SH, Xie S, Xiao LL, Fu XB (2014) Helium-neon laser irradiation promotes the proliferation and migration of human epidermal stem cells in vitro: proposed mechanism for enhanced wound re-epithelialization. Photomed Laser Surg 32:219–225
Mei HL, Zhao LP, Li W, Zheng ZW, Tang DM, Lu XL, He YZ (2020) Inhibition of ferroptosis protects House Ear Institute-Organ of Corti 1 cells and cochlear hair cells from cisplatin-induced ototoxicity. J Cell Mol Med 24:12065–12081
Meng SS, Xu XP, Chang W, Lu ZH, Huang LL, Xu JY, Liu L, Qiu HB, Yang Y, Guo FM (2018) LincRNA-p21 promotes mesenchymal stem cell migration capacity and survival through hypoxic preconditioning. Stem Cell Res Ther 9:11
Park E, Chung SW (2019) ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis 10:10
Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, Torii S (2017) Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 108:2187–2194
Shu WT, Baumann BH, Song Y, Liu YR, Wu XW, Dunaief JL (2019) Iron accumulates in retinal vascular endothelial cells but has minimal retinal penetration after IP iron dextran injection in mice. Invest Ophthalmol vis Sci 60:4378–4387
Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, Han X, Xiang Y, Huang X, Lin H, Xie T (2018) RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol 9:1371. https://doi.org/10.3389/fphar.2018.01371
Tomita K, Takashi Y, Ouchi Y, Kuwahara Y, Igarashi K, Nagasawa T, Nabika H, Kurimasa A, Fukumoto M, Nishitani Y, Sato T (2019) Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer Sci 110:2856–2866
Ud-Din S, Wilgus TA, McGeorge DD, Bayat A (2021) Pre-emptive priming of human skin improves cutaneous scarring and is superior to immediate and delayed topical anti-scarring treatment post-wounding: a double-blind randomised placebo-controlled clinical trial. Pharmaceutics 13:37
Wang DQ, Cheng F, Zhu HT (2021) Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ 28:1222–1236
Wang YF, Wu YN, Li T, Wang XY, Zhu CL (2019) Iron metabolism and brain development in premature infants. Front Physiol 10:13
Wang K, Zhang ZY, Tsai HI, Liu YF, Gao J, Wang M, Song L, Cao XF, Xu ZX, Chen HB, Gong AH, Wright JA, Richards T, Srai SKS (2014) The role of iron in the skin and cutaneous wound healing. Front Pharmacol 5:8
Wuyun G, Hu Y, He ZH, Li YC, Yan X (2019) The short tandem repeat of the DMT1 gene as a molecular marker of elite long-distance runners. Int J Genomics 2019:9
Yin H, Chen CY, Liu YW, Tan YJ, Deng ZL, Yang F, Huang FY, Wen C, Rao SS, Luo MJ, Hu XK, Liu ZZ, Wang ZX, Cao J, Liu HM, Liu JH, Yue T, Tang SY, Xie H (2019) Synechococcus elongatus PCC7942 secretes extracellular vesicles to accelerate cutaneous wound healing by promoting angiogenesis. Theranostics 9:2678–2693
Zhang JL, Chen XH, Hong JJ, Tang AF, Liu Y, Xie N, Nie GH, Yan XY, Liang MM (2021) Biochemistry of mammalian ferritins in the regulation of cellular iron homeostasis and oxidative responses. Sci China-Life Sci 64:352–362
Zhang M, Cao YQ, Li XH, Hu LZ, Taieb SK, Zhu XL, Zhang J, Feng YQ, Zhao R, Wang MQ, Xue WJ, Yang ZJ, Wang YB (2018) Cd271 mediates proliferation and differentiation of epidermal stem cells to support cutaneous burn wound healing. Cell Tissue Res 371:273–282
Zhang M, Zhang R, Li XH, Cao YQ, Huang KF, Ding J, Liu MY, Feng Z, Yin SY, Ma JX, Zhang HY, Wang YB (2020) CD271 promotes STZ-induced diabetic wound healing and regulates epidermal stem cell survival in the presence of the pTrkA receptor. Cell Tissue Res 379:181–193
Zhang M, Zhang YH, Ding J, Li XH, Zang CY, Yin SY, Ma JX, Wang YB, Cao YQ (2018) The role of TrkA in the promoting wounding-healing effect of CD271 on epidermal stem cells. Arch Dermatol Res 310:737–750
Funding
This study was supported by the National Natural Science Foundation of China (81972947), Natural Science Foundation of Shandong Province of China (Major Basic Research Program) (ZR2019ZD38), “Academic Elevation Program” of Shandong First Medical University (Shandong Academy of Medical Sciences) (2019LJ005), and Jinan Clinical Research Center for Tissue Engineering Skin Regeneration and Wound Repair.
Author information
Authors and Affiliations
Contributions
Conceptualization: GZ, SY, YW; data curation: GZ, SY, JM, ZW, GC; formal analysis: GZ, SY, JM, ZW; funding acquisition: YW; Investigation: GZ, SY, RS, JM, GC, ZW; methodology: GZ, SY, ZW; project administration: GZ, SY, CL, YW; Resources: CL, YW; supervision: YW, SY; validation: RS, AC; visualization: GZ; writing–original draft: GZ; writing–review and editing: CL, SY, YW.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Consent to participate
Not applicable.
Additional information
Co-first authors: Guang Zhang, Jiaxu Ma
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Ma, J., Wu, Z. et al. ACOT7 protects epidermal stem cells against lipid peroxidation. In Vitro Cell.Dev.Biol.-Animal 58, 549–557 (2022). https://doi.org/10.1007/s11626-022-00703-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-022-00703-9