
Abstract
Escalating oil consumption has resulted in an increase in accidental spills of petroleum hydrocarbons, causing severe environmental degradation, notably in vulnerable regions like the Niger Delta. Complex mixture of these hydrocarbons particularly long-chain alkanes presents unique challenges in restoration of polluted environment due to their chemical properties. This study aimed to investigate the long-chain hydrocarbon-degrading bacterial communities within long-term chronically polluted soil in Ogoniland, by utilizing both traditional cultivation methods and modern culture-independent techniques. Results revealed that surface-polluted soil (SPS) and subsurface soil (SPSS) exhibit significantly higher total organic carbon (TOC) ranging from 5.64 to 5.06% and total petroleum hydrocarbons (TPH) levels ranging from 36,775 ppm to 14,087 ppm, compared to unpolluted soil (UPS) with 1.97% TOC and 479 ppm TPH, respectively. Analysis of carbon chain lengths reveals the prevalence of longer-chain alkanes (C20-28) in the surface soil. Culture-dependent methods, utilizing crude oil enrichment (COE) and paraffin wax enrichment (PWE), yield 47 bacterial isolates subjected to a long-chain alkane degradation assay. Twelve bacterial strains demonstrate significant degradation abilities across all enriched media. Three bacterial members, namely Pseudomonas sp. (almA), Marinomonas sp. (almA), and Alteromonas (ladA), exhibit genes responsible for long-chain alkane degradation, demonstrating efficiency between 50 and 80%. Culture-independent analysis reveals that surface SPS samples exhibit greater species richness and diversity compared to subsurface SPSS samples. Proteobacteria dominates as the phylum in both soil sample types, ranging from 22.23 to 82.61%, with Firmicutes (0.2–2.22%), Actinobacteria (0.4–3.02%), and Acidobacteria (0.1–3.53%) also prevalent. Bacterial profiles at genus level revealed that distinct variations among bacterial populations between SPS and SPSS samples comprising number of hydrocarbon degraders and the functional predictions also highlight the presence of potential catabolic genes (nahAa, adh2, and cpnA) in the polluted soil. However, culture-dependent analysis only captured a few of the dominant members found in culture-independent analysis, implying that more specialized media or environments are needed to isolate more bacterial members. The findings from this study contribute valuable information to ecological and biotechnological aspects, aiding in the development of more effective bioremediation applications for restoring oil-contaminated environments.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In an era marked by rapid technological advances and rising affluence, the global demand for fossil fuels, particularly oil and coal, has surged significantly. This escalating consumption has led to an increase in accidental spills of petroleum hydrocarbons, causing severe environmental degradation, notably in vulnerable regions like the Niger Delta (Nwilo and Badejo 2006; Ite et al. 2013; Okon and Ogba 2018). Crude oil is a complex mixture, predominantly composed of four categories: aliphatic (saturated) hydrocarbons, aromatics, asphaltenes, and resins (Cheng et al. 2014; Song et al. 2018). Within the aliphatics, alkanes exhibit a range of structural variants such as linear (n-alkanes), cyclic (cyclo-alkanes), and branched (iso-alkanes). Depending on the crude oil’s origin, alkanes can constitute a substantial 20–50% of its composition. Intriguingly, alkanes are also biosynthesized by diverse organisms like plants, algae, and bacteria for various biological functions such as waste management, structural support, and chemical signaling (Van Beilen et al. 2003). However, the degradation of these compounds, particularly long-chain alkanes, presents unique challenges due to their low solubility, minimal reactivity, and high solidification points, complicating both environmental remediation and efficient fossil fuel utilization (Rojo 2009; El Mahdi et al. 2016).
In the face of ongoing environmental pollution, the quest for sustainable and effective remediation techniques has never been more critical. Bioremediation emerges as an attractive and environmentally friendly alternative to traditional chemical and physical remediation methods, ensuring minimal disruption to the environment (Azubuike et al. 2016). This approach combines the natural capabilities of microorganisms to degrade pollutants into less harmful forms, prioritizing economic efficiency (Wu et al. 2023b). Among these microorganisms, bacteria play a pivotal role due to their metabolic versatility, capability for rapid reproduction, and resilience in extreme environmental conditions (Mrozik and Piotrowska-Seget 2010). Specifically, certain bacterial communities possess distinctive enzymatic systems capable of degrading hydrocarbons. For example, recent studies have explored the use of surfactants as a competitive alternative to enhance the removal of n-alkanes. While these systems have proven effective in the removal of various short-chain n-alkanes (Wu et al. 2022, 2023a) their efficacy for long-chain n-alkanes remains uncertain due to their low water solubility and bioavailability. This uncertainty highlights the ongoing challenge of detoxifying polluted soils and waters..The biodegradation of long-chain alkanes and paraffins has recently gained significant attention, particularly in diverse extreme environments, including long-term polluted soils. Therefore, understanding the composition and functional capabilities of these bacterial communities can open up avenues for efficient, cost-effective, and sustainable remediation strategies for long chain alkanes. Despite the urgency for effective bioremediation techniques, a significant gap exists in the comprehensive study of hydrocarbon-degrading bacterial communities, particularly in areas with a long history of chronic pollution. Existing research often leans either toward culture-dependent or culture-independent methodologies, failing to provide a holistic view of microbial diversity and functionality in contaminated environments (Das and Chandran 2011; Hedgpeth et al. 2021). Moreover, the mechanisms by which these microbial communities collectively degrade a broad spectrum of hydrocarbons, especially the more recalcitrant long-chain alkanes, are not fully understood (Head et al. 2006; Rojo 2009). Additionally, the genetic basis for hydrocarbon degradation, including the role of specific monooxygenase genes, is not fully elucidated. These gaps in knowledge signify a crucial need for an integrated approach that could offer more effective and adaptable solutions for soil remediation in chronically polluted areas.
Ogoniland, part of Nigeria’s Niger Delta, has been plagued by chronic pollution due to prolonged exposure to oil spills and leaks. This environmental catastrophe has not only jeopardized the local ecosystem but also the livelihoods of communities who depend on these lands for subsistence (Ogri 2001; Kponee et al. 2015). Given the complexity and resilience of hydrocarbon contaminants, especially long-chain alkanes, there is a pressing need to explore innovative and efficient cleanup strategies. One promising avenue is the study of naturally occurring hydrocarbon-degrading bacterial communities. These microbial assemblages in petroleum-contaminated environments have evolved mechanisms to utilize hydrocarbons as a carbon source (Austin and Callaghan 2013). This adaptation influences nutrient cycling, energy flow, and biogeochemical processes, presenting a potential bioremediation tool for contaminated sites (Chicca et al. 2022; Adetitun and Tomilayo 2023). The present work aimed to investigate the long-chain hydrocarbon-degrading bacterial communities within long-term chronically polluted soil in Ogoniland, by utilizing both traditional cultivation methods and modern culture-independent techniques. By combining enrichment strategies and genetic analysis of 16S rRNA gene sequences, the identification of hydrocarbonoclastic bacterial isolates was achieved. Additionally, selected bacterial strains were evaluated for their long-chain hydrocarbon degradation capabilities, alongside the characterization of flavin-binding monooxygenase genes (alma and ladA). In addition, we report bacterial strains proficient in efficiently breaking down a wide spectrum of hydrocarbons, including long-chain alkanes common in oil-polluted soils. Moreover, targeted amplicon analysis provided insights into the total bacterial populations and their functions in polluted and unpolluted soils, facilitating a comparative understanding of microbial communities in distinct environments.
Materials and methods
Sampling area
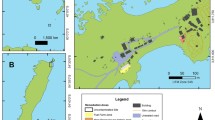
Soil samples contaminated with crude oil were collected from four different points of North, South, West, and East—at a pollution site in Gio community, Tai Local Government Area, Niger Delta, Nigeria, as depicted in Fig. 1A. The selected sampling points is a zone of area heavily impacted by oil contamination, a consequence of accidental oil spill incidents. Sterilized soil augers were used for sample collection. Surface soil samples (SPS) were obtained from a depth ranging between 0 and 0.15 m, whereas subsurface soil samples (SPSS) were taken from a depth of 1 m, as illustrated in Fig. 1B–C. For control purposes, unpolluted soil samples (UPS) were collected approximately 1 km (km) away from the contaminated site (Supplementary Fig. 1). All samples were placed in sterile polyethylene bags and transported to the lab for analysis within 6 h, maintained at a temperature of 4 °C.

A Map highlighting the sample collection area in the Gio community of Niger Delta region, Nigeria, B surface soil impacted by oil contamination, and C sample collection from the subsurface soil layer (about 1 m depth)
Soil chemical properties
Soil chemical properties were analyzed for the collected soil samples. pH levels were monitored with a Metrohn automated probe analyzer (692, Herisau, Switzerland). Moisture content was measured by drying the soil at 105 °C for 12 h. The concentrations of nitrate (NO3−) and phosphate (PO4−), as well as toxic elements such as lead (Pb), nickel (Ni), and cadmium (Cd), were quantified following standard ASTM procedures and using ICP-OES, respectively. The electrical conductivity (EC) was determined using a conductivity meter, and the total organic carbon content was evaluated using the Walkley and Black (1934) method. The petroleum hydrocarbon content in the oil-contaminated soil was quantified using gas chromatography-flame ionization detector (GC-FID) analysis, adhering to the protocol given by Chikere et al. (2019). Briefly, for the extraction process, soil samples weighing 1 g each were combined with 20 ml of n-pentane solvent. This mixture allowed for ultra-sonication for 15 min, after which the samples were left to allow organic phase separation at 30 °C for 60 min. Subsequently, 1 ml of the separated organic phase was collected, transferred to GC vials, and prepared for analysis. The GC-FID apparatus was equipped with an HP 7673 FID detector, an autosampler, and a specialized fused silica capillary column. Both detector and injector were calibrated to temperatures of 320 °C and 250 °C, respectively. The oven’s temperature was set to start at 40 °C for 3 min, and then ramped up to 300 °C. Helium was used as the carrier gas, at a velocity of 38 cm/s. All data was processed using the Agilent Chemstation chromatography software (v10). The quantification of TPHs was attained by identifying the peak area, employing forced line integration via the Agilent Chemstation, spanning from n-C6 (n-hexane) to n-C35 (pentatriacontane).
Enrichment, isolation, and biochemical characterization of hydrocarbon-degrading bacteria
To cultivate bacteria capable of degrading hydrocarbon, in particular long-chain alkanes in oil-contaminated soil, a 10 g soil sample was combined with 200 mL of modified Bushnell Hass (BH) Medium. This medium included the following components per liter: 0.2 g of magnesium sulfate (MgSO4), 0.02 g of calcium chloride (CaCl2), 1 g of monopotassium phosphate (KH2PO4), 1 g of dipotassium phosphate (K2HPO4), 1 g of ammonium nitrate (NH4NO3), and 0.05 g of ferric chloride (FeCl3) and 1 mL of trace element solution. The trace element solution, added at a rate of 1 mL per liter, consisted of 50 mg of MnCl2·H2O, 300 mg of H3BO3, 1.1 mg of FeSO4·7H2O, 190 mg of CoCl2·6H2O, 2 mg of CuCl2·2H2O, 24 mg of NiCl2·6H2O, 18 mg of NaMoO4·2H2O, 42 mg of ZnCl2·7H2O, and 1 mL of a vitamin solution (Pfennig and Trüper 1992). The pH of the medium was adjusted to 7.1, and it was sterilized at 121 °C for 20 min before adding the filter-sterilized vitamin solution. To further enrich the BH medium, 1 mL of Heavy Bonny crude oil (COE) and 1 g of paraffin wax (PWE) were added separately containing BH media to isolate bacteria capable of degrading long-chain alkanes. The mixture was then incubated at 30 °C and 100 rpm for 72 h. Cultures with an optical density at 660 nm > 1.0 were subsequently plated on agar containing BH medium and LB medium and incubated under the same conditions to obtain individual bacterial colonies of total hydrocarbon utilizing bacteria (THUB) and total heterotrophic bacteria (THB) respectively and the respective colony forming units (cfu) were calculated. Colonies displaying distinct morphological characteristics were selected and further purified through subculturing on Luria–Bertani (LB) plates. Isolates exhibiting distinct morphological traits, obtained from two separate enrichment media, were selected for a comprehensive array of biochemical analyses. These analyses encompassed assessments of catalase, oxidase, Methyl Red Voges Proskauer (MR-VP), citrate utilization, urease activity, and hydrogen sulfide (H2S) production. In addition, sugar fermentation tests, including glucose, lactose, and sucrose, were conducted to identify the bacterial members. The frequency of occurrence (%) was then calculated for both the COE and PWE sets of isolates. All isolated strains were preserved at -20 °C in liquid cultures containing 20% glycerol (v/v).
Degradation assay for long-chain alkanes
To screen for hydrocarbon degradation capabilities, a little modified method adapted from Selvarajan et al. (2017a). The bacterial isolates, initially cultivated in enrichment media, were subsequently grown in BH media supplemented with glucose (10 g/L) and yeast extract (1.0 g/L). After a 72-h incubation period at 25 °C with continuous shaking at 120 rpm, cultures demonstrating robust growth were subjected to centrifugation at 10,000 × g for 10 min. The resulting pellet was resuspended in phosphate buffer, and this was followed by another centrifugation step to eliminate residual culture medium components. The final pellet was resuspended in phosphate buffer to an optical density matching the McFarland 0.5 standard. A 2,6-dichlorophenolindophenol (DCPIP) indicator assay was set up in a sterile 96-well microplate. Each well contained 20 µL of the bacterial suspension, 168 µL of BH medium, 12 µL of DCPIP, and 2 µL of a hydrocarbon source rich in long-chain alkanes (paraffin wax, hexadecane, and crude oil). In addition to the test wells, both glucose (10%) positive controls and blank negative controls were also added. These plates were incubated at 30 °C, and photometric readings at 600OD were taken after 72, 144, and 288 h of incubation.
Molecular characterization and detection of potential gene in selected strains
Selected bacterial strains from both enrichment processes were subjected to molecular characterization for identification and testing their phytogenic relationship. DNA extraction from each culture was executed using the Quick g-DNA extraction kit (Zymo Research Corporation). This was followed by PCR amplification employing the 16S universal bacterial primers (27F and 1492R). The thermal cycling conditions included an initial denaturation at 95 °C for 5 min, then 32 cycles each of 95 °C denaturation for 30 s, 52 °C annealing for 30 s, and 72 °C elongation for 1 min, and with a final extension at 72 °C for 10 min. The PCR products were purified using gel purification kit and sent to Inqaba Biotech (Pretoria, South Africa) for sanger sequencing. The obtained sequences were then analyzed via BLAST to ascertain the identity of the isolates. Phylogenetic assessment employing the maximum-likelihood method was performed using the MEGA-X software (Centre for Evolutionary Medicine and Informatics, Tempe, AZ, USA) (Kumar et al. 2018). Functional gene identification (almA and ladA gene) was carried out by following the method given by Wang and Shao (Wang and Shao 2012) and Tourova et al. (Tourova et al. 2016) respectively.
Total DNA extraction and Illumina sequencing
The genomic DNA from soil samples was extracted using the Power Soil™ DNA extraction kit as per the manufacturer’s instructions. To assess the quantity of extracted DNA, a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA) was used. For DNA amplification, the PCR primer pairs 341F (CCTACGGGNGGCWGCAG) and 805R (GGACTACHVGGGTWTCTAAT) were employed, targeting the V3-V4 hypervariable region of the 16S rRNA gene. The DNA amplicons generated by PCR were confirmed using agarose gel electrophoresis. Initial purification of the amplicons was carried out using AMPure XP beads (Beckman Coulter, Brea, CA, USA) following the manufacturer’s instructions. After uniquely indexing the amplicons and adding Illumina sequencing adapters, an additional purification step was performed using AMPure XP beads. The resulting purified product was then normalized to ensure equal concentration, denatured, and loaded onto a MiSeq V3 cartridge for paired-end sequencing using the Illumina MiSeq sequencer (Illumina Inc., San Diego, CA, USA).
Bioinformatics and diversity analysis
The raw Illumina MiSeq sequences underwent processing using QIIME2 (Bolyen et al. 2018) Platform. Before the QIIME2 analysis, the primers and adapter sequences were trimmed using cutadapt 3.1 (Martin 2011) and the quality of the sequences was assessed using FastQC (Andrews 2010). DADA2 (Callahan et al. 2016) was utilized for sequence denoising, eliminating low-quality reads, marginal sequences, and clustering sequences into amplicon sequence variants (ASVs). Then the ASVs were clustered with 100% similarity based on the representative sequences. The SILVA v138 database (Quast et al. 2012) served as the reference dataset for this process. Diversity analyses, including alpha and beta diversities, were calculated based on ASVs using QIIME2. To investigate the microbial community functional attributes in both polluted and unpolluted soil, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2) (Douglas et al. 2020) was employed. PICRUSt2 enables the prediction of functional profiles using 16S rRNA data from the community. The prediction outputs encompass enzymatic gene families and MetaCyc pathway profiles associated with the 16S rRNA representative sequences. Statistical analysis and visualizations were performed using Qiime2, Origin 2022, and R 4.0.2. All raw sequencing data have been deposited in the NCBI Sequence Read Archive (NCBI-SRA) under BioProject accession number PRJNA1037324.
Results
Chemical characteristics of polluted and unpolluted soil samples
The chemical characteristics were assessed across three distinct soil types: surface-polluted soil (SPS), subsurface-polluted soil (SPSS), and unpolluted soil (UPS), as detailed in Table 1. Among the pH levels, SPSS exhibited the acidic condition (pH 4.82), followed by SPS (pH 5.03), whereas UPS displayed relatively lesser acidity with a pH of 6.12. Temperature demonstrated slight variations between polluted and unpolluted soils. Regarding nutrient composition, nitrate concentrations were most prominent in SPS (8.33 mg/kg), followed by a decrease in SPSS (6.67 mg/kg), and the lowest levels were observed in UPS (3.84 mg/kg). Conversely, phosphate levels exhibited an inverse relationship, being highest in UPS (20.52 mg/kg) and lowest in SPS (8.11 mg/kg). Electrical conductivity (EC) reached its peak in UPS (185.46 µs/cm), succeeded by SPS (141.1 µs/cm) and SPSS (140.73 µs/cm). The trend of total organic carbon (TOC) inclined from SPS (5.64%) to SPSS (5.068%), reaching the lowest value in UPS (1.97%). Moisture content displayed its highest value in SPS (7.93%), followed by SPSS (6.76%), and the lowest content was recorded in UPS (4.88%). Concentrations of heavy metals like nickel, lead, and cadmium were generally more elevated in the polluted soils, though some exceptions were noted, such as nickel having the highest concentration in UPS (0.68 mg/kg).
Hydrocarbons
The levels of total petroleum hydrocarbon (TPH) and polycyclic aromatic hydrocarbons (PAHs) in different soil samples were quantified (Fig. 2A); the results revealed that SPS showed the highest contamination, with TPH levels at 36,775 ppm and PAHs at 12,209.3 ppm. These were significantly higher than the levels in SPSS, which were 14,087.8 ppm for TPH and 3,248.75 ppm for PAHs. UPS exhibited minimal contamination, with TPH at 479.67 ppm and PAHs at 22.72 ppm, both far below the levels in polluted soils. For comparison, the petroleum resource standard (PRS) values were (5000 ppm for TPH and 40 ppm for PAHs) used and the findings indicate severe contamination in the surface- and subsurface-polluted soils, especially in the SPS, compared to both the unpolluted soil and established petroleum resource standards. An in-depth analysis of individual polycyclic aromatic hydrocarbons (PAHs) in both surface and subsurface contaminated soils were quantified, revealing a uniform pattern of elevated contamination levels in the surface soil. Significantly, benzo(a)anthracene was found to be the most abundant in the surface soil, with concentrations reaching 1120.2 ppm. In the subsurface soil, acenaphthylene was the most prevalent, recorded at 278.624 ppm. In contrast, the lowest levels were detected for benzo(a)pyrene in the surface soil, at 341.3 ppm, and for anthracene in the subsurface soil, measuring at 116.323 ppm (Fig. 2B).

A Measured concentration (ppm) of total petroleum hydrocarbons (TPH) and polyaromatic hydrocarbons (PAHs) in oil polluted surface (SPS) and subsurface (SPSS) soil samples compared with unpolluted soil samples (UPS) and petroleum resource standard (PRS), B measured concentrations (ppm) of different PAHs, and C distribution of various carbon chain lengths in collected soil samples
The distribution of various carbon chain lengths (C8 to C34) in surface- and subsurface-polluted soils was investigated. Notably, the highest concentration in the surface-polluted soil was observed for C20, which recorded a level of 864.615 ppm, followed closely by C28 at 798.78 ppm. In contrast, the subsurface-polluted soil had its highest concentration at C18, registering at 398.126 ppm. Most notably, the surface soil consistently displayed higher concentrations of long-chain alkanes compared to the subsurface soil (Fig. 2C). Additionally, certain long-chain alkanes such as C19, C22, C25, C27, C29, C31, C32, and C33 were absent in both soil types, highlighting the specificity of alkane degradation or accumulation in these environments.
Culture-dependent analysis
Bacterial enrichment, isolation, and characterization
The bacterial populations in collected soil samples were cultivated using two distinct enrichment methods: crude oil enrichment (COE) and paraffin wax enrichment (PWE). In SPS, the colony-forming units (cfu) of total hydrocarbon utilizing bacteria (THUB) were found to be 8.9 cfu/g and 6.8 cfu/g for COE and PWE, respectively. This was accompanied by a total of 9 cfu/g of total heterotrophic bacteria (THB). In SPSS, the counts were slightly lower, with THUB at 8 cfu/g (COE) and 6 cfu/g (PWE), alongside 8.6 cfu/g of THB. The UPS samples showed markedly lower bacterial counts, with THUB at 3.95 cfu/g (COE) and 5 cfu/g (PWE), and THB at 3.6 cfu/g (Fig. 3A–C).

A Hydrocarbon utilization bacterial counts from crude oil enrichment (COE); B hydrocarbon utilization bacterial counts from paraffin wax enrichment; C the total heterotrophic bacterial counts (THB) of collected soils; D frequency of occurrence for bacterial isolates for paraffin wax enrichment; E frequency of occurrence for bacterial isolates for crude oil enrichment
Tables 2 and 3 present the biochemical characterization of bacterial isolates derived from enrichment media of crude oil and paraffin wax, respectively. A total of 47 pure bacterial colonies were isolated and subjected to a series of biochemical tests. The results shows that Pseudomonas sp. was the predominant bacterial type in the crude oil-enriched medium, followed by Rhodococcus sp., Corynebacterium sp., Bacillus sp., and Micrococcus sp. Conversely, Proteus sp., Salmonella sp., E. coli, Serratia sp., Achromobacter sp., Providencia sp., and Vibrio sp. were identified with lower frequency. Similarly, in the paraffin wax-enriched samples, Pseudomonas sp. emerged as the most frequently observed bacterial member, with subsequent occurrences of Bacillus sp., and Achromobacter sp. Other bacterial members, namely Aeromonas sp., Rhodococcus sp., Serratia sp., and Acinetobacter sp., were identified less frequently. In soil samples, variations in the frequency of bacterial members were evident depending on the specific enrichment conditions. In both types of enriched samples, the presence of bacterial members such as Bacillus sp., Pseudomonas sp., and Citrobacter sp. was consistently detected. Notably, paraffin wax-enriched samples displayed a distinctive set of bacterial members including Aeromonas sp., Proteus sp., and Klebsiella sp. (Fig. 3D), while crude oil-enriched media revealed a prevalence of different bacterial members, encompassing Micrococcus sp., E. coli, Rhodococcus sp., Staphylococcus sp., and Vibrio sp. (Fig. 3E). Additionally, surface samples consistently exhibited a higher frequency of bacterial members when compared to sub-surface and unpolluted soil samples. Among the identified bacterial members, Pseudomonas sp. demonstrated as the dominant species, present not only in enriched media but also across all collected samples.
Following biochemical characterization, the bacterial strains were tested for initial growth on BH media enriched with glucose to optimize growth conditions. Out of the 47 isolates, 16 exhibited robust growth and were selected for subsequent molecular characterization and studies on the degradation of long-chain hydrocarbons. The analysis of PCR-amplified 16S rDNA sequences of these selected isolates, when compared with a reference species database from NCBI, revealed their affiliation with various bacterial genera. These genera included Bacillus pumilis, Bacillus stratosphericus, Bacillus pacificus, Bacillus cereus, Pseudomonas aeruginosa, Streptromonas atlantica, Marinomonas atlantica, Proteus vulgaris, and Providencia rettgeri. Importantly, all isolates exhibited a high nucleotide sequence similarity, ranging from 99 to 100%, with these known species. A phylogenetic tree, generated using MegaX software, is depicted in Fig. 4. This tree highlights that most of the isolates are classified into two phyla: Firmicutes and Proteobacteria. Within the Firmicutes cluster, isolates from the Bacillaceae family are notably prominent. Additionally, the phylogenetic tree highlights that all the isolates closely align with their respective genetic relatives.

Phylogenetic tree was constructed using maximum likelihood based on 16S rDNA gene sequences, illustrating the bacterial isolates’ phylogenetic relationships. Each dot on the tree represents an isolated strain from this study. Strains labelled with codes “P” were isolated from paraffin wax enrichment, while those labelled “C” were isolated from crude oil enrichment
Degradation potential of long-chain alkanes
To evaluate the ability to degrade long-chain alkanes, about 20 bacterial strains were selected based on their growth conditions. These were then subjected to a degradation assay using BH medium with the DCPIP indicator, enriched with hydrocarbon sources rich in long-chain alkanes: paraffin wax (POL), hexadecane (HDX), and heavy crude oil (HCO). The experiment also included positive controls and incubated for a period of 288 h. The results revealed that 12 of the bacterial strains exhibited significant degradation abilities across all enriched media (Supplementary Table 1, Fig. 5A). However, a few bacterial members demonstrated weak degradation abilities in HCO-enriched media. Notably, strains like Proteus and Bacillus pacificus displayed no degradation capabilities against long-chain alkanes. To identify the functional genes responsible for degrading long-chain alkanes, the strains were subjected to targeted PCR analysis, specifically looking for the almA and ladA genes. Out of all the strains, only three bacterial members exhibited these target genes: almA was found in Pseudomonas aeruginosa and Marinomonas atlantica, whereas ladA was identified in Alteromonas confluentis. A subsequent analysis of these three strains’ degradation abilities revealed Marinomonas (POL (80%), HCO (66.2%) and HDX: (40%) as the most capable at degrading paraffin wax and crude oil. In contrast, Pseudomonas exhibited strong degradation efficiency (POL: 56%, HCO: 50%, HDX: 64.7%) against Hexadecane (Fig. 5B).

Long chain hydrocarbon degradation potential by selected bacterial members enriched with (A) paraffin oil, Hexadecane, and crude oil as sole carbon source—the presence of functional genes (almA and ladA) indicated in yellow with respect to the bacterial members. B Percentage of degradation by the bacterial members possessing functional genes
Culture-independent analysis
In total 8 soil samples collected from surface and subsurface of the oil polluted sites were sequenced using illumina platform. To evaluate community richness and diversity, α-diversity indices at the amplicon sequence variant (ASV) level were analyzed using Qiime 2. Examining the Chao1 diversity indices, surface samples exhibited greater species richness compared to subsurface samples (Fig. 6a). However, it is worth noting that the observed differences in bacterial richness did not reach statistical significance (p-value: 0.70559). Similarly, the Shannon index (Fig. 6b) indicated higher species diversity in surface soil samples compared to subsurface soil (p-value: 0.11533). Analyzing the Bray–Curtis dissimilarity between soil bacterial communities revealed differences in the community structure between surface and subsurface soil (Fig. 6c). Notably, subsurface samples tended to cluster together, while surface samples displayed some variations and clustered separately. Additionally, the observed differences in bacterial community composition between surface and subsurface samples in multivariate space were not highly significant (PERMANOVA F-value: 3.051; R-squared: 0.33709; p-value: 0.082).

Alpha and beta diversity indices A Chao1 and B Shannon index, and C principal coordinate analysis (PCoA) plot based on Bray–Curtis dissimilarities of collected surface and subsurface soil samples
Proteobacteria emerged as the dominant phylum in both soil sample types, ranging from 22.23 to 66.71% in surface samples and from 57.49 to 82.61% in subsurface samples. In the surface soil samples, following Proteobacteria, Acidobacteria was prevalent, ranging from 0.1 to 3.53%. Actinobacteria (0.4 to 3.02%), Firmicutes (0.29 to 2.22%), and AD3 (0.1 to 1.56%) were also notably abundant (Fig. 7a). Conversely, in the subsurface soil samples, the abundance of Firmicutes (0.9 to 1.88%), Actinobacteria (0.36 to 1.38%), and Acidobacteria (0.1%) was comparatively lower than in the surface soil samples. Notably, both soil samples showed a high number of unclassified reads, indicating the potential presence of novel microbes in these polluted areas. Within the Proteobacteria phylum, the class Betaproteobacteria dominated in surface soil samples, ranging from 1.5 to 60.04%. It was followed by Alphaproteobacteria, which accounted for 4.76 to 37.46%, and Gammaproteobacteria, with an abundance ranging from 1.2 to 27.09%. In contrast, subsurface soil samples showed a higher abundance of Betaproteobacteria, ranging from 39.63 to 61.43%, compared to surface soil. The higher abundance of Betaproteobacteria in subsurface soil could be influenced by specific adaptations to subsurface conditions or the availability of substrates favoring the proliferation of this bacterial class. Alphaproteobacteria accounted for 5.06 to 30.65%, and Gammaproteobacteria were present at lower abundance, ranging from 1.58 to 17.96%, compared to surface soil samples. Additionally, both soil samples contained notable classes such as Acidobacteriia and Bacilli in a considerable abundance compared to other bacterial classes (Fig. 7b) suggests their potential ecological significance in hydrocarbon-contaminated environments, possibly playing roles in bioremediation processes or responding to the presence of long-chain alkanes.

Abundance of phylotypes at the taxonomic rank A Phylum and B Class level observed in surface and subsurface soil samples
Clear distinctions between surface and subsurface soil samples were evident at the genus level. Surface samples exhibited a notable presence of genera including Burkholderia, Pedomicrobium, Methyloversatilis, Rothia, Granulicatella, Deflivibacter, Rhodococcus, Rhodobacter, Agrobacterium, Novosphingbium, Bacillus, and Mycobacterium. In contrast, subsurface samples were characterized by the prevalence of genera such as Sphingomonas, Acinetobacter, Arcobacter, Hyphomicrobium, Halomonas, Mycoplana, Ralstonia, Pseudomonas, and Lactococcus. It is worth noting that several genera were common to both sample types imply their versatility and adaptability to different soil layers, showcasing their potential role as key players in hydrocarbon degradation across the entire soil profile. The heatmap representation visually highlights the distribution patterns of these major genera (Fig. 8), emphasizing their relative abundance and providing a comprehensive overview of the microbial landscape in response to chronic pollution.

Heat map showing the dominant bacterial members (genus level) in the surface and subsurface soil samples
To assess the degradative capabilities and functional traits of bacterial communities in oil-polluted sites, we conducted a comparative analysis of predicted functional genes and enzymes between the polluted and unpolluted sites. Notably, genes like nahAa (naphthalene 1,2 dioxygenase ferredoxin reductase), adh2 (alcohol dehydrogenase), eutE (aldehyde dehydrogenase), and cpnA (cyclopentanol dehydrogenase) were solely detected in bacterial communities from the oil-polluted samples and were absent in the unpolluted samples. These genes play pivotal roles in catabolic pathways responsible for breaking down aromatic and hydrocarbon compounds. Additionally, various genes involved in multiple metabolic pathways, including catE, catA, dmpB, aldB, ssuD, dhaA, and alkB, were identified in both soil communities. However, it is noteworthy that the oil-polluted soils exhibited a relatively higher abundance of these genes compared to the unpolluted soils (Fig. 9).

Hydrocarbon catabolic enzymes, genes, and pathways predicted in the polluted and unpolluted soil samples (differences in the overall abundance were significant; p = 0.02)
Discussion
Soil environments in regions that have been chronically exposed to crude oil contamination are home to a diverse range of microorganisms with unique capabilities for degrading recalcitrant compounds. These microorganisms hold significant potential for application in the oil industry and areas affected by accidental oil spills, offering an effective solution of addressing and reducing the environmental toxicity caused by such incidents through bioremediation techniques. In this study, surface and subsurface soil samples were collected from Gio community, Niger Delta, Nigeria (Fig. 1A), specifically targeting sampling locations significantly affected by oil contamination due to accidental oil spill incidents. The chemical analysis of the soil indicated that the unpolluted soil samples exhibited a relatively higher pH compared to the polluted soil. Notably, the polluted surface soil samples were more acidic than the subsurface soil samples. These findings align with the results of a study by Shaoping et al. (2021), which showed that exposure to petroleum and crude oil compounds tends to decrease soil pH. However, it is worth noting that the extent of pH change may vary depending on the initial soil chemical properties and the concentration of oil present in the soil. When the soil pH becomes acidic, certain reactions, such as the adsorption of heavy metals onto soil organic matter, can become more evident (Jabbarov et al. 2019). This study also revealed that the organic matter content in both surface- and subsurface-polluted soils was relatively high compared to unpolluted soil (Table 1). This increased organic matter content contributed to the adsorption of heavy metals like lead and cadmium from the crude oil, resulting in the enrichment of toxic elements in the soil. This aligns with the observations of Aigberua et al. (2017), where concentrations of heavy metals were noted to be comparatively higher in soils contaminated with crude oil. Notably, the concentration of nickel was higher in unpolluted soil compared to polluted soil (Table 1). However, elevated levels of Ni in different soil layers may be influenced by both natural soil-forming processes and human activities (Pivková et al. 2022).
As expected, both the surface-polluted soil (SPS) and subsurface soil (SPSS) showed significantly higher total organic carbon (TOC) levels compared to the unpolluted soil (UPS). This difference can be attributed to the prolonged exposure of SPS to hydrocarbons in contrast to the relatively limited exposure of UPS. Consistent with the TOC findings, the concentrations of total petroleum hydrocarbons (TPH) were notably higher (p < 0.05) in the polluted soils when compared to the unpolluted ones (Fig. 2A). This increase in TOC content within the oil-contaminated areas can be linked to the higher levels of TPH present in the soil. It is important to report that there was a positive correlation between TOC content and TPH content in the soils, highlighting the feasibility of using TOC as an effective tool for monitoring petroleum hydrocarbon contamination due to the fact that TOC encompasses all weight fractions of TPH (Schreier et al. 1999; Kuppusamy et al. 2020). The analysis of carbon chain lengths in polluted soils showed that longer-chain alkanes, particularly C20, were more abundant on the surface soil. Similarly, a study conducted in China petrol station soil samples reported relatively higher concentrations of long-chain alkanes compared to other n-alkane concentrations (Li et al. 2021). This could be because these larger molecules are less mobile than shorter alkanes. In the layers beneath the surface, C18 alkanes were more common, indicating that smaller alkanes might sink deeper into the soil, potentially due to them being more soluble (Chen et al. 2022). The absence of certain long-chain alkanes (C19, C22, C25, C27, C29, C31, C32, and C33) in both the surface and subsurface soils (Fig. 2C) may point to the selective utilization of microbes or the unique characteristics of the oil spill. This study, therefore, focused on the indigenous microbes that catabolize these longer chains. The observed distribution of alkanes in the soil highlights the complex nature of environmental oil contamination, highlighting the influence of physical, chemical, and biological dynamics on the fate of pollutants within the soil matrix (Si-Zhong et al. 2009; Kuppusamy et al. 2020).
In recent era, studies utilizing culture-independent methods have been pivotal in shedding light on microbial populations in extreme environments (Ramganesh et al. 2018; Selvarajan et al. 2018b, a; Sibanda et al. 2018; Ubani et al. 2022). Nevertheless, the role of culture-dependent techniques in extracting and isolating microbes from such extreme conditions remains crucial. These indigenous microbes, having adapted to survive in extreme conditions, could harbor metabolic potential valuable for biotechnological advancement (Ramganesh et al. 2014; Selvarajan et al. 2017b; Sibanda et al. 2017). In this study, an integrated approach of both culture-independent and culture-dependent methods was adopted to explore the capabilities of bacterial communities residing in soils that have been chronically exposed to oil contamination. To isolate bacteria adept at this condition and catabolize long-chain alkanes, soil samples were enriched with crude oil (COE) and paraffin wax (PWE), which are rich in these complex hydrocarbons. Tables 2 and 3 outline the biochemical characteristics of bacterial isolates obtained from enrichment media containing crude oil and paraffin wax. In degradation assays using the DCPIP indicator, approximately 20 bacterial strains were tested for their ability to break down long-chain alkanes. Marinomonas emerged as the most proficient in degrading both paraffin wax and crude oil. Previous studies have shown that Marinomonas can degrade hydrocarbons to some extent (Melcher et al. 2002; Gontikaki et al. 2018; Gidudu and Chirwa 2022), and a recent study by Zannotti et al. (2023) found this bacterium associated with the Antarctic marine ciliate Euplotes focardii, has potential for degrading hydrocarbons commonly found in diesel oil. Moreover, Marinomonas can thrive in both marine and terrestrial environments and possesses a genome with sequences encoding various enzymes for benzene and naphthalene degradation (John et al. 2020). Following Marinomonas, the Pseudomonas demonstrated a strong ability for degrading Hexadecane. This observation aligns with findings from other studies (Zhong et al. 2016; He et al. 2019) that have similarly reported Pseudomonas as an efficient degrader of this hydrocarbon. However, it is worth noting that in those previous studies, the degradation rate of n-hexadecane reached 78% after 7 days, whereas our strains exhibited a slightly lower rate of 65% degradation. To provide further insights, we identified the potential genes responsible for long-chain alkane degradation, namely almA, in both Marinomonas and Pseudomonas (Fig. 5A). These genes have the ability to regulate the expression of the almA oxygenase, which is known for its involvement in the oxidation of super long-chain alkanes (> C30) (Wentzel et al. 2007). Additionally, the presence of gene ladA was confirmed in Alteromonas, which displayed a high degradation capacity, closely with Marinomonas and Pseudomonas (Fig. 5B). Several hydrocarbon-degrading bacteria belonging to the genus Pseudomonas or other genera like Rhodococcus, Gordina, and Acinetobacter carries alkB gene (Kubota et al. 2008). However, to best of our knowledge, this is the first documented study of the almA gene being reported in cultured Alteromonas bacteria, as it had previously only been observed in uncultured bacteria from the North Atlantic Ocean (Vázquez Rosas Landa et al. 2023).
Novel technologies continue to expand our understanding of microbial diversity and community structure; this study integrated 16 s amplicon analysis using next-generation sequencing technology along with culture-dependent analysis to comprehensively understand the microbial richness and composition of long-term oil contaminated soil microbial communities. Alpha diversity analysis revealed that surface samples exhibited greater species richness and diversity compared to subsurface samples, similar pattern was observed in bacterial communities of soil contaminated with five different oil refineries in China (Jiao et al. 2016). Bray–Curtis dissimilarity indices indicated distinct differences in bacterial community structures between surface and subsurface soils, with surface samples exhibiting specific variations that led to their separate clustering (Fig. 6). These variations could be traced back to dominant taxa, which accounted for a significant share of the dissimilarity in the overall community, as noted by Shade et al. (2014). Furthermore, despite its significant beta diversity, the minor sub-community had a minimal impact on the differences observed within the overall community structure. This observation aligns with findings by Jiao et al. (2017), who reported similar diversity patterns in soils contaminated by oil. In the taxonomic analysis, Proteobacteria emerged as the dominant phylum in both soil sample types (Fig. 7). These findings align with similar results reported by Khan et al. (2018), where Proteobacteria were identified as the most abundant phyla among the 45 phyla observed in petroleum-contaminated soils collected from South Australia. This phylum was well-represented by the β-, α-, and γ-Proteobacterial classes, a pattern commonly observed in hydrocarbon-amended soils. The prevalence of Proteobacteria in these environments is unsurprising, as it is recognized for its ability to metabolize both aliphatic and aromatic compounds. This increased abundance of Proteobacteria in response to hydrocarbon pollution has been well documented in various geographical locations (Baruah et al. 2017; Chikere et al. 2019; Cabral et al. 2022; Gao et al. 2022). Acidobacteria was the second most dominant phylum followed by Actinobacteria in surface soil, while Firmicutes and Actinobacteria dominated in subsurface soil. These results corroborate a previous study by Sutton et al. (2013), which identified Proteobacteria, Firmicutes, Actinobacteria, Acidobacteria, and Chloroflexi as major bacterial groups in long-term diesel-contaminated soil. While Firmicutes are well-known for their significant role in breaking down plant polymers, they are not commonly associated with broad metabolic activity involving aromatic and/or aliphatic hydrocarbons (Brzeszcz and Kaszycki 2018).
Analysis of bacterial profiles at genus level revealed that distinct variations among bacterial populations between surface and subsurface oil polluted soil samples. Surface samples exhibited Burkholderia, Pedomicrobium, Methyloversatilis, Rothia, Granulicatella, Deflivibacter, Rhodobacter, Rhodococcus, Agrobacterium, Novosphingbium, Bacillus, and Mycobacterium. In contrast, subsurface samples were characterized with Sphingomonas, Acinetobacter, Arcobacter, Hyphomicrobium, Halomonas, Mycoplana, Ralstonia, Pseudomonas, and Lactococcus (Fig. 8). The genus Burkholderia has been recognized for its ability to produce a stable biosurfactant with robust emulsification properties for crude oil, showcasing its potential for use in commercial bioremediation applications for oil-contaminated soils (Almatawah 2017). The obligate methylotrophs within the Methyloversatilis group have been noted for their role in the biodegradation of Benazolin-Ethyl in activated sludge and methane production (Cai et al. 2011; Fenibo et al. 2023b). Pseudomonas species, as well documented in various studies, can degrade hydrocarbons such as naphthalene, phenanthrene, anthracene, and diesel (Rentz et al. 2004; Shukor et al. 2009; Chikere et al. 2019). These species are also known for producing biosurfactants that enhance the desorption and breakdown of petroleum-derived hydrocarbons (Taccari et al. 2012). Rhodococcus, another genus draw attention for its capabilities, efficiently breaks down long-chain n-alkanes and crude oil, making it an advantageous representative for cleaning up areas with significant oil pollution (Whyte et al. 2002). Other genera such as Sphingomonas, Acinetobacter, Arcobacter, Hyphomicrobium, and Halomonas have also been identified as degraders of hydrocarbons, especially long-chain alkanes (Miyashita 2015; Kumar et al. 2020; Ehiosun et al. 2022). Comparative analysis of potential functional genes and enzymes from both polluted and unpolluted sites highlighted an array of metabolic and catabolic genes implicated in hydrocarbon degradation (Fig. 9). Certain functions (data not shown), such as amino acid transport and metabolism, energy production and conversion, and replication, recombination, and repair, could play a role in adapting to the stress from petroleum hydrocarbons. Previous studies have suggested that having sufficient energy is crucial not only for supporting normal microbial growth and reproduction processes but also for the transportation and metabolism of petroleum hydrocarbons (Xu et al. 2017). In addition, genes such as nahAa, adh2, eutE, and cpnA were solely found in the oil-contaminated soil samples. The presence of these genes aligns with recent findings of microbial communities in Arctic Sea Ice, which possess genes capable of producing enzymes that break down a variety of hydrocarbons and aromatic compounds (Peeb et al. 2022). Furthermore, genes that contribute to various metabolic processes like catE, catA, dmpB, aldB, ssuD, dhaA, and alkB were present in greater abundance in the contaminated soils. These particular genes are known for their role in the production of enzymes that convert alkanes into alcohols. These alcohols are then further processed into fatty acids and finally broken down via the bacterial β-oxidation pathway (Rojo 2009; Fenibo et al. 2023a). In this study, all genes related to degradation were identified, suggesting that microorganisms capable of degrading petroleum hydrocarbons might display a biogeographic distribution of functional genes associated with degradation. Indeed, these genes linked to the degradation of petroleum hydrocarbons have been identified and detected across different environmental media (Wang et al. 2010; Long et al. 2017). Therefore, considerable efforts should be made to isolate indigenous microbes for a comprehensive exploration of these degradation-related genes. Nevertheless, a comparison of the outcomes from culture-independent analyses reveals that current laboratory isolation efforts have successfully identified only a limited number of predominant members, including Bacillus, Halomonas, Pseudomonas, Rhodococcus, and Lactococcus. These bacteria can thrive under standard laboratory conditions without specialized media or environments. Nonetheless, there is a continuous need to cultivate other significant members in the lab, those that have shown promise in degrading long-chain hydrocarbons.
Conclusion
In conclusion, this investigation into the microbiota of long-term oil-contaminated soils highlights a diverse array of microorganisms capable of degrading various hydrocarbons. The combined approach, utilizing both culture-dependent and culture-independent methods, has provided a comprehensive understanding of the microbial community structure and potential metabolic functions in these environments. The results indicate a distinct microbial population variation between surface and subsurface soils, with surface soils exhibiting higher microbial richness and diversity. Notably, total organic carbon (TOC) levels in surface-polluted soil (SPS) and subsurface soil (SPSS) range from 5.64 to 5.06%, compared to 1.97% in unpolluted soil (UPS). Similarly, total petroleum hydrocarbons (TPH) levels in SPS and SPSS range from 36,775 ppm to 14,087 ppm, contrasting with UPS at 47,936 ppm. Carbon chain analysis reveals a prevalence of longer-chain alkanes (C20-28) in surface soil. Culture-dependent methods yield 47 bacterial isolates, with 12 strains displaying significant degradation abilities. Culture-independent analysis demonstrates higher species richness in surface samples, dominated by Proteobacteria. Specific bacterial genera (e.g., Pseudomonas, Marinomonas, Alteromonas) exhibit 50 to 80% degradation efficiency. Furthermore, the study has also shed light on specific bacterial genera and catabolic genes that contribute to hydrocarbon and long-chain alkanes degradation pathways, offering new insights into the metabolic capabilities present in contaminated sites. However, the results also stress the need for ongoing efforts to isolate and study a broader spectrum of microorganisms under laboratory conditions, to further unlock the bioremediation potential of indigenous soil bacteria that may aid in the development of more effective bioremediation applications for restoring oil-contaminated environments.
Data availability
The raw sequence data available publicly in the NCBI-SRA under the BioProject ID PRJNA1037324.
References
Adetitun DO, Tomilayo R (2023) Ecological implications of bacterial degradation of alkanes in petroleum-contaminated environments: a review of microbial community dynamics and functional interactions. Glob J Pure Appl Sci 29:133–144. https://doi.org/10.4314/gjpas.v29i2.4
Aigberua AO, Ekubo AT, Inengite AK, Izah SC (2017) Assessment of some selected heavy metals and their pollution indices in an oil spill contaminated soil in the Niger Delta: a case of Rumuolukwu community. Biotechnol Res 3:11–19
Almatawah Q (2017) An indigenous biosurfactant producing Burkholderia cepacia with high emulsification potential towards crude oil. J Env Anal Toxicol 7:525–2161
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 10 Jan 2020
Austin RN, Callaghan AV (2013) On the origins of deep hydrocarbons. Front Microbiol 4:1–2. https://doi.org/10.2138/rmg.2013.75.14
Azubuike CC, Chikere CB, Okpokwasili GC (2016) Bioremediation techniques–classification based on site of application: principles, advantages, limitations and prospects. World J Microbiol Biotechnol 32:1–18. https://doi.org/10.1007/s11274-016-2137-x
Baruah R, Mishra SK, Kalita DJ et al (2017) Assessment of bacterial diversity associated with crude oil-contaminated soil samples from Assam. Int J Environ Sci Technol 14:2155–2172
Bolyen E, Rideout JR, Dillon MR et al (2018) QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr 9–10. https://doi.org/10.7287/peerj.preprints.27295v2
Brzeszcz J, Kaszycki P (2018) Aerobic bacteria degrading both n-alkanes and aromatic hydrocarbons: an undervalued strategy for metabolic diversity and flexibility. Biodegradation 29:359–407
Cabral L, Giovanella P, Pellizzer EP et al (2022) Microbial communities in petroleum-contaminated sites: structure and metabolisms. Chemosphere 286:131752
Cai T, Qian L, Cai S, Chen L (2011) Biodegradation of benazolin-ethyl by strain Methyloversatilis sp. cd-1 isolated from activated sludge. Curr Microbiol 62:570–577
Callahan BJ, McMurdie PJ, Rosen MJ et al (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Chen Y, Wang Y, Yu K et al (2022) Occurrence characteristics and source appointment of polycyclic aromatic hydrocarbons and n-alkanes over the past 100 years in southwest China. Sci Total Environ 808:151905
Cheng L, Shi S, Li Q et al (2014) Progressive degradation of crude oil n-alkanes coupled to methane production under mesophilic and thermophilic conditions. PLoS One 9:e113253
Chicca I, Becarelli S, Di Gregorio S (2022) Microbial involvement in the bioremediation of total petroleum hydrocarbon polluted soils: challenges and perspectives. Environ - MDPI 9. https://doi.org/10.3390/environments9040052
Chikere CB, Mordi IJ, Chikere BO et al (2019) Comparative metagenomics and functional profiling of crude oil-polluted soils in Bodo West Community, Ogoni, with other sites of varying pollution history. Ann Microbiol 69. https://doi.org/10.1007/s13213-019-1438-3
Das N, Chandran P (2011) Microbial degradation of petroleum hydrocarbon contaminants: an overview. Biotechnol Res Int 2011:1–13. https://doi.org/10.4061/2011/941810
Douglas GM, Maffei VJ, Zaneveld JR et al (2020) PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38:685–688
Ehiosun KI, Godin S, Urios L et al (2022) Degradation of long-chain alkanes through biofilm formation by bacteria isolated from oil-polluted soil. Int Biodeterior Biodegradation 175:105508
El Mahdi AM, Aziz HA, Abu Amr SS et al (2016) Isolation and characterization of Pseudomonas sp. NAF1 and its application in biodegradation of crude oil. Environ Earth Sci 75:1–11
Fenibo EO, Selvarajan R, Abia ALK, Matambo T (2023a) Medium-chain alkane biodegradation and its link to some unifying attributes of alkB genes diversity. Sci Total Environ 877:162951
Fenibo EO, Selvarajan R, Wang H et al (2023b) Untapped talents: insight into the ecological significance of methanotrophs and its prospects. Sci Total Environ 903:166145
Gao Y, Yuan L, Du J et al (2022) Bacterial community profile of the crude oil-contaminated saline soil in the Yellow River Delta Natural Reserve, China. Chemosphere 289:133207
Gidudu B, Chirwa EM (2022) Diversity of marine biosurfactant-producing bacteria and their role in the degradation of heterogenous petrochemical hydrocarbons. In: Marine Surfactants. CRC Press, pp 163–183
Gontikaki E, Potts LD, Anderson JA, Witte U (2018) Hydrocarbon-degrading bacteria in deep-water subarctic sediments (Faroe-Shetland channel). J Appl Microbiol 125:1040–1053
He S, Ni Y, Lu L et al (2019) Enhanced biodegradation of n-hexane by Pseudomonas sp. strain NEE2. Sci Rep 9:16615
Head IM, Jones DM, Röling WF (2006) Marine microorganisms make a meal of oil. Nat Rev Microbiol 4:173–182. https://doi.org/10.1038/nrmicro1348
Hedgpeth BM, McFarlin KM, Prince RC (2021) Crude oils and their fate in the environment. In: Petrodiesel Fuels, pp 891–910
Ite AE, Ibok UJ, Ite MU, Petters SW (2013) Petroleum exploration and production: past and present environmental issues in the Nigeria’s Niger Delta. Am J Environ Prot 1:78–90
Jabbarov Z, Abdrakhmanov T, Pulatov A, Kováčik P, Pirmatov K (2019) Change in the parameters of soils contaminated by oil and oil products. Agriculture (Pol’nohospodárstvo) 65(3):88–98
Jiao S, Liu Z, Lin Y et al (2016) Bacterial communities in oil contaminated soils: biogeography and co-occurrence patterns. Soil Biol Biochem 98:64–73
Jiao S, Chen W, Wei G (2017) Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol Ecol 26:5305–5317
John MS, Nagoth JA, Ramasamy KP et al (2020) Horizontal gene transfer and silver nanoparticles production in a new Marinomonas strain isolated from the Antarctic psychrophilic ciliate Euplotes focardii. Sci Rep 10:10218
Khan MAI, Biswas B, Smith E et al (2018) Microbial diversity changes with rhizosphere and hydrocarbons in contrasting soils. Ecotoxicol Environ Saf 156:434–442
Kponee KZ, Chiger A, Kakulu II et al (2015) Petroleum contaminated water and health symptoms: a cross-sectional pilot study in a rural Nigerian community. Environ Heal 14:1–8
Kubota K, Koma D, Matsumiya Y et al (2008) Phylogenetic analysis of long-chain hydrocarbon-degrading bacteria and evaluation of their hydrocarbon-degradation by the 2,6-DCPIP assay. Biodegradation 19:749–757. https://doi.org/10.1007/s10532-008-9179-1
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547
Kumar S, Zhou J, Li M et al (2020) Insights into the metabolism pathway and functional genes of long-chain aliphatic alkane degradation in haloarchaea. Extremophiles 24:475–483
Kuppusamy S, Maddela NR, Megharaj M, Venkateswarlu K, Kuppusamy S, Maddela NR, ... Venkateswarlu K (2020) Fate of total petroleum hydrocarbons in the environment. In: Total Petroleum Hydrocarbons: Environmental Fate, Toxicity, and Remediation, pp 57–77
Li J, Xu Y, Song Q et al (2021) Polycyclic aromatic hydrocarbon and n-alkane pollution characteristics and structural and functional perturbations to the microbial community: a case-study of historically petroleum-contaminated soil. Environ Sci Pollut Res 28:10589–10602. https://doi.org/10.1007/s11356-020-11301-1
Long H, Wang Y, Chang S et al (2017) Diversity of crude oil-degrading bacteria and alkane hydroxylase (alkB) genes from the Qinghai-Tibet Plateau. Environ Monit Assess 189:1–14
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12
Melcher RJ, Apitz SE, Hemmingsen BB (2002) Impact of irradiation and polycyclic aromatic hydrocarbon spiking on microbial populations in marine sediment for future aging and biodegradability studies. Appl Environ Microbiol 68:2858–2868
Miyashita NT (2015) Contrasting soil bacterial community structure between the phyla Acidobacteria and Proteobacteria in tropical Southeast Asian and temperate Japanese forests. Genes Genet Syst 90:61–77. https://doi.org/10.1266/ggs.90.61
Mrozik A, Piotrowska-Seget Z (2010) Bioaugmentation as a strategy for cleaning up of soils contaminated with aromatic compounds. Microbiol Res 165:363–375
Nwilo PC, Badejo OT (2006) Impacts and management of oil spill pollution along the Nigerian coastal areas. Adm Mar Spaces Int Issues 119:1–15
Ogri OR (2001) A review of the Nigerian petroleum industry and the associated environmental problems. Environmentalist 21:11–21
Okon IE, Ogba CO (2018) The impacts of crude oil exploitation on soil in some parts of Ogoni region, Rivers State, Southern Nigeria. Open Access Libr J 5:1–20
Peeb A, Dang NP, Truu M et al (2022) Assessment of hydrocarbon degradation potential in microbial communities in Arctic sea ice. Microorganisms 10:328
Pfennig N, Trüper HG (1992) The family chromatiaceae. In: The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications. Springer New York, pp 3200–3221
Pivková I, Kukla J, Hniličková H et al (2022) Content of cadmium and nickel in soils and assimilatory organs of park woody species exposed to polluted air. Life 12:2033
Quast C, Pruesse E, Yilmaz P et al (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Ramganesh S, Maredza AT, Tekere M (2014) Microbial exploration in extreme conditions: metagenomic analysis and future perspectives. In: Benedetti C (ed) Metagenomics - Methods, Applications and Perspectives. Nova Science Publishers Inc, New York, pp 157–181
Ramganesh S, Timothy S, Memory T (2018) Thermophilic bacterial communities inhabiting the microbial mats of ‘indifferent’ and chalybeate (iron-rich) thermal springs: diversity and biotechnological analysis. Microbiol Open 7:1–12
Rentz JA, Alvarez PJJ, Schnoor JL (2004) Repression of Pseudomonas putida phenanthrene-degrading activity by plant root extracts and exudates. Environ Microbiol 6:574–583
Rojo F (2009) Degradation of alkanes by bacteria. Environ Microbiol 11:2477–2490
Schreier CG, Walker WJ, Burns J, Wilkenfeld R (1999) Total organic carbon as a screening method for petroleum hydrocarbons. Chemosphere 39:503–510
Selvarajan R, Sibanda T, Tekere M et al (2017a) Diversity analysis and bioresource characterization of halophilic bacteria isolated from a South African saltpan. Molecules 22:1–20. https://doi.org/10.3390/molecules22040657
Selvarajan R, Sibanda T, Tekere M et al (2017b) Thermophilic bacterial communities inhabiting the microbial mats of ‘indifferent’ and chalybeate (iron-rich) thermal springs: diversity and biotechnological analysis. Microbiol Open. https://doi.org/10.1002/mbo3.560. (Accepted in Press)
Selvarajan R, Venkatachalam S, Meddows-Taylor S et al (2018b) Defunct gold mine tailings are natural reservoir for unique bacterial communities revealed by high-throughput sequencing analysis. Sci Total Environ 650:2199–2209. https://doi.org/10.1016/j.scitotenv.2018.09.380
Selvarajan R, Sibanda T, Venkatachalam S et al (2018a) Industrial wastewaters harbor a unique diversity of bacterial communities revealed by high-throughput amplicon analysis. Ann Microbiol 1–14. https://doi.org/10.1007/s13213-018-1349-8
Shade A, Jones SE, Caporaso JG et al (2014) Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. Mbio 5:10–1128
Shaoping K, Zhiwei D, Bingchen W et al (2021) Changes of sensitive microbial community in oil polluted soil in the coastal area in Shandong, China for ecorestoration. Ecotoxicol Environ Saf 207:111551
Shukor MY, Hassan NAA, Jusoh AZ et al (2009) Isolation and characterization of a Pseudomonas diesel-degrading strain from Antarctica. J Environ Biol 30:1–6
Sibanda T, Selvarajan R, Tekere M (2017) Synthetic extreme environments: overlooked sources of potential biotechnologically relevant microorganisms. Microb Biotechnol. https://doi.org/10.1002/mrd.22357
Sibanda T, Selvarajan R, Tekere M (2018) Targeted 16S rRNA amplicon analysis reveals the diversity of bacterial communities in carwash effluents. Int Microbiol. https://doi.org/10.1007/s10123-018-00038-0
Si-Zhong Y, Hui-Jun JIN, Zhi WEI et al (2009) Bioremediation of oil spills in cold environments: a review. Pedosphere 19:371–381
Song W-F, Wang J-W, Yan Y-C et al (2018) Shifts of the indigenous microbial communities from reservoir production water in crude oil-and asphaltene-degrading microcosms. Int Biodeterior Biodegradation 132:18–29
Sutton NB, Maphosa F, Morillo JA et al (2013) Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microbiol 79:619–630. https://doi.org/10.1128/AEM.02747-12
Taccari M, Milanovic V, Comitini F et al (2012) Effects of biostimulation and bioaugmentation on diesel removal and bacterial community. Int Biodeterior Biodegradation 66:39–46
Tourova TP, Sokolova DS, Semenova EM et al (2016) Detection of n-alkane biodegradation genes alkB and ladA in thermophilic hydrocarbon-oxidizing bacteria of the genera Aeribacillus and Geobacillus. Microbiology 85:693–707
Ubani O, Atagana HI, Selvarajan R, Ogola HJ (2022) Unravelling the genetic and functional diversity of dominant bacterial communities involved in manure co-composting bioremediation of complex crude oil waste sludge. Heliyon 8:e08945. https://doi.org/10.1016/j.heliyon.2022.e08945
Van Beilen JB, Li Z, Duetz WA et al (2003) Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci Technol 58:427–440
Vázquez Rosas Landa M, De Anda V, Rohwer RR, Angelova A, Waldram G, Gutierrez T, Baker BJ (2023) Exploring novel alkane-degradation pathways in uncultured bacteria from the North Atlantic Ocean. Msystems 8(5):e00619–e00623
Wang W, Shao Z (2012) Diversity of flavin-binding monooxygenase genes (almA) in marine bacteria capable of degradation long-chain alkanes. FEMS Microbiol Ecol 80:523–533
Wang L, Wang W, Lai Q, Shao Z (2010) Gene diversity of CYP153A and AlkB alkane hydroxylases in oil-degrading bacteria isolated from the Atlantic Ocean. Environ Microbiol 12:1230–1242
Wentzel A, Ellingsen TE, Kotlar H-K et al (2007) Bacterial metabolism of long-chain n-alkanes. Appl Microbiol Biotechnol 76:1209–1221
Whyte LG, Smits THM, Labbe D et al (2002) Gene cloning and characterization of multiple alkane hydroxylase systems in Rhodococcus strains Q15 and NRRL B-16531. Appl Environ Microbiol 68:5933–5942
Wu X, Lin Y, Wang Y et al (2022) Enhanced removal of hydrophobic short-chain n-alkanes from gas streams in biotrickling filters in presence of surfactant. Environ Sci Technol 56:10349–10360
Wu X, Lin Y, Wang Y, Yang C (2023b) Interactive effects of dual short-chain n-alkanes on removal performances and microbial responses of biotrickling filters. Chem Eng J 461:141747
Wu X, Lin Y, Wang Y, Wu S, Yang C (2023a) Volatile organic compound removal via biofiltration: influences, challenges, and strategies. Chem Eng J 144420. https://doi.org/10.1016/j.cej.2023.144420
Xu J, Zhang L, Hou J et al (2017) iTRAQ-based quantitative proteomic analysis of the global response to 17β-estradiol in estrogen-degradation strain Pseudomonas putida SJTE-1. Sci Rep 7:41682
Zannotti M, Ramasamy KP, Loggi V et al (2023) Hydrocarbon degradation strategy and pyoverdine production using the salt tolerant Antarctic bacterium Marinomonas sp. ef1. RSC Adv 13:19276–19285
Zhong H, Wang Z, Liu Z et al (2016) Degradation of hexadecane by Pseudomonas aeruginosa with the mediation of surfactants: relation between hexadecane solubilization and bioavailability. Int Biodeterior Biodegradation 115:141–145
Acknowledgements
Authors would like to thank UNISA (Florida campus) for providing Illumina sequencing platform for sequencing our samples. Furthermore, the authors would also like to thank the Centre for High Performance Computing (CHPC) facility, South Africa, for providing computational support for sequence data analysis.
Funding
Open access funding provided by University of South Africa. Full sponsorship/scholarship by world Bank for PhD program in Africa Centre of Excellence-Centre for Oilfield Chemicals Research, (ACE-CEFOR), University of Port Harcourt, Port Harcourt, Rivers State. Nigeria (2017–2020).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.B.C., G.C.O., and R.S.; methodology: C.B.C.; software: R.S.; validation: R.S., C.B.C., and K.M.; formal analysis: A.U.O.; investigation: A.U.O.; resources: K.M.; data curation: R.S.; writing—original draft preparation: A.U.O. and R.S.; writing—review and editing: C.B.C. and K.M.; visualization: R.S.; supervision: C.B.C., G.C.O., and R.S. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
All participants provided written informed consent.
Consent for publication
All enrolled subjects signed informed consent regarding publishing their data.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Robert Duran
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Amara Ukamaka Okoye is deceased before the submission of this manuscript, which forms part of her doctoral thesis.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okoye, A.U., Selvarajan, R., Chikere, C.B. et al. Characterization and identification of long-chain hydrocarbon-degrading bacterial communities in long-term chronically polluted soil in Ogoniland: an integrated approach using culture-dependent and independent methods. Environ Sci Pollut Res 31, 30867–30885 (2024). https://doi.org/10.1007/s11356-024-33326-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-024-33326-6