
Abstract
Simple reaction procedure, boiling of an equimolar mixture of [Re(Cl)(CO)3(dppv)] (dppv—cis-1,2-bis(diphenylphosphino)-ethene), 2-(2-pyridyl)benzimidazole (pbi), and TlClO4 in o-dichlorobenzene solution, leads to two, chromatographically separable, isomers of [Re(CO)2(dppv)(pbi)] complex. The obtained species, the yellow-emissive [OC-6–54] and the orange-emissive [OC-6–53] isomers, have been identified using FT-IR, 1H NMR, and 31P NMR spectroscopy and their structures confirmed by means of X-ray investigations. According to obtained crystallography results, the investigated isomers differ mainly in the arrangement of the 2-(2-pyridyl)benzimidazole anion in their structures. Diverse arrangement of the dppv and pbi ligand around the central cis-[Re(CO)2]+ core results in well-pronounced changes in luminescence properties of the studied luminophores. According to results from performed DFT and TDA-DFT computations, the [OC-6–54] isomer emission occurs from the intra-ligand excitation localized mainly within the pbi ligand. In contrary to that, the [OC-6–53] isomer emission indicates some metal-to-ligand charge-transfer character associated with the charge transfer from the Re(CO)2(dppv)+ fragment to the pbi ligand. Moreover, both isomers exhibit different character of their emission decays, mono-exponential and bi-exponential for the [OC-6–54] and [OC-6–53] ones, respectively. In the view of obtained DFT/TDA-DFT results, this has been explained by the presence of an additional excited dark state, operative only in the case of the [OC-6–53] isomer and associated with the charge transfer from the Re(CO)2(pbi) fragment to the dppv ligand.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhenium(I) carbonyl complexes represent a fascinating class of molecules with interesting photochemical and photophysical properties. The first reports on the photophysical properties of strongly emissive rhenium(I) tricarbonyl diimine complexes were published almost 50 years ago [1, 2]. Since then, this class of compounds has been extensively studied and many luminescent fac-[Re(CO)3(N^N)(L)]0/+ complexes that exhibit interesting and diverse photochemical and photophysical properties have been described [2-12]. The broad structural variations of the N^N, as well as the axial L ligands, affect the nature and energy order of the low-lying excited states and thus the spectroscopy, photophysics, photochemistry, and electrochemistry of the Re(I)-based luminophores. This opens the way to numerous applications of [Re(L)(CO)3(N^N)]0/+ complexes such as photocatalysts for CO2 reduction [13-17] or for the production of H2 [18] organic light emitting devices [19-22] as well as bio-molecular agents [23-27].
Compared to the [Re(L)(CO)3(N^N)]0/+ complexes, the photochemical and photophysical properties of the complexes containing the Re(I) dicarbonyl cis-Re(CO)2+ core are much less extensively characterized. Those described in the literature contain mainly in their structure symmetric polypyridine ligands and monodentate or bidentate phosphines [28-35]. However, photochemical and photophysical behavior of the complexes containing fac-[Re(CO)3]+ as well as cis-[Re(CO)2]+ core is governed by the relative energy position and the interaction of the closely lying excited states. The lowest excited state can possess distinctly different character. In some cases, the electronic excitation is localized within individual ligand/ligands (ligand-centered, LC) or on the metal (metal-centered, MC). In addition to these states, states involving charge separation, either charge transfer from the metal to the ligand (MLCT) or ligand-to-metal charge-transfer (LMCT), as well as ligand-to-ligand charge-transfer (LLCT) states may play a crucial role in determining the photophysical properties of these organometallic rhenium(I) luminophores. Due to the large splitting of the rhenium(I) d orbitals caused by the presence of three or two strong ligand-field CO ligands in fac-[Re(CO)3]+ and cis-[Re(CO)2]+ core, respectively, the excited MC states are usually located energetically well above the excited LC or MLCT states [8]. In turn, because of the distinctly smaller energy gap between the excited *LC or *MLCT states, there is an efficient electron interaction between them. In consequence, these electronic states are mixed with each other by configuration interactions and strong spin–orbit coupling effect. The former is responsible for mixing of states with the same spin multiplicities and the latter for relatively fast radiative and non-radiative deactivation of the lowest excited triplet states [11, 36]. The mixing of these excited states causes monotonic changes in the nature of emitters as it was reported for several series of complexes. These include the neutral [11] [Re(CO)3(N^N)(Cl)] or cationic [35] [Re(CO)3(N^N)(CH3CN)]+ and [Re(CO)3(N^N){P(C6H5)3}]+ with fac-[Re(CO)3]+ core as well as [Re(CO)2(dppv)(N^N)]+ and [Re(CO)2(N^N){P(C6H5)3}2]+ with cis-[Re(CO)2]+ core [34, 35] The results obtained from the DFT and TD-DFT computation and the emission band-shape analyses revealed some similarities and differences in the spectroscopic and photophysical properties of these five series of complexes. The emission characteristics for all complexes, regardless of the fac-[Re(CO)3]+ or cis-[Re(CO)2]+ cores, are strongly affected by the nature of coordinated α-diimine N^N ligands. Furthermore, all five series of complexes have a similar nature of the lowest excited state. In both types of complexes with fac-[Re(CO)3]+ and cis-[Re(CO)2]+ cores, their phosphorescence in solutions at room temperature occurs from the lowest triplet excited state with a distinct metal to ligand charge-transfer (MLCT) character. However, when the measurement temperature is lowered from 298 to 77 K, some intrinsic differences between them are clearly seen. For the cis-[Re(CO)2]+ complexes, the emissions in solid matrices exhibit still MLCT character, whereas for those with fac-[Re(CO)3]+ core, the emission is structured and the character of emission changes from MLCT to LC.
Taking into account the above considerations, which showed the decisive influence of the N^N ligand on the emission properties of the rhenium(I) complexes, we have decided to replace this neutral ligand with the negatively charged pbi ligand (i.e., 2-(2-pyridyl)-benzimidazole anion). As pbi contains only nitrogen donor atoms, one can consider it as “bipyridine analogue,” but having worse π-accepting properties compared to the neutral α-diimines. On the other hand, due to the asymmetry of the pbi chelating ligand, any heteroleptic complexes bearing this ligand may exist in different geometric isomers with presumably different spectroscopic and photophysical properties. In fact, this has been observed in the case of the previously studied by us [Os(Cl)(CO)(P^P)(pbi)] complexes [37, 38] As the P^P and pbi ligands are both the electron donating/withdrawing species, two directions of the intramolecular charge shift, from Os(Cl)(CO)(P^P)+ to pbi or from Os(Cl)(CO)(pbi) to P^P, are essentially possible in the [Os(Cl)(CO)(P^P)(pbi)] molecules. This particular option is realized in the case of the [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] isomer where the presence of two states close in their energies results in the observed delayed emission. Since one can expect that such unusual behavior may be commonly characteristic for other metal complexes bearing dppv and pbi ligands, we have decided to synthesize and investigate the rhenium(I) isomeric [Re(CO)2(dppv)(pbi)] complexes in order to explore this issue in more detail.
Materials
Commercially available analytical grade ligands 2-(2-pyridyl)-benzimidazole and cis-1,2-bis(diphenylphosphino)-ethene as well as other required chemicals (Trimen, Acros Organics, Sigma-Aldrich, or Alfa Aesar) were used, without any additional purification, in the performed syntheses and purification of the investigated [Re(CO)2(dppv)(pbi)] isomers. Solvent used in UV–vis absorption and emission studies, toluene, tetrahydrofuran, acetonitrile, methanol, and ethanol, specially designed for spectroscopic studies of the highest spectral purity Uvasol®, were purchased from Merck.
Synthesis
The investigated isomers of [Re(CO)2(dppv)(pbi)] complex were synthesized in a manner very similar to the procedures reported in the literature [28, 33]. However, we have used a simplified variant of the synthesis (cf. Scheme 1) that worked well when we had been obtaining a series of the [Re(CO)2(dppv)(N^N)]+ complexes (N^N – α-diimines ligands) [34]. In the reaction procedure, 0.18 g (0.5 mmol) of Re(CO)5Cl and 0.20 g (0.5 mmol) of dppv were refluxed under argon atmosphere in 10 ml of o-dichlorobenzene (ODB) for ca. 2 h. After completing conversion of Re(CO)5Cl into intermediate [Re(CO)3(dppv)(Cl)] in the first step of the synthetic procedure, the dehalogenation agent in the form of TlPF6 salt (in the 1.15 molar excess) was added to the reaction mixture and precipitation was observed. Then, 0.19 g (0.55 mmol) of pbiH was added in 8 ml of ODB. The solution was further refluxed for next ca. 5 h in dark resulting in a yellow-orange color. After this time, the precipitated TlCl was filtered off from the hot reaction mixture. The solution was then cooled to room temperature and the final products as a mixture of isomers of the [Re(CO)2(dppv)(pbi)] complex were precipitated with an excess of diethyl ether/hexane mixture added to the filtrate.
The isolated precipitate was dried and then pre-purified by means of flash column chromatography on silica gel using dichloromethane followed by CH2Cl2/acetone mixture as further eluents for removal of unreacted substrates and separation of luminescent products, i.e., the mixture of isomers obtained, respectively. The collected luminescent fraction was concentrated to dryness on a rotary evaporator. In order to obtain spectroscopically pure isomers, separation of the reaction products has been done by means of preparative thin-layer chromatography carried out on basic TLC aluminum oxide plates with ethyl acetate as an eluent. The applied separation procedure enabled the final isolation and purification of two synthesized luminescent isomers of [Re(CO)2(dppv)(pbi)] complex, exhibiting yellow and orange emission during illumination of their solid samples. These isomers were further analyzed by means of FT-IR, 1H NMR, and 31P NMR spectroscopy to confirm their chemical nature and purity. The final yield of the synthesis in relation to the yellow-emissive [OC-6–54]-isomer was 13%, while in relation to the orange-emissive [OC-6–53]-isomer was five times higher and amounted to 65%. The synthetic route of the investigated [Re(CO)2(dppv)(pbi)] isomers is shown in Scheme 1.

Scheme 1 The synthetic procedure applied. Both reaction steps performed in refluxed o-dichlorobenzene for ca. 2 and 5 h, respectively. Two obtained yellow-emissive and orange-emissive [Re(CO)2(dppv)(pbi)] complexes are designated throughout the paper as [OC-6-54] and [OC-6-53] isomers, respectively. Their published previously green-emissive and yellow-emissive [Os(Cl)(CO)(dppv)(pbi)] analogues [37] are designated as [OC-6-24] and [OC-6-23], respectively
[OC-6–54]-[Re(CO)2(dppv)(pbi)]:
C40H30N3O2P2Re; Elemental Anal. Calc.: C, 57.69; H, 3.63; N, 5.05. Found: C, 57.04; H, 3.79 N, 4.95%.1H-NMR (400 MHz, CD2Cl2, ppm), δ: 9.34 (d, J = 5.6 Hz, 1H), 8.28 (ddd, J = 46.9, 9.6, 8.1 Hz, 1H), 8.15 (d, J = 7.9 Hz, 1H), 8.05–8.12 (m, 2H), 7.66–7.89 (m, 2H), 7.48–7.60 (m, 3H), 7.35–7.47 (m, 5H), 7.20–7.35 (m, 5H), 7.07–7.17 (m, 2H), 6.88 (td, J = 7.5, 1.3 Hz, 1H), 6.81 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.50 (td, J = 7.9, 2.2 Hz, 2H), 6.39 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 6.17 (d, J = 8.3 Hz, 1H), 5.96 (ddd, J = 10.4, 8.3, 1.2 Hz, 2H), 30 protons in total (8 from the pbi ligand and 22 from dppv); 31P-NMR (162 MHz, CD2Cl2, ppm), δ: 50.90 (d, J = 18.8 Hz), 37.87 (d, J = 18.8 Hz); FT-IR (solid samples, cm−1): νCO: 1925, 1840; UV–Vis (acetonitrile), λmax (εM) [nm (M−1 cm−1)]: 344 (1.0 × 104), 309 (1. × 104), 217sh (~ 5.1 × 104).
[OC-6–53]-[Re(CO)2(dppv)(pbi)]:
C40H30N3O2P2Re; Elemental Anal. Calc.: C, 57.69; H, 3.63; N, 5.05. Found: C, 57.11; H, 3.88; N, 4.74%.1H-NMR (400 MHz, CD2Cl2, ppm), δ: 8.41 (ddd, J = 45.9, 9.6, 7.5 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.94–8.02 (m, 2H), 7.80 (ddd, J = 45.9, 9.6, 6.4 Hz, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.69 (m, 3H), 7.53–7.61 (m, 3H), 7.31–7.48 (m, 7H), 7.22–7.30 (m, 4H), 7.18 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.02 (td, J = 7.4, 1.3 Hz, 1H), 6.80 (td, J = 7.9, 2.2 Hz, 2H), 6.38 (ddd, J = 7.3, 5.7, 1.5 Hz, 1H), 6.07–6.18 (m, 2H), 30 protons in total (8 from the pbi ligand and 22 from dppv); 31P-NMR (162 MHz, CD2Cl2, ppm), δ: 50.18 (d, J = 19.4 Hz), 37.53 (d, J = 19.9 Hz); FT-IR (solid samples, cm−1): νCO: 1931, 1859; UV–Vis (acetonitrile), λmax (εM) [nm (M−1 cm−1)]: 400sh (~ 2.9 × 103), 350 (1.6 × 104), 223 (6.6 × 104).
Crystallography
For both isomers under study, their X-ray quality crystals were grown by slow evaporation of acetonitrile solutions. X-ray diffraction measurements were carried out with a Rigaku Oxford Diffraction XCALIBUR EOS CCD diffractometer equipped with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The unit cell determination and data integration were carried out using the CrysAlis package of Oxford Diffraction [39]. The structures were solved by Intrinsic Phasing using Olex2 software [40] with the SHELXT [41] structure solution program, and refined by full-matrix least-squares on F2 with SHELXL-2018/3 [42] using an anisotropic model for non-hydrogen atoms. Hydrogen atoms were calculated at their idealized positions and were refined as riding atoms with isotropic thermal parameters based upon the corresponding bonding atom (Uiso = 1.2Ueq, Uiso = 1.5Ueq for water hydrogens).
Crystal data for [OC-6–54]-[Re(CO)2(dppv)(pbi)]
C40H30N3O2P2Re, M = 832.81 g mol−1, pale yellow prism, 0.10×0.10×0.10 mm. Monoclinic, space group P21/n, a = 9.6496(2) Å, b = 17.0408(3) Å, c = 20.6314(4)° Å, β = 99.643(2)°, V = 3344.6(1) Å3, Z = 4, Dc = 1.654 g/cm3. F000 = 1648, T = 160(2) K, 2 \({\theta }_{\mathrm{max}}\) = 48.5°, 27789 reflections collected, 7894 unique (Rint = 0.043). Final GooF = 1.08, R = 0.032, wR = 0.053, R indices based on 6547 reflections with I > 2σ(I) (refinement on F2), 433 parameters, 0 restraints. Lp and absorption corrections applied, μ = 3.770\({\mathrm{mm}}^{-1}\).
Crystal data for [OC-6–53]-[Re(CO)2(dppv)(pbi)]
C40H30N3O2P2Re × 1.25H2O, M = 855.33 g mol−1, pale yellow prism, 0.10×0.10×0.15 mm. Monoclinic, space group P21/n, a = 11.4823(2) Å, b = 40.2880(7) Å, c = 15.0648(2) Å, β = 91.737(1)°, V = 6965.8(2) Å3. Z = 8, Dc = 1.631 g/cm3. F000 = 3396, T = 160(2) K, 2\({\theta }_{\mathrm{max}}\) = 48.9°, 56543 reflections collected, 16,242 unique (Rint = 0.050). Final GooF = 1.12, R = 0.042, wR = 0.059, R indices based on 13,315 reflections with I > 2σ(I) (refinement on F2), 901 parameters, 0 restraints. Lp and absorption corrections applied, μ = 3.625mm-1.
Instrumentation
Elemental analysis (CHN) for carbon, hydrogen, and nitrogen was performed on an Elementar Vario Micro Cube analyzer. 1H-NMR and 31P-NMR spectra were recorded in CD2Cl2 solutions using VARIAN MR-400 spectrometer. The chemical shifts (δ) of 1H-NMR were referenced to residual solvent shift in the deuterated solvent. FT-IR spectra were acquired with Shimadzu IRAffinity-1 (with the powder-ATR sampling accessory) spectrometer in the 4000 to 600 cm−1 region. UV–vis absorption spectra were measured with Shimadzu UV 3100 spectrometer in the range of 200–600 nm in a series of the spectroscopically pure grade organic solvents (acetonitrile, toluene, tetrahydrofuran, and MeOH/EtOH 1:1). The corrected steady-state luminescence spectra were recorded with Gilden Photonics FluoroSense fluorimeter. The measurements at 298 K were performed in 1 × 1 cm fused silica cells fulfilled with the investigated complex solutions thoroughly deaerated by the prolonged saturation with preliminary purified and dried argon. The emission quantum yields ϕem (with an estimated error of ca. ± 10%) were determined in relation to [Ru(bpy)3]2+ in aerated acetonitrile solutions served as a reference luminophore with ϕem = 0.018 [43]. The 77 K measurements were performed in NMR-like fused silica tubes with 3 mm inner diameter by placing them in a liquid nitrogen-filled Dewar flask with a bottom transparent finger. The emission decays were recorded with FluoroSense-P fluorimeter especially designed to the time-resolved measurements in the microsecond range with the temporal resolution of 0.01 μs. In the performed lifetime measurements, the investigated samples were excited at 350 nm whereas the emission signals were recorded at the wavelengths corresponding to the maxima of the steady-state emissions. The experimental decay curves were analyzed by the single-curve method using the reference convolution based on the Marquardt algorithm [44] with the \({\chi }^{2}\) values (close to unity), the distributions of residuals, and the adjusted coefficients of determination R2 values (at least 0.995) served as the criteria in the evaluation of the fit quality. The lifetime τem values were determined with an estimated error of ca. ± 10%.
Computational details
Density functional theory (DFT) calculations within this work were carried out using the Gaussian 09 software [45] supported by GaussView 5.0. The calculations were performed using B3LYP functional [46, 47] with a “double-\(\zeta\)” basis set consisting of the Hay and Wadt effective core potentials LANL2DZ [48] and 6-31G(d,p) basis set [49, 50] employed for the rhenium and light atoms, respectively. Full optimization for each stationary structure in the ground S0 and the lowest triplet T1 electronic states in vacuum +was done by means of the DFT computations at the spin-restricted and spin‐unrestricted B3LYP level with a spin multiplicity of 1 and 3, respectively. The obtained geometries were further used for optimization performed in solutions with accounting the solvation effects by means of the polarizable continuum solvation model (PCM) [51]. Calculation of the vertical electronic transitions have been performed at the optimized S0 and T1 geometries using the time‐dependent generalization of the density functional theory [52, 53] within the framework of the Tamm‐Dancoff (TDA‐DFT) approximation [54]. Ground state geometry optimizations were performed starting from the respective crystal structures. The optimized S0 geometries show a good agreement with the X-ray data with the differences between computed and experimental bond lengths typically smaller than 0.03 Å. Only the computed Re-P lengths were found to be somewhat longer (up to 0.07 Å) than the experimental ones. Taking into account the level of the applied computation, the obtained agreement can be regarded as satisfactory. For the optimized structures of S0 geometry, the vibrational frequency calculations were also performed to ensure that the optimized geometries represent the local minima and there are only positive eigenvalues. In similar way, only positive vibrational frequencies were obtained for optimized T1 excited state geometries.
Results and discussion
Identification of the investigated isomers
The synthesized [OC-6–54] and [OC-6–53] isomers can be easily differentiated by 31P NMR, 1H NMR, and FT-IR spectroscopic techniques. In the 31P NMR spectra, recorded for the crude reaction mixture before isolation of individual isomers, two sets of doublet signals with the 1:5 ratio of the signal integrals have been observed, corresponding to two different phosphine-containing species. However, only two coupled doublet signals are observed in the 31P NMR spectra of the already separated isomers. The chemical shifts of signals and the values of JP−P coupling constants differ significantly for both isomers (see experimental section). These findings confirm the presence of one phosphine moiety in the structure of each isomer with two magnetically different phosphorus atoms of dppv. The signal positions and values of the coupling JP−P constants are very similar to those obtained for analogous rhenium(I) complexes with α-diimines ligands [34].
In turn, the FT-IR spectra confirm the presence of two non-equivalent CO molecules in the structures of the examined isomeric complexes. For both isomers, two single, sharp, and intense absorption bands in the \({\nu }_{c\equiv 0}\)stretching region are observed. They are located at 1840 and 1925 cm−1 or 1859 and 1931 cm−1 for [OC-6–54] and [OC-6–53] isomers, respectively. There is a particularly pronounced difference between the positions of the \({\nu }_{c\equiv 0}\) band characterizing the two isomers. Very similar effect has been observed for the analogous isomeric osmium(II) complexes containing CO, dppv, and pbi ligands in the structures of the [Os(Cl)(CO)(dppv)(pbi)] isomers [37]. In the case of the green emissive [OC-6–24] isomer, in which the CO molecule is trans-located with respect to the benzimidazole fragment of the pbi ligand, the \({\nu }_{c\equiv 0}\) band is observed at lower energies in comparison to the analogues band of the yellow-emissive [OC-6–23] isomers with the CO molecule cis-located in relation to the same fragment of the pbi ligand. This finding can be rationalized supposing the same ligand arrangements in the respective [OC-6–54]-[Re(CO)2(dppv)(pbi)] and [OC-6–24]-[Os(Cl)(CO)(dppv)(pbi)] or [OC-6–53]-[Re(CO)2(dppv)(pbi)] and [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] pairs.
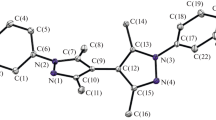
The 1H NMR spectra, although rather complex due to the presence of many overlapping signals reflecting the large number of magnetically non-equivalent protons in the studied species, allow to prove the presence of one dppv as well as one pbi ligand in the molecules of both isomers. In any case, it is possible to discern some common features and some differences in the spectra of the two isomeric forms. Clearly, the integrated signal intensities in the 1H NMR spectra reproduce the expected number of protons in both [Re(CO)2(dppv)(pbi)] isomer structures. In the spectra of both [OC-6–54] and [OC-6–53] isomers, the characteristic set of two signals from non-equivalent vinyl protons of dppv ligand in the range of δ = 7.80–8.40 ppm is observed. This set of two doublet of doublet of doublets with the values of the coupling constants for these signals of about J = 46–47 Hz, J = 9.6 Hz, and J = 7.5–8.1 Hz results from the interactions between the given proton and the second proton of the − CH = CH − group and the two 31P nuclei associated with the vinyl carbon atoms. A similar set of signals with close values of coupling constants was observed in the 1H spectra of analogous rhenium(I) complexes with dppv and polypyridyl ligands [34], as well as in the spectra of osmium(II) isomers containing the same phosphine and pbi ligand [37]. Another common feature of the 1H spectra of both isomers is the occurrence of signals sets, three multiplets with 2:1:2 intensity ratio located at 5.96–6.18, 6.38–6.39, and 6.50–6.80 ppm that can be assigned to one of the phenyl group from dppv ligand. These multiplets are clearly shifted toward lower \(\delta\) values relative to the proton signals of the other three phenyl rings of the dppv ligand (integrated to 15H), which are located in the range of chemical shift values from 7.1 to 7.6 ppm. Hence, it can be postulated that these three multiplet signals originate from this phenyl group of the dppv ligand, which is located in the closest vicinity to the 2-(2-pyridyl)-benzimidazole ligand present in the investigated isomers of the [Re(CO)2(dppv)(pbi)] complex. The remaining signals in the 1H NMR spectra can be attributed to the protons of the pbi ligand coordinated in the isomeric molecules. In the case of the [OC-6–53] isomer, these signals are concentrated in a narrow \(\delta\) range from ~ 7 to ~ 8.1 ppm, while in the spectrum of [OC-6–54] isomer, they are located over a much wider range from ~ 6.2 up to ~ 9.3 ppm. Similarly, as it was observed in the case of analogues [OC-6–24] isomer of the osmium(II) complex [37], the signal from one of the protons of the [OC-6–54]-[Re(CO)2(dppv)(pbi)] complex is located at the highest value of the chemical shift. Most likely this is a proton adjacent to the nitrogen atom of the pyridyl ring in the pbi ligand. Based on the compatible analysis of 31P NMR, 1H NMR, and FT-IR spectra, the presence of one pbi ligand, one dppv ligand, and two carbonyl ligands in the structures of the two analyzed isomers can be inferred. These conclusions remain in excellent agreement with the resolved X-ray structures presented in Fig. 1. In Table 1, the selected bond distances are listed together with those for their [OC-6–24]-[Os(Cl)(CO)(dppv)(pbi)] and [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] analogues. Both isomeric complexes [OC-6–54]-[Re(CO)2(dppv)(pbi)] and [OC-6–53]-[Re(CO)2(dppv)(pbi)] exhibit the expected octahedral coordination environment around rhenium(I) ion, which is slightly distorted.

X-ray structures (50% probability ellipsoids) of the yellow-emissive [OC-6–54]-[Re(CO)2(dppv)(pbi)], the orange-emissive [OC-6–53]-[Re(CO)2(dppv)(pbi)], the green emissive [OC-6–24]-[Os(Cl)(CO)(dppv)(pbi)], and the yellow-emissive [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] complexes (from left to right). The atoms forming the presented structures are colored as follows: C – gray, N – blue, Cl – green, P – orange, O – red, and Re or Os – tale. The hydrogen atoms and solvent molecules omitted for clarity. Structures of osmium complexes taken from refs [37] and [38]
In the equatorial plane, in coordination to the central metal ion, the pbi ligand containing negatively charged benzimidazole and neutral pyridine fragments is involved. One of the CO ligands that is also bounded in the equatorial position is trans-located with respect to the negatively charged benzimidazole or neutral pyridine fragment of the pbi molecule in the [OC-6–54] or [OC-6–53] isomer, respectively. The fourth position is occupied by the phosphorus atom of dppv ligand with acceptor properties, which is in trans arrangement to the pyridine. The different distribution of ligands in these isomer molecules, with different donor–acceptor properties, affects the bond distances to a greater or lesser extent. In both Re(I) isomers, [OC-6–54] and [OC-6–53], the corresponding Re-P and Re-C bond distances are very similar. Analogously, in the case of Os(II) isomers, [OC-6–24] and [OC-6–23], the corresponding of Os-P, Os-C, and Os-Cl does not show significant differences [37, 38]. However, clear differences are observed in the Re–N(bim) and Re–N(pyr) as well as Os-N(bim) and Os-(pyr) bond distances. In the case of [OC-6–53] and [OC-6–23] isomers, in which the P donor atom of the dppv ligand is coordinated in trans-position with respect to the benzimidazole moiety of pbi ligand, the corresponding Re–N(bim) or Os-N(bim) bond distances are shorter (of about 0.03–0.04 Å) and Re–N(pyr) or Os-N(pyr) are longer (of about 0.05–0.07 Å) than analogous bond distances in the structures of their [OC-6–54] and [OC-6–24] isomeric counterparts. Very likely different ligand arrangement and/or metal–ligand distances are responsible for differences in the photophysical properties of the investigated isomers (see the “Nature of the excited triplet states” section).
UV–vis absorption
UV–vis absorption spectra recorded in acetonitrile solutions for the investigated [OC-6–54] and [OC-6–53] isomers are shown in Fig. 2, respectively. The spatial orientation of the pbi ligand has a quite pronounced effect on the UV–vis spectroscopic properties of the investigated isomers. In the recorded spectra for both isomers, strong absorption at high energies at wavelengths shorter than 250 nm is observed which, taking into account its shape and intensity, can be assigned to the \(\pi \to \pi\)* transitions localized within CO and/or dppv ligands [55]. The low-energy bands can be tentatively assigned to the electronic transitions involving the coordinated pbi anion [56, 57]. However, in the lower energy region, the spectra differ clearly, as the [OC-6–54] isomer spectrum shows two maxima of similar intensity in the 310–344 nm range, the [OC-6–53] isomer spectrum shows only one broad band at around 350 nm of medium intensity and a much less intense shoulder around 400 nm.

UV–vis absorption spectra in acetonitrile solutions of the yellow-emissive [OC-6–54]-[Re(CO)2(dppv)(pbi)] (left) and the orange-emissive [OC-6–53]-[Re(CO)2(dppv)(pbi)] (right) isomers. Insert presents the expanded low-energy part of the spectra with the red vertical bars denoting the positions and relative intensities of the electronic transitions from TDA-DFT computations
The shapes, positions, and intensities of the observed absorption bands for the [OC-6–54] and [OC-6–53] isomers reported in this work are similar to their osmium(II) counterparts [37, 38]. Furthermore, due to different location of the ligands in the molecules of the respective isomers of the discussed complexes, the low-energy absorption band of the [OC-6–53] isomer is shifted toward longer wavelengths in comparison to [OC-6–54] one. Similar effect was observed in the case of their [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] and [OC-6–24]-[Os(Cl)(CO)(dppv)(pbi)] counterparts [37]. This seems to be associated with the charge redistribution within the electronically excited pbi anion that is somewhat modified by the different (in relation to the benzimidazole and pyridine subunits of pbi) location of the ligands (CO molecule and = CH2–P(C6H5)2 fragment) possessing different electron accepting and donating properties [37]. It is also noteworthy to note that the recorded spectra do not show distinct features characteristic for the previously investigated cationic complexes [Re(CO)2(dppv)(N^N)]+ complexes with α-diimine ligands possessing relatively intense \(\left({\varepsilon }_{\mathrm{M}}=3.1-9.6\times {10}^{3^\circ }{\mathrm{M}}^{-1}{\mathrm{cm}}^{-1}\right)\) broad low-energy bands located in the 350–500 nm region of the UV–vis radiation and assigned to the S0 → 1*MLCT transitions [34]. The observed difference is most likely due to the presence of negatively charged pbi ligand in the molecules of the studied [Re(CO)2(dppv)(pbi)] isomers, having worse π-accepting properties as compared to the neutral N^N α-diimine ligands in the [Re(CO)2(dppv)(N^N)]+ cations [34].
For the investigated [OC-6–54] and [OC-6–53] isomers, the TDA-DFT computations for the optimized ground state geometry S0 were performed to obtain a somewhat deeper insight into their UV–vis absorption spectra (cf. data in Table 2 and Fig. 3). The computed positions of the S0 → Sn transitions remain in quite good agreement with the experimentally observed absorption profiles (cf. Figure 2). In the case of both isomers, the lowest S0 → S1 transitions involve the HOMO and LUMO orbitals, both localized mainly on the pbi ligand with some involvement of the metal orbital to the HOMO one.

Shapes and energies of the HOMO-2, HOMO-1, HOMO, LUMO, and LUMO + 1 orbitals for the yellow-emissive [OC-6–54] (top) and the orange-emissive [OC-6–53] (bottom) isomers. Data from the DFT computations performed for the ground state optimized geometries of [Re(CO)2(dppv)(pbi)] complexes in acetonitrile solutions
Thus, the lowest excited S1 state can be assigned as the mixed LC/MLCT excitation with the intrinsic intra-ligand contribution localized within the pbi ligand. For the [OC-6–54] isomer, one can classify the S0 → S2 transition (involving the HOMO-2 and LUMO orbitals) as the MLCT transitions, whereas the S0 → S3 transition (involving HOMO and LUMO + 1 orbitals) as corresponding to the ligand-to-ligand electron transfer (from the pbi to dppv). Similar situation is characteristic for the [OC-6–53] isomer where the HOMO → LUMO + 1 and HOMO-2°→ LUMO transitions characterize the excited S2 and S3 states. For both isomers, the discussed CT transitions are possessing rather low oscillator strength values and their contributions to the low-energy parts of the UV–vis spectra are correspondingly small. For the [OC-6–53] isomer, the HOMO-1°→ LUMO transition (S0°→ S6 at 340 nm with the oscillator strength f = 0.391) is dominating.
For the [OC-6–54] isomer, however, the multifaceted S0 → S4 transition (at 360 nm with f = 0.137) together with the S0 → S1 one (at 380 nm with f = 0.035) seems to intrinsically participate to its lowest UV–vis absorption band.
Emission spectra and decays
The geometrical isomerism results also in significant differences in the emission properties of the yellow-emissive [OC-6–54] and the orange-emissive [OC-6–53] isomers under study. They are luminescent both in room temperature solutions and in 77 K matrices (cf. Figure 4). The maxima of the emission bands of a given isomer to a small extent depends on the type of solvent; the respective \(\bar{\nu}_{em}\) values are similar and differ within 850 cm−1 for [OC-6–54] and only within 150 cm−1 for the [OC-6–53] isomer, respectively (cf. data in Table 3). At room temperature, the investigated complexes are rather weak emitters. Depending on the solvent, the emission quantum yields are as low as 0.0061–0.028 for the [OC-6–54] isomer and ca. one order of magnitude lower, 0.00029–0.0029 for the [OC-6–53] isomer, respectively (cf. data in Table 3). Regardless of the type of solvent used, the luminescence spectra recorded in solutions at 298 K are typically broad and show usually no vibronic structure (cf. Figure 4).

Room temperature (red lines) and 77 K (blue lines) emission spectra of [OC-6–54]-[Re(CO)2(dppv)(pbi)] (top) and [OC-6–53]-[Re(CO)2(dppv)(pbi)] (bottom) complexes. Data for acetonitrile (dotted lines) and MeOH/EtOH 1:1 (solid lines) solutions
At 77 K, however, their luminescence behavior is different and the significant changes in the emission profiles of both isomers are observable. In this temperature regime, the [OC-6–54] isomer displays highly structured emission band with quite well-pronounced vibrational progression, whereas the emission of the [OC-6–53] isomer remains broad with only some shoulders. For both isomers, the lowering of temperature causes hypsochromic shift of their emission bands. The observed rigidochromism effects [58] are rather small (ca. 1000 cm−1) and similar for both isomers.
Taking into account the steady-state luminescence properties of the yellow-emissive [OC-6–54] and the orange-emissive [OC-6–53] isomers, one can emphasize the well-visible differences between these species. This is also reflected in their emission decays (cf. Figure 5). Whereas at 77 K both complexes exhibit mono-exponential decays with relatively long lifetimes of their emissions (23.7 and 14.6 μs, respectively), the room temperature decays are distinctly shorter (cf. data in Table 3). The [OC-6–54] complex still exhibits mono-exponential decays of its 298 K emissions with the lifetimes within the range of 0.32–1.28 μs depending on the solvent type.

The room temperature emission decays recorded for the yellow-emissive [OC-6–54]-[Re(CO)2(dppv)(pbi)] (left) and the orange-emissive [OC-6–53]-[Re(CO)2(dppv)(pbi)] (right) isomers in toluene (cyan line), tetrahydrofuran (green line), and acetonitrile (red lines) solutions, excitation pulse profile (gray line). The dashed black lines present the fitted mono-exponential and bi-exponential decay curves for isomer [OC-6–54] and [OC-6–53], respectively
The photophysical behavior of the [OC-6–53] isomer is, however, distinctly more multifaceted with bi-exponential decays usually observed in solutions at 298 K (cf. Figure 5). The recorded decays consist of fast and slow components with their contributions to the overall emission intensity depending on the solvent polarity. The less polar the solvent in which the emission decay of this isomer is recorded, the greater the contribution from the long-lived emission component is observed (cf. data in Table 3). In the low-polar toluene solution, the long-lived component of the emission dominates, while in the moderately polar tetrahydrofuran solvent, both emission components are present with comparable contributions. Consistently, in the more polar acetonitrile solvent, the short-lived emission contributes most to the overall emission intensity. In the mostly polar medium, MeOH/EtOH (1:1) mixture, the recorded emission decay is essentially mono-exponential. However, one cannot exclude that the emission decay in this particular case consists also two components, but, due to the probably very low intensity of the long-lived one, it was not possible to record it. The occurrence of these bi-exponential decays observed at room temperature for the [OC-6–53] isomer is most likely caused by the presence of two energetically close excited states with an energy gap between them depending on the polarity of the measurement medium, similarly as it was observed for the previously investigated [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] chelate [37].
Nature of the excited triplet states
In order to better understand the nature of the excited triplet states of the investigated [OC-6–54] and [OC-6–53] isomers, the TDA-DFT computations were carried out for the ground S0 state geometries. In the case of both isomers, the lowest S0 → T1 transitions involve mainly the HOMO and LUMO orbitals (cf. data in Table 2). Thus, analogously to the lowest excited S1 states, their T1 counterparts can be assigned as the mixed LC/MLCT excitation with the dominated intra-ligand contribution localized within the pbi ligand.
The calculation carried out for the yellow-emissive [OC-6–54] isomer in the optimized geometry of the lowest excited triplet state revealed that the nature of the HOMO and LUMO orbitals remains the same for both S0 and T1 geometries. At both geometries, the electronic S0 → T1 transition involves the LUMO and HOMO orbitals localized primarily on the pbi ligand. The analysis of orbital shapes and spin density redistribution (cf. Figure 6) within the lowest excited 3*[OC-6–54]-[Re(CO)2(dppv)(pbi)] state allows classifying this isomer as the LC type luminophore exhibiting almost “pure” S0 ← 3*LC emission with negligibly small MLCT contribution.

Shapes and energies of the HOMO, LUMO, and LUMO + 1 orbitals of the yellow-emissive [OC-6–54] isomer in the singlet Franck–Condon state reached in the S0 ← LC emission (left) and the spin density redistribution in the triplet state (right). Data from DFT computations performed for the excited triplet state optimized geometry in acetonitrile solutions
The DFT results obtained for the orange-emissive [OC-6–53] isomer are essentially different. Results from the performed geometry optimization of its triplet state point to the presence of two energetically close states with the LC/MLCT and LL’CT natures, respectively. The computed energy difference between these states depends on the medium polarity, changing from − 0.12 eV in vacuum to 0.10 eV in methanol, the most polar solvent. Correspondingly, the LC/MLCT and LL’CT states are isoenergetic in the low-polar toluene. Both excitations involve HOMO orbitals with their natures nearly the same for both LC/MLCT and LL’CT geometries (cf. Figure 7). In contrary to that, the LUMO orbitals involved in the S0 → LC/MLCT or S0 → LL’MLCT transitions are essentially different. The lowest excited LC/MLCT state exhibits substantial 3*LC nature with some admixture of the MLCT character with the amount of the charge transferred being ca. 0.2 e− as it can be deduced considering the Mulliken charges on the central rhenium ion and the attached ligands. Thus, one can classify the discussed state as the LC/MLCT emitter with the charge transfer from the Re(CO)2(dppv)+ fragment of the complex molecule to the pbi ligand. Significant contribution from the metal orbitals to the HOMO orbital of the [OC-6–53] isomer results in somewhat more pronounced changes between the S0 and LC/MLCT geometries, which is reflected in the observed broadening of its emission spectra. The computed energies of this excited triplet state are nearly independent of the medium polarity and they are only ca. 0.07–0.15 eV lower than found for their [OC-6–54] analogue. In contrary to the excited LC/MLCT state, the excited LL’CT state of the [OC-6–53] isomer is associated with relatively large charge transfer (ca. 0.5 e−) from the Re(CO)2(pbi) fragment to the dppv ligand and the spin redistribution over the whole molecule (cf. Figure 7).

Shapes and energies of the HOMO, LUMO, and LUMO + 1 orbitals of the orange-emissive [OC-6–53] isomer in the singlet Franck–Condon state reached in the S0 ← LC/MLCT (top) and S0 ← LL’CT (bottom) emissions (left) and the spin density redistribution in the triplet LC/MLCT and LL’CT states (right). Data from DFT computations performed for the excited triplet state optimized geometries in acetonitrile solutions
The obtained DFT results remain in nice agreement with the experimentally observed photophysical behavior of the discussed orange-emissive complex. Taking into account the character of the excited LC/MLCT and LL’CT states of the [OC-6–53] isomer, one can attribute its experimentally observed emission as originating from the S0 ← LC/MLCT transition. Such assignment is based on quite similar energies of the LC/MLCT and LL’CT states and very different energies of the Franck–Condon states reached in the S0 ← LC/MLCT and S0 ← LL’CT transitions (0.33–0.43 eV and 0.98–1.07 eV, respectively). Thus, eventually possible, presumably low intensity S0 ← LL’CT emission should be located at distinctly longer wavelengths as compared to the S0 ← LC/MLCT one. Depending on the solvent, the computed S0 ← LC/MLCT and S0 ← LL’CT transitions are located within the ranges of 630–670 nm and 820–1100 nm, respectively. Since limited spectral range of our fluorimeter, the latter emission could not be observed in the performed experiments.
With the DFT and TDA-DFT data obtained in this work, one can also construct the energy level diagram (cf. Figure 8) describing the triplet manifold of the studied complexes at their optimized singlet and triplet geometries. Taking into account differences in the energies of the given geometries in their singlet states, one can straightforwardly estimate the excited state energies from the vertical transition energies provided by the TDA-DFT computations. All values presented in Fig. 8 were obtained in such a way, despite that the T1 state energies are also available from the DFT computations performed during geometry optimizations. Comparing the DFT and TDA-DFT outcomes, one can conclude quite well coincident values of the excited T1 state energies with differences not larger than 0.1 eV. The observed discrepancies seem to be, however, acceptable.

Excited triplet state energies for the orange-emissive [OC-6–53]-[Re(CO)2(dppv)(pbi)] (left) and the yellow-emissive [OC-6–54]-[Re(CO)2(dppv)(pbi)] (right) complexes at the optimized geometries of the ground state (blue bars), the excited triplet LC/MLCT or LC states (red bars), and the excited LL’CT state (green bars), respectively. Gray bars present anticipated energies of the excited LC/MLCT and LL’CT states for the [OC-6–54] isomer at the LL’CT geometry. Back dotted lines connect states with the same orbitals assignment characterizing the excited triplet states with the LC/MLCT or LC and LL’CT character, correspondingly. Data from the TDA-DFT computations performed for acetonitrile solutions
The energy level diagram shown in Fig. 8 allows better understanding of the experimentally available photophysical data. In the case of the [OC-6–53] isomer, the presence of two different triplet states with different equilibrated molecular geometries but with similar energies (as required for the delayed emission) seems to be unquestionable.
Similarly, as it was previously reported for the [OC-6–23]-[Os(Cl)(CO)(dppv)(pbi)] complex [37, 38], one can attribute the observed phenomenon to the presence of the prompt (S0 ← LC/MLCT) and delayed (S0 ← LC/MLCT ← LL’CT) emissions. Thus, the presence of the LL’CT state is crucial for the bi-exponential decays of the [OC-6–53] emissions observed at room temperature. Since the energy gap between the involved LC/MLCT and LL’CT depends on the medium polarity, the relative contributions of the prompt and delayed emission are different.
Pursuing the close analogy between both investigated isomers, one can expect the presence of the LL’CT-like state also in the case of the [OC-6–54] isomers. However, the performed triplet geometry optimization of the [OC-6–54] isomer resulted, independently of the initial structure applied in the DFT computations, only in one energy minimum corresponding to the LC/MLCT-like state. Thus, in the case of the [OC-6–54] isomers, the LL’CT-like triplet seems to be located at distinctly higher energies as compared to the experimentally observed LC/MLCT one. Results from the performed TDA-DFT computations confirm this hypothesis. For the [OC-6–53] isomer at its equilibrated LC/MLCT geometry, the third excited triplet state (T3 located ca. 0.86 eV above the LC/MLCT) possesses the charge and spin redistributions characteristic for the LL’CT one. In analogous way, at the equilibrated LL’CT geometry of the [OC-6–53] isomer, its third excited triplet state (located ca. 0.67 eV above the LL’CT) relates with the LC/MLCT one. Relaxations of the mentioned T3 states, with the released energies of ca. 0.77 eV, result in the equilibrated LC/MLCT or LL’CT states, respectively. In the case of the [OC-6–54] isomer in the LC/MLCT geometry, however, its excited T5 state (with the charge transfer from the Re(CO)2(pbi) moiety to the dppv ligand) is located ca. 1.14 eV above its lowest excited T1 triplet. Assuming that the relaxation of the excited T5 state from the LC/MLCT geometry to the anticipated LL’CT conformation is associated with the same amount of the released energy as for the [OC-6–53] isomer, the excited LL’CT state of the [OC-6–54] isomer will be localized energetically well above (by ca. 0.37 eV) its excited LC/MLCT state. Thus, one can only hardy expect that the S0 ← 3*[OC-6–54]-[Re(CO)2(dppv)(pbi)] transition, in contrary to the S0 ← 3*[OC-6–53]-[Re(CO)2(dppv)(pbi)] one, will be affected by the presence of the excited LL’CT state.
The above presented picture neglects the presence of the excited T2 state. However, this seems to be justified considering quite similar LC or LC/MLCT nature of the S0 → T1 and S0 → T2 excitations as well as the energy gaps between the T1 and T2 states in their Franck–Condon states reached in the absorption. Since one can expect similar energies released in the relaxations from the Franck–Condon states to the equilibrated T1 and T2 geometries, the relaxed T2 states are expected to be located above the relaxed T1 ones for both isomers under study. Preliminary results from the performed TD-DFT optimizations of the T1 an T2 geometries remain in agreement with such expectations.
Conclusions
Due to dissymmetry of the anionic pbi ligand, the investigated [Re(CO)2(dppv)(pbi)] complex exists in the form of two isomers, affordable by means of simple, one pot reaction and chromatographically separable. Both investigated isomers exhibit different photophysical properties arising from the different orientation of the ligand attached to the rhenium(I) core. For both isomers, the performed DFT and TDA-DFT computations point to the presence of two excited triplet states with LC/MLCT or LL’CT character. Energy gap between these states is the main factor affecting photophysical behavior of the reported [OC-6–53] and [OC-6–54] isomers. In the latter one, the excited LL’CT state is energetically located well above the lowest excited triplet state with dominant LC nature. The lowest excited 3*[OC-6–54]-[Re(CO)2(dppv)(pbi)] state deactivates according to simple superposition of the radiative and non-radiative S0 ← 3*[OC-6–54]-[Re(CO)2(dppv)(pbi)] transitions. In contrary to that, the [OC-6–53] isomer exhibits more complex deactivation mechanism. Since its excited LC/MLCT and LL’CT states are close in their energies, two deactivation channels take part in the S0 ← 3*[OC-6–53]-[Re(CO)2(dppv)(pbi)] process. The first one is the simple S0 ← LC/MLCT transition, whereas the second one engages the LL’CT state through the thermally activated LL’CT → LC/MLCT → S0 process. This leads to the presence of the delayed emission phenomenon. Since the thermally activated LC/MLCT → LL’CT process is not operative at 77 K, only the S0 ← LC/MLCT transition is observable in the low-temperature regime where the [OC-6–53] isomer, similarly to its [OC-6–54] counterpart, displays only mono-exponential decays. On the other hand, the presence of the LL’CT → S0 deactivation channel leads to lowering of the emission quantum yields at room temperature, the [OC-6–53] isomer is distinctly less emissive as compared with the [OC-6–54] one.
The presence of the excited LL’CT state in the reported molecules seems to be associated with an ambivalent nature of the bidentate pbi ligand, containing negatively charged benzimidazole and neutral pyridine fragments, with the charge donating and charge accepting properties, respectively. This results in two possible direction of charge transfer, to and from the pbi ligand, the latter possible if the given complex molecule contains an additional ligand with relatively well-pronounced electron acceptor properties (e.g., dppv in the case of reported isomers). Since analogous behavior was also observed in the case of structurally similar [Os(CO)(Cl)(dppv)(pbi)] isomers, one can expect this as a more general rule in any other transition metal complexes bearing asymmetric negatively charged ligand. The presence of the excited LL’CT state with energies possibly close to the excited LC/MLCT states in such complexes may strongly affect their emissive properties. Since the energy gap between these states depends on the ligand arrangement around the central metal ion, one can also expect the isomeric effects for any complexes similar to the [Re(CO)2(dppv)(pbi)] or [Os(CO)(Cl)(dppv)(pbi)] molecules.
Considering the emissive properties of both isomers reported in this work, one can tentatively attribute the observed differences to somewhat dissimilar character of their molecular orbital. Despite that the same ligands are attached to the central rhenium(I) ion, the relatively small structural changes lead to different energies and composition of the molecular orbitals in the isomers under study. Particularly, as it appears in the results from DFT computations, the nature of the LUMO and LUMO + 1 orbitals in the studied molecules depends on the geometry of the lowest triplet state. Very likely all of these differences are associated with divergent splitting of the rhenium(I) 5d orbitals, caused by the ligand orientation–induced changes in the ligand-field strength. This, more or less plausible, hypothesis requires, however, further more systematic experimental and theoretical studies of any complexes similar to the [Re(CO)2(dppv)(pbi)] and [Os(CO)(Cl)(dppv)(pbi)] examples.
Availability of data and material
1H NMR, 31P NMR, and FT-IR spectra are available as electronic supplementary material and crystallographic data (CIF files) for [OC-6–53]-Re(CO)2(dppv)(pbi)] and [OC-6–54]-[Re(CO)2(dppv)(pbi)] complexes). CCDC 2,153,573 and CCDC 2,153,574 contain the crystallographic data for [OC-6–53]-Re(CO)2(dppv)(pbi)] and [OC-6–54]-Re(CO)2(dppv)(pbi)] complexes, respectively. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223–336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Wrighton M, Morse DL (1974) The nature of the lowest excited state in tricarbonylchloro-1,10-phenanthrolinerhenium(I) and related complexes. J Am Chem Soc 96:998–1003
Luong JC, Nadjo L, Wrighton MS (1978) Ground and excited state electron transfer processes involving factricarbonylchloro(1,10-phenanthroline)rhenium(I). Electrogenerated chemiluminescence and electron transfer quenching of the lowest excited state. J Am Chem Soc 100:5790–5795
Westmoreland TD, Le Bozec H, Murray RW, Meyer TJ (1983) Multiple-state emission and intramolecular electron-transfer quenching in rhenium(I) bipyridine based chromophore-quencher complexes. J Am Chem Soc 105:5952–5954
Worl LA, Duesing R, Chen P, Della Ciana L, Meyer TJ (1991) Photophysical properties of polypyridyl carbonyl complexes of rhenium(I). J Chem Soc Dalton Trans 849–858
Stufkens DJ, Vlček A Jr (1998) Ligand-dependent excited state behaviour of Re(I) and Ru(II) carbonyl−diimine complexes. Coord Chem Rev 177:127–179
Vogler A, Kunkely H (2000) Excited state properties of organometallic compounds of rhenium in high and low oxidation states. Coord Chem Rev 200–202:991–1008
Striplin DR, Crosby GA (2001) Photophysical investigations of rhenium(I)Cl-(CO)3(phenanthroline) complexes. Coord Chem Rev 211:163–175
Kirgan RA, Sullivan BP, Rillema DP (2007) Photochemistry and photophysics of coordination compounds: rhenium. Top Curr Chem 281:45–100
Vlček A Jr (2009) Ultrafast excited-state processes in Re(I) carbonyl-diimine complexes: from excitation to photochemistry. Top Organomet Chem 29:115–158
Kumar A, Sun S-S, Lees AJ (2009) Photophysics and photochemistry of organometallic rhenium diimine complexes. Top Organomet Chem 29:37–71
Woźna A, Kapturkiewicz A (2015) The luminescence properties of the heteroleptic [Re(CO)3(N∩N)Cl] and [Re(CO)3(N∩N)(CH3CN)]+ complexes in view of the combined Marcus-Jortner and Mulliken-Hush formalism. Phys Chem Chem Phys 17:30468–30480
Villafañe F (2017) ReI(CO)3 complexes with diimine ligands synthesized in situ. Coord Chem Rev 339:128–137
Takeda H, Ishitani O (2010) Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord Chem Rev 254:346–354
Windle CD, Perutz RN (2012) Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord Chem Rev 256:2562–2570
Yamazaki Y, Takeda H, Ishitani O (2015) Photocatalytic reduction of CO2 using metal complexes. J Photochem Photobiol C 25:106–137
Merillas B, Cuéllar E, Diez-Varga A, Torroba T, García-Herbosa G, Fernández S, Lloret-Fillol J, Martín-Alvarez JM, Miguel D, Villafañe F (2020) Luminescent rhenium(I)tricarbonyl complexes containing different pyrazoles and their successive deprotonation products: CO2 reduction electrocatalysts. Inorg Chem 59:11152–11165
Takeda H, Koike K, Morimoto T, Inumaru H, Ishitani O (2011) Photochemistry and photocatalysis of rhenium(I) diimine complexes. Inorg Photochem 63:137–186
Zarkadoulas A, Koutsouri E, Kefalidi C, Mitsopoulou CA (2015) Rhenium complexes in homogeneous hydrogen evolution. Coord Chem Rev 304–305:55–72
Lundin NJ, Blackman AG, Gordon KC, Officer DL (2006) Synthesis and characterization of a multicomponent rhenium(I) complex for application as an OLED dopant. Angew Chem Int Ed 45:2582–2584
Yersin H, Rausch AF, Czerwieniec R, Hofbeck T, Fischer T (2011) The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs. Coord Chem Rev 255:2622–2652
Zhao G-W, Zhao J-H, Hu Y-X, Zhang D-Y, Li X (2016) Recent advances of neutral rhenium(I) tricarbonyl complexes for application in organic light-emitting diodes. Synth Met 212:131–141
Gonçalves MR, Benvenho ARV, Frin KPM (2019) Electrical and optical properties of organic light-emitting diodes with rhenium(I) complexes using DC and AC methods. Opt Mater 94:206–212
Lo KK-W (2010) Exploitation of luminescent organometallic rhenium(I) and iridium(III) complexes in biological studies. Top Organomet Chem 29:73–114
Lo K-W, Zhang KY, Li SP-Y (2011) Recent exploitation of luminescent rhenium(I) tricarbonyl polypyridine complexes as biomolecular and cellular probes. Eur J Inorg Chem 3551–3568
Hostachy S, Policar C, Delsuc N (2017) Re(I) carbonyl complexes: multimodal platforms for inorganic chemical biology. Coord Coord Chem Rev 351:172–188
Lee LC-C, Leung K-K, Lo KK-W (2017) Recent development of luminescent rhenium(I) tricarbonyl polypyridine complexes as cellular imaging reagents, anticancer drugs, and antibacterial agents. Dalton Trans 46:16357–16380
Konkankit CC, Marker SC, Knopf KM, Wilson JJ (2018) Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium. Dalton Trans 47:9934–9974
Ishitani O, George MW, Ibusuki T, Johnson FPA, Koike K, Nozaki K, Pac C, Turner JJ, Westwell JR (1994) Photophysical behavior of a new CO2 reduction catalyst, Re(CO)2(bpy){P(OEt)3}2+. Inorg Chem 33:4712–4717
Schutte E, Helms JB, Woessner SM, Bowen J, Sullivan BP (1998) A new class of luminescent polypyridinecomplexes of rhenium(I) containing cis-carbonyl ligands. Inorg Chem 37:2618–2619
Shen Y, Maliwal B, Lakowicz J (2001) Long-lived luminescent Re(I) complexes containing cis-carbonyl and bidentate phosphine ligands. J Fluoresc 11:315–318
Reece Y, Nocera DG (2005) Direct tyrosine oxidation using the MLCT excited states of rhenium polypyridyl complexes. J Am Chem Soc 127:9448–9458
Tsubaki H, Sekine A, Ohashi Y, Koike K, Takeda H, Ishitani O (2005) Control of photochemical, photophysical, electrochemical, and photocatalytic properties of rhenium(I) complexes using intramolecular weak interactions between ligands. J Am Chem Soc 127:1554–15555
Smithback JL, Helms JB, Schutte E, Woessner SM, Sullivan BP (2006) Preparative routes to luminescent mixed-ligand rhenium(I) dicarbonyl complexes. Inorg Chem 45:2163–2174
Kamecka A, Prachnio K, Kapturkiewicz A (2018) The luminescence properties of [Re(CO)2(P^P)(N^N)]+ complexes: comparison with their [Re(CO)3(N^N)(Cl)] analogues. J Lumin 203:409–419
Kapturkiewicz A, Kamecka A, Grochowska O (2020) Heteroleptic Re(CO)2+ and Re(CO)3+ complexes with α-diimines: similarities and differences in their luminescence properties. RSC Adv 10:29642–29658
Striplin DR, Crosby GA (2001) Photophysical investigations of rhenium(I)Cl(CO)3(phenanthroline) complexes. Coord Chem Rev 211:163–175
Kamecka A, Suwińska K, Kapturkiewicz A (2019) Heteroleptic [Os(Cl)(CO)(P^P)(pbi)] complexes bearing bidentate phosphine and 2-(2-pyridyl)-benzimidazolate ligands: impact of isomerism on their luminescence properties. Phys Chem Chem Phys 21:17746–17758
Kamecka A, Kapturkiewicz A, Suwińska K (2018) Luminescent osmium(II) complexes with 2-(2-pyridyl)-benzimidazolate anion. Inorg Chem Commun 89:27–31
CrysAlisPro Software System (2015) Version 1.171.38.46, Rigaku Corporation: Oxford, UK
Dolomanov V, Bourhis JJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 42:339–341
Sheldrick GM (2015) SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr A 71:3–8
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr C 71:3–8
Suzuki K, Kobayashi A, Kaneko S, Takehira K, Yoshihara T, Ishida H, Shiina Y, Oishi S, Tobita S (2009) Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector. Phys Chem Chem Phys 11:9850–9860
Marquardt DW (1963) An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math 11:431–444
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J and Fox DJ (2013) Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Guillemole J-F, Barone V, Joubert L, Adamo C (2002) A Theoretical investigation of the ground and excited states of selected Ru and Os polypyridyl molecular dyes. J Phys Chem A 106:11354–11360
Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations - potentials for K to Au including the outermost core orbitals. J Chem Phys 82:299–310
Hariharan P, Pople JA (1973) The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chim Acta 28:213–222
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, Defrees DJ, Pople JA (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3094
Scalmani G, Frisch MJ, Mennucci B, Tomasi J, Cammi R, Barone V (2006) Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J Chem Phys 124:94107
Stratmann RE, Scuseria GE, Frisch MJ (1998) An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J Chem Phys 109:8218–8224
Hirata S, Head-Gordon M (1999) Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem Phys Lett 314:291–299
Kamecka A, Kapturkiewicz A (2015) The luminescence properties of heteroleptic [OsCl(CO)(N∩N)(P∩P)]+ complexes - radiative and non-radiative deactivation of the excited 3*MLCT state. Phys Chem Chem Phys 17:23332–23345
Rodriguez Prieto F, Mosquera M, Novo M (1990) Dual fluorescence of 2-(2‘-pyridyl)benzimidazole in aqueous solution due to photoinduced proton-transfer processes. J Phys Chem 94:8536–8542
Burai TN, Mukherjee TK, Lahiri P, Panda D, Datta A (2009) Early events associated with the excited state proton transfer in 2-(2′-pyridyl)benzimidazole. J Chem Phys 131:034504
Lees AI (1995) The luminescence rigidochromic effect exhibited by organometallic complexes: rationale and applications. Comments Inorg Chem 17:319–346
Funding
The research leading to these results received funding from the institution affiliated by the authors.
Author information
Authors and Affiliations
Contributions
A. K. and A. K.: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing—original draft, writing—review and editing, visualization.
S. S.: methodology, formal analysis, investigation, data curation, validation, writing—review and editing. K. S.: methodology, formal analysis, investigation, data curation, validation, writing—review and editing.
Corresponding author
Ethics declarations
Ethics approval
Not applicable; no human or animal studies have been carried out.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kamecka, A., Kapturkiewicz, A., Shova, S. et al. The influence of structural isomerism on luminescence properties of [Re(CO)2(dppv)(pbi)] complexes containing cis-1,2-bis(diphenylphosphino)-ethene and 2-(2-pyridyl)benzimidazole ligands. Struct Chem 34, 1641–1655 (2023). https://doi.org/10.1007/s11224-023-02177-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02177-1