
Abstract
This article is the first attempt to present different influence of substituent effects on double and triple bonds and, conversely, to present the impact of these bonds on the electronic structure of substituents. For this purpose, quantum-mechanical calculations were made for X-substituted derivatives of ethene and acetylene with 27 diverse substituents representing a wide spectrum of electronic properties, from strongly electron-accepting to strongly electron-donating ones. In addition to these systems, their boranyl derivatives are also investigated. It turns out that the Hammett substituent constants do not correctly describe changes in the CC bond length in any of the considered family of systems. However, the relationships with the CB bond length are significantly better. It is shown that the triple bond in acetylene derivatives is much more resistant to external perturbations than the double bond in the analogs containing an ethene unit. As a consequence, in acetylene derivatives, the substituent effects on CC bond length are about half of the substituent effects in ethene derivatives. We suggest that the observed lack of a clear linear correlation between the length of the CC triple bond in acetylene derivatives and the value of electron density on this bond is due to the disturbing additional interaction between the structure of the X substituent in the xy plane and the π bond being in the same plane in the acetylene unit—on the contrary, this interaction is not possible in ethene analogs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Quantitative approaches to the problems of substituent effects (SE) in organic chemistry originate from the fundamental works by L. P. Hammett [1, 2] by introduction of substituent constants:
where KX and K are dissociation constants for para/meta-X-substituted and unsubstituted benzoic acids, respectively, and the acid-base reaction reads as follows:
The relationship between the influence of substituents X on the physicochemical properties P (e.g., kinetic constants, equilibrium constants, spectral or electrochemical properties for benzene derivatives) and σp/m constants is described by the Hammett equation:
where ρ is the so-called reaction constant that describes the sensitivity of the reaction under study (physicochemical process) to the effect of X. Hammett substituent constants represent the numerical characteristics of electron accepting/donating properties of substituents and have become a very convenient way for describing most of substituent effects encountered in physical organic chemistry. Then, an avalanche of papers has appeared that have undertaken these problems presented and summarized in a numerous reviews and monographs [3,4,5,6,7,8,9,10,11].
In most cases, the substituted systems were either benzene or some aromatic system derivatives, and much less frequently unsaturated systems or those with triple bonds. Moreover, usually the problem considered was rather the substituent effect on some physicochemical properties of the systems with a particular group fixed in the series like X–R–Y and rather rarely concentrated on the changes of electronic structure of the substituents which have participated in this kind of interactions. To some extent, it was due to the method of interpretation of the SE by use of the Hammett substituent constants σm, σp which could take into account usually these aspects and in most cases in standard situations as in benzoic acids or alike [12]. In other cases, differently defined substituent constants like σ+, σ−, R, I, etc., were in use [11].
Applications of the quantum chemistry methods [13] for describing SE have changed the situation substantially. An atomic charge computed for a given substituent, q(X), allows showing how its electronic structure changes in dependence on the molecule to which it is attached as well as on the position of the attachment. In some cases, a useful way is to apply charge of the substituent active region, abbreviated as cSAR(X), equal to the sum of atomic charges of all atoms of the substituent X and the substituted carbon atom Cipso [14,15,16]:
This way of presentation of electronic structure of the substituent has substantially opened possibilities to learn about changeability of the substituent electronic properties in dependence on the environment of its attachment. Undoubtedly, cSAR(X) values depend on the kind of carbon atom to which the substituent is attached and comparisons are allowed only if these atoms are strongly alike. It may be assumed that cSAR(X) for substituted benzene and e.g. butadiene may be still allowed to be compared, but this kind of comparison for acetylene and ethene derivatives seems to be not acceptable.
Unlike the earlier mentioned systems, acetylene derivatives have not been subject of too many systematic studies with substituent effects as a main topic. Some more important papers have to be mentioned. Most likely the first ab initio studies on substituted acetylene derivatives were performed by Powell et al. [17] in 1983 and concerned the description of the influence of substituent effects on the acidity of an acetylene hydrogen atom. Then, substituent effects on acetylene stability were studied using isodesmic methyl exchange reactions [18]. Detailed NMR studies of substituted acetylene derivatives were performed by Kamienska-Trela et al. [19] and Wiberg et al. [20]. The heats of formation of some haloacetylenes [21, 22] and polyacetylenes [23] were also computed. The substituent effects in acetylenes and ethylenes were also studied [24] by Quantum Theory of Atoms in Molecules (QTAIM) [25]. Quite recently, we have performed a comparative analysis of the electronic structure in substituted acetylene and diacetylene derivatives [26]. Various spectroscopic parameters for the latter group of compounds were calculated by Roman et al. [27]. Most of the important information on chemistry of acetylene and its derivatives are gathered in monograph by Diederich, Stang, and Tykwinski [28].
As already mentioned, Hammett substituent constants and their further modifications were introduced for para/ meta-X-substituted benzoic acid [1, 2] and for this reason they were used to describe the substituent effects only in more or less complex benzene derivatives, while there are no studies on the applicability of these constants in non-aromatic systems, but containing double or triple bonds or some conjugated variants thereof. The simplest representatives of compounds containing such bonds are, respectively, ethylene and acetylene. Comparing the π-electron structure in both of these molecules, it can be expected that the electronic effects related to the presence of the X substituent should be different in them, which results from the fact that in ethylene the double bond occurs only in one plane, while in acetylene both π bonds are in two planes perpendicular to each other. This difference in the π-electron structure should manifest itself in some way during the interaction with the substituent X. It should be emphasized here that the simple theoretical parameter cSAR(X) [14,15,16], used to describe the substituent effects, cannot be utilized in this case due to the fact that the carbon atoms are too different in both these molecules. Therefore, we have undertaken, what is the main purpose of this article, to perform a comparative analysis between substituent effects on ethene and acetylene derivatives. To some degree, this is continuation and substantial extension of the studies presented in our former article [26] in which there were presented substituent effects on acetylene and diacetylene derivatives themselves.
In addition to X-substituted ethylene and acetylene, we also investigate substituent effects on their boranyl derivatives. The boranyl group, as having an empty 2pz orbital, has very specific electronic properties [29,30,31], as it can act as a strong electron charge acceptor. At the same time, the flatness of this group enables easy coupling with the adjacent π-electron system, creating good conditions for charge relocation. The situation is thus somewhat similar to that which characterizes the amino group: although the population of the 2pz orbital in both groups is opposite, there is as an electron pair vacancy in the former, whereas the lone electron pair in the latter. Thus, in terms of electronic properties, both of these groups, i.e., -NH2 and -BH2, are opposite ends of the same stick. This makes studies on the effect of the type of CC bond on the interaction of the -BH2 group with various substituents particularly interesting.
In the studies presented here, 27 X substituents with the full spectrum of electronic properties are used, from strongly electron-withdrawing to strongly electron donating, as described by the Hammett σp constant. As for historical reasons [1, 2], the substituent constants are commonly used only to describe substituent effects on various benzene derivatives [3,4,5,6,7,8,9,10,11,12], one of the main and most interesting aims of this research is to investigate the applicability of the substituent constant σp in describing substituent effects in X-substituted ethene and acetylene derivatives. This is the first study of this issue. It is worth recalling at this point that the use of cSAR(X) [14,15,16] seems to be unacceptable here because of too much different ipso-carbons in both types of compounds investigated. As part of the presented research, the following issues will be discussed:
-
1.
How much the X⋯Y (Y = H, BH2) interaction changes delocalization in double-bonded systems in comparison with the triple-bonded ones
-
2.
How double-bonded moieties change electron structure of substituents and the -BH2 group in comparison with the triple-bonded moieties
-
3.
A difference in ability of charge transfer via double and triple bond(s)
Methodology
All calculations were performed at the ω B97X-D/6-311++G(2df,2p) level of theory, that is utilizing the ω B97X-D exchange-correlation functional [32, 33] of Density Functional Theory (DFT) [34,35,36] and the 6-311++G(2df,2p) basis set [37,38,39,40,41], including both polarization and diffuse functions.
At the beginning, the geometries of X-substituted (X ∈{-NO2, -CN, -CHO, -CFO, -CClO, -CMeO, -CONH2, -COOH, -COOMe, -CF3, -OCN, -NCO, -SCN, -NCS, -BH2, -CCH, -CCF, -H, -F, -Cl, -Me, -OH, -SH, -OMe, -NH2, -NHMe, -NMe2}, where Me = CH3) ethene, acetylene, and their respective boranyl derivatives (i.e., having Y = BH2) were fully optimized. To increase the accuracy of the optimization procedure and numerical integration, cutoffs on forces and step size that are used to determine convergence were additionally tightened (0.000015 and 0.000010 for maximum force and its root mean square, respectively, and 0.000060 and 0.000040 for maximum displacement and its root mean square, respectively) and integration grid was increased to the (99, 590) one (UltraFine) having 99 radial shells and 590 angular points per shell. The fact of being true minima on the potential energy hypersurface has been confirmed by the lack of imaginary frequencies in vibrational analysis. These calculations were performed by means of the Gaussian 09 program [42]. In the case of X-substituted boranylethene derivatives, the more extended trans conformation was used. For simplicity, the -C=C- and -C≡C- transmitting units in XRY molecules will be labeled as En and Ac, respectively. Fully optimized structures of boranyl derivatives are shown in Figs. 1 (XEnBH2) and 2 (XAcBH2) for reader’s convenience. Of course, they are derivatives of their counterparts in which instead of the -BH2 group there is a hydrogen atom, i.e., Y = H. The latter are therefore easy to imagine.

Fully optimized structures of XEnBH2 (X = -NO2 (1), -CN (2), -CHO (3), -CFO (4), -CClO (5), -CMeO (6), -CONH2 (7), -COOH (8), -COOMe (9), -CF3 (10), -NCS (11), -OCN (12), -SCN (13), -BH2 (14), -NCO (15), -CCH (16), -CCF (17), -NH2 (18), -NHMe (19), -NMe2 (20), -OH (21), -SH (22), -Me (23), -H (24), -F (25), -Cl (26), -OMe (27)

Fully optimized structures of XAcBH2 (X = -NO2 (1), -CN (2), -CHO (3), -CFO (4), -CClO (5), -CMeO (6), -CONH2 (7), -COOH (8), -COOMe (9), -CF3 (10), -OCN (11), -NCO (12), -SCN (13), -NCS (14), -BH2 (15), -CCH (16), -CCF (17), -Me (18), -H (19), -F (20), -Cl (21), -OH (22), -SH (23), -NH2 (24), -NHMe (25), -NMe2 (26), -OMe (27)
The values of electron density at critical points [25] of CC bonds were computed using the AIMAll program [43]. Electron densities at bond critical points are often used [25] as descriptors of the strength of various chemical bonds and intermolecular interactions.
Atomic charges were computed utilizing Multiwfn [44] program and the Voronoi Deformation Density (VDD) method [45,46,47]. Earlier it was shown that VDD atomic charges are highly reliable [31, 47, 48] and, importantly, almost independent of the computational method used to determine them [47]. The charge difference ΔqXH= q(H) − q(X), i.e., determined for the terminal hydrogen atom and group X, was used in the analysis of charge distribution in the examined systems. This parameter should be identified with the polarization of the XRY molecule rather than with the X→Y charge transfer effect, which was instead determined using the most recently introduced definition [26]:
where
In Eq. 6, qX(XY) and qX(XX) denote the charge of the X substituent in the XRY and XRX systems, respectively. The same applies to the charge of group Y in Eq. 7. Importantly, the definition given by Eq. 5 takes into account the following facts: (i) group Y cannot accept more charge from X than this group donates (ii) both groups, i.e., X and Y, already had some charge before the charge transfer occurs (iii) the “own group charges” can be determined from the symmetrical XRX and YRY systems, in which the charge transfer effect is not possible [26].
Results and discussion
When CC bond lengths (dCC) in X-substituted acetylene (XAcH) and X-substituted acetyleneborane (XAcBH2) derivatives are compared (see Table 1), it results in the range of observed CC bond lengths being somewhat greater for the latter (0.023 Å) than for the former (0.017 Å), giving an increase by 0.006 Å. Comparisons made for the X-substituted etheneboranes and ethenes result in 0.043 Å and 0.024 Å, respectively, giving an increase by 0.019 Å.
Thus, the effects of substituent on CC bond lengths are significantly smaller for substituted acetylene than ethene derivatives and in both cases the boranyl functional group increases clearly the observed effects, in particular in the case of ethenes. This may indicate a greater resistance of the triple CC bonds than of the double ones for external perturbations and most likely results from much higher force constants of the former compared to the latter.
It is important to indicate that any relationships are not observed between the CC bond lengths and the characteristics of substituents described by the substituent constants σp, F, and R (see Fig. S1). For example, the linear regressions of dCC on σp lead to determination coefficients R2< 0.35 for acetylenes and ethenes and significantly better for their boranyl derivatives, but still not sufficient to be subject of any deeper analyses. It may be concluded that the substituent constants while very effective in interpretation of manifold physicochemical properties of X-R-Y fail in the case of describing the substituent effects on CC bond lengths of substituted ethene and acetylene derivatives.
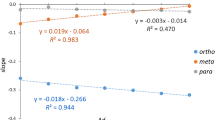
Situation is clearly better for dependence of CB bond lengths (dCB) on the σp, R (Fig. 3) and F (Fig. S2) substituent constants.

Relationships between dCB and either σp or R
In the case of σp, the determination coefficients for XEnBH2 and XAcBH2 are 0.805 and 0.745, respectively. Interestingly, for substituents with σp < 0 (i.e., electron-donating) both linear regressions are clearly better since determination coefficients improve to 0.861 and 0.937, respectively (see the light blue and black fitting lines in the upper subfigure of Fig. 3). However, this may result from a smaller variety of electron-donating substituents in our set of X. For the constant R, the values of the determination coefficients are somewhat better than those for σp and amount to 0.885 and 0.780 for XEnBH2 and XAcBH2, respectively. This result suggests that the interaction between the X substituent and the CB bond is mainly resonance in nature.
Interesting dependence is observed when CC bond lengths in XAcH and XAcBH2 are plotted against these bond lengths for their ethene-based analogs, i.e., XEnH and XEnBH2, as presented in Fig. 4. The appropriate linear regressions are as follows: \(d_{\text {CC}}^{\text {XAcH}}\) = 0.569 \(d_{\text {CC}}^{\text {XEnH}}\) + 0.442 with R2 = 0.815 and \(d_{\text {CC}}^{\text {XAcBH}_{2}}\) = 0.542 \(d_{\text {CC}}^{\text {XEnBH}_{2}}\) + 0.484 with R2 = 0.792. In spite of the somewhat poor quality of linear regression, it may be concluded that in acetylene derivatives the substituent effect on the CC bond length is ca. 0.5 of that observed in ethene derivatives, and the boranyl group slightly decreases the substituent effects.

Relationships between dCC
Very interestingly, the linear regressions between the CC bond distances, dCC, and the electron density computed at the critical points [25] of these bonds (ρCC) have been found (in Fig. 5) to feature somewhat unexpected characteristics. Namely, the regression lines are well acceptable with R2 ≥ 0.916 for XEnH and XEnBH2, i.e., the X-substituted ethene and etheneborane derivatives, whereas for both the X-substituted acetylenes and the X-substituted acetyleneboranes a lack of any correlation is observed since R2 ≤ 0.118. The interpretation may be as follows. Assuming the C-X direction as the x axis (see Fig. 6), two kinds of interactions should be taken into account: (i) a direct interaction of electronic structure of the X substituent with its one π-electron bond in acetylene in xz plane, and (ii) an interaction of the planar structure of the substituent in xy plane with another π-electron bond in the acetylene unit. The latter interaction is most probably the reason of perturbation that causes lack of the simple relationship observed for acetylene derivatives. In contrast, such a perturbation is not possible in ethene derivatives, where the π-electron interaction can take place only in one plane. Obviously, the detailed characteristics of the interaction between the py orbital of the acetylene unit and the planar electronic structure of the substituent in the xy plane depend on the type (type, number, and spatial arrangement of atoms) of the substituent.

Relationships between ρCC and dCC

Illustration of the interaction between the electronic structure of the X substituent and the p-orbitals of the acetylene unit in XAcY. The red arrows refer to point (ii) in the text
Of course, dispersion of points relative to the vertical axis dCC visible in Fig. 5 confirms the previously discussed largest range of dCC values obtained for XEnBH2 (see also Table 1). It is also seen that the values of electron density determined at the critical point of the CC double bond in the analyzed ethenes are from 0.327 to 0.368 au and are significantly lower than the corresponding values for the CC triple bond in the acetylene derivatives in question (0.400–0.429 au). As a consequence, this significant difference makes the triple bond much stronger (having a greater force constant) and therefore less susceptible to external perturbations, such as substituent effects resulting from the presence of group X.
It is also worth mentioning about good, though slightly worse than for dCC vs ρCC for the investigated ethene derivatives (Fig. 5), correlations between the length of the CB bond (dCB) and the value of electron density at the critical point of this bond (ρCB). Values of R2 amount to 0.807 and 0.917 for XEnBH2 and XAcBH2, respectively (see Fig. 7).

Relationships between ρCB and dCB
The somewhat smaller slope (− 3.669) for the latter family of systems shows a slightly weaker relationship between ρCB and dCB, i.e., the length of CB bond, which is most likely due to the disturbing axial charge relocation in the acetylene unit compared to the single-plane delocation in the ethene unit.
When charges of the substituents q(X) are plotted against σp, F and R substituent constants [9], the situation is better than that observed for the CC bond length (see Fig. 8).

Relationships between q(X) and σp, F and R
In the case of substituent constants σp and R, these correlations are also better in the case of acetylene derivatives than in the case of ethene derivatives. However, the situation is reversed when the F substituent constant in considered instead. Interestingly, the presented data clearly show that for X-substituted ethene and acetylene derivatives, R2 values are the largest for the substituent constant F (0.840 and 0.766, respectively). On the contrary, for their boranyl counterparts, R2 values are the largest for the substituent constant σp (0.812 and 0.844, respectively). This result suggests that the electronic properties of the X substituent described by its charge, i.e., q(X), are best suited to the inductive effect of this substituent, but the presence of the boranyl group increases the proportion of resonance interactions.
It was particularly interesting to study the effect of changing the double bond in the X-substituted ethenes on the triple bond in the X-substituted acetylenes on the electronic state of both the X substituent and terminal hydrogen atom. These states can be sampled using the charges q(X) and q(H) as well as their changes upon the step XEnH→XAcH. The corresponding values are listed in Table 2.
It is easy to see that the XEnH→XAcH step, i.e., the replacement of the double bond in XEnH by the triple bond in XAcH, increases the charge of the substituent X on the absolute scale (i.e., Δq(X) > 0). The same effect is also visible for the terminal hydrogen atom (Δq(H) > 0). Thus, compared to the double bond in the ethylene unit, the triple bond in the acetylene unit additionally pulls the electron charge from both the X substituent and the hydrogen atom. It is worth noting that, as a consequence, the hydrogen atom in substituted acetylenes is more acidic than in substituted ethenes.
Like the relationships between dCC values (Fig. 4), where data for X-substituted acetylene and acetyleneborane are plotted against the appropriate data for the ethene derivatives, the relationships between q(X) values are also characterized by large R2 values (see Fig. 9).

Relationships between q(X)
Moreover, they are clearly larger (R2 ≥ 0.925) than for dCC. The appropriate linear regression equations are as follows: q(X)XAcH = 0.8352 q(X)XEnH + 0.0764 and q(X)\(^{\text {XAcBH}_{2}}\) = 0.8749 q(X)\(^{\text {XEnBH}_{2}}\) + 0.0680. In both cases, the slopes are less than 1.0, indicating that the acetylene moiety affects the charge of the substituent X weaker than the ethene moiety does. Moreover, since the slope for boranyl derivatives is slightly larger (0.875) than for the other systems (0.835), it may suggest that the boranyl functional group stimulates this increasing effect via resonance through the triple bond stronger than via a double bond.
The conductivity through a given system of bonds can also be studied by analyzing the value of the transferred charge from X to Y in the XRY system. This quantity is usually known as the charge transfer. Starting from the fact that the acceptor group Y cannot accept from donor X more charge than it donates, we have very recently proposed [26] calculating the value of the charge transfer by means of Eq. 5. Using this expression, it was interesting to compare the values of the transferred charge through double and triple bonds. This analysis was performed on the H2NRNO2 (R = En, Ac) systems containing a strongly electron-donating amino group on one end and a strongly electron-accepting nitro group on the other. The appropriate values of X and H charges as well as the transferred charge from the NH2 group to the NO2 group are shown in Table 3. The obtained results suggest that the charge transferred via the triple bond in H2NAcNO2 (0.122 au) is ca. 7% smaller than the charge transferred via the double bond in H2NEnNO2 (0.131 au).
Conclusions
The Hammett substituent constants do not describe properly substituent effects on CC bond length, neither in ethene nor in acetylene, nor in their boranyl derivatives. Relatively better are these relationships for CB bond lengths. We have found a much greater resistance against external perturbation in the case of CC triple bonds than in the case of CC double ones, so that in acetylene derivatives the substituent effect on bond length is approximately half of that observed in ethene derivatives. The substituent effects are then somewhat weakened when the BH2 boranyl group is at the opposite end of the molecule to the X substituent.
In the case of both the ethene and etheneborane derivatives, the relationships between the CC bond distances, dCC, and the charge density at the critical point of these bonds, ρCC, are well described by linear equations, whereas for their acetylene-based analogs lack of these dependences is observed, and is most probably due to an interaction of the planar structure of the substituent in xy plane with another π-electron bond in acetylene.
Compared to the double bond in the ethylene unit, the triple bond of the acetylene unit additionally draws an electron charge from both the X substituent and the terminal hydrogen atom, making this atom more acidic. The NH\(_{2}\!\rightarrow \textit {NO}_{2}\) charge transfer via the triple bond in H2N-C≡ C-NO2 is ca. 7% smaller than the amount of charge transferred via the double bond in H2N-CH=CH-NO2.
References
Hammett LP (1937) The effect of structure upon the reactions of organic compounds. Benzene derivatives. J Am Chem Soc 59:96–103
Hammett LP (1940) Physical organic chemistry. McGraw-Hill, New York
Jaffé HH (1953) A reëxamination of the Hammett equation. Chem Rev 53:191–261
Zuman P (1967) Substituent effects in organic polarography. Plenum Press, New York
Exner O (1972) . In: Chapman N (ed) Advances in linear free energy relationships. Plenum Press, London
Charton M (1973) Substituent effects in nonaromatic unsaturated systems. Progr Phys Org Chem 10:81–204
Katritzky AR, Topsom RD (1977) Infrared intensities: a guide to intramolecular interactions in conjugated systems. Chem Rev 77:639–658
Shorter J (1991) . In: Zalewski RI, Krygowski TM, Shorter J (eds) Similarity models in organic chemistry biochemistry and related fields. Elsevier, Amsterdam
Hansch C, Leo A, Taft RW (1991) A survey of Hammett substituent constants and resonance and field parameters. Chem Rev 91:165–195
Krygowski TM, Stȩpień BT (2005) Sigma- and pi-electron delocalization: focus on substituent effects. Chem Rev 105:3482–3512
Exner O, Böhm S (2006) Theory of substituent effects: recent advances. Curr Org Chem 10:763–778
Johnson CD (1973) The Hammett equation. Cambridge University Press, Cambridge
Piela L (2020) Ideas of quantum chemistry, 3rd edn. Elsevier, Amsterdam
Sadlej-Sosnowska N (2007) On the way to physical interpretation of Hammett constants: How substituent active space impacts on acidity and electron distribution in p-substituted benzoic acid molecules. Polish J Chem 81(5-6):1123–1134
Sadlej-Sosnowska N (2007) Substituent active region – a gate for communication of substituent charge with the rest of a molecule: monosubstituted benzenes. Chem Phys Lett 447:192–196
Krygowski TM, Sadlej-Sosnowska N (2011) Towards physical interpretation of Hammett constants: charge transferred between active regions of substituents and a functional group. Struct Chem 22:17–22
Powell MF, Peterson MR, Csizmadia IG (1983) Substituent effects on the acidity of the acetylenic proton: an ab initio study. J Mol Struct:, THEOCHEM 92:323–335
Furet P, Hallak G, Matcha RL, Fuchs R (1985) Substituent effects on acetylene stability. A comparison of STO-3G, 6-31G, 6-31G**, and 6-311G** calculations. Can J Chem 63:2990– 2994
Kamienska-Trela K, Biedrzycka Z, Machinek R, Knieriem B, Lüttke W (1984) Substituent effects on nuclear spin-spin carbon-carbon coupling constants in derivatives of acetylene. Org Magn Reson 22:317–322
Wiberg KB, Hammer JD, Zilm KW, Keith TA, Cheeseman JR, Duchamp JC (2004) NMR chemical shifts. Substituted acetylenes. J Org Chem 69:1086–1096
Parthiban S, Martin JML, Liebman JF (2002) The heats of formation of the haloacetylenes XCCY [X, Y = H, F, Cl]: basis set limit ab initio results and thermochemical analysis. Mol Phys 100:453–464
Rayne S, Forest K (2011) Thermochemistry of mono- and disubstituted acetylenes and polyynes at the Gaussian-4 level of theory. Comput Theor Chem 970:15–22
Rogers DW, Zavitsas AA, Matsunaga N (2005) G3(MP2) enthalpies of hydrogenation, isomerization, and formation of extended linear polyacetylenes. J Phys Chem A 109:9169–9173
Grabowski SJ, Walczak MA, Krygowski TM (2004) The substituent effect in ethylenes and acetylenes – AIM analysis. Chem Phys Lett 400:362–367
Bader RFW (1990) Atoms in molecules a quantum theory. Clarendon Press, Oxford
Jabłoński M, Krygowski TM (2020) Changes in electron structure of the triple bond in substituted acetylene and diacetylene derivatives. ChemPhysChem 21:1847–1857
Roman M, JCz Dobrowolski, Baranska M (2011) Theoretical modeling of molecular spectra parameters of disubstituted diacetylenes. J Chem Inf Model 51:283–295
Diederich F, Stang PJ, Tykwinski RR (2005) Acetylene chemistry: chemistry, biology, and material science. Wiley-VCH, Weinheim
Jabłoński M (2017) Strength of Si–H⋯B charge-inverted hydrogen bonds in 1-silacyclopent-2-enes and 1-silacyclohex-2-enes. Struct Chem 28:1697–1706
Jabłoński M (2018) Hydride-triel bonds. J Comput Chem 39:1177–1191
Jabłoński M, Krygowski TM (2020) Study of the influence of intermolecular interaction on classical and reverse substituent effects in para-substituted phenylboranes. New J Chem 44:9656–9670
Chai J -D, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Mardirossian N, Head-Gordon M (2017) Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol Phys 19 :2315–2372
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138
Parr RG, Yang W (1989) Density-Functional Theory of atoms and molecules. Oxford University Press, New York
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650–654
McLean AD, Chandler GS (1980) Contracted Gaussian basis sets for molecular calculations. I. second row atoms, Z= 11–18. J Chem Phys 72:5639–5648
Curtiss LA, McGrath MP, Blandeau J -P, Davis NE, Binning RC Jr, Radom L (1995) Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J Chem Phys 103:6104–6113
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Clark T, Chandrasekhar J, Spitznagel GW, von Ragué Schleyer P (1983) Efficient diffuse function-augmented basis sets for anion calculations. III.⋆ the 3-21+G basis set for first-row elements, Li–F. J Comput Chem 4:294–301
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE Jr, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian, Inc., Wallingford
Keith TA (2015) AIMAll (Version 15.05.18), TK Gristmill Software, Overland Park KS, USA, aim.tkgristmill.com
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Rousseau B, Peeters A, Van Alsenoy C (2001) Atomic charges from modified Voronoi polyhedra. J Mol Struct (Theochem) 538:235–238
Bickelhaupt FM, van Eikema Hommes NJR, Fonseca Guerra C, Baerends EJ (1996) The carbon-lithium electron pair bond in (CH3Li)n (n = 1, 2, 4). Organometallics 15:2923–2931
Fonseca Guerra C, Handgraaf J-W, Baerends EJ, Bickelhaupt FM (2004) Voronoi deformation density (VDD) charges: assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J Comput Chem 25:189–210
Stasyuk OA, Szatylowicz H, Fonseca Guerra C, Krygowski TM (2015) Theoretical study of electron-attracting ability of the nitro group: classical and reverse substituent effects. Struct Chem 26:905–913
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jabłoński, M., Krygowski, T.M. On differences in substituent effects in substituted ethene and acetylene derivatives and their boranyl analogs. Struct Chem 32, 285–296 (2021). https://doi.org/10.1007/s11224-020-01666-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01666-x