
Abstract
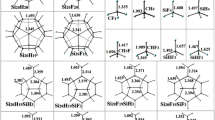
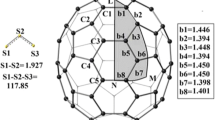
We applied density functional theory calculations to study reactions NH2 •+ SinHn fullerenes (n = 4, 6, 8, 10, 20, 24, 30, 36, and 50). The reactions between SinHn fullerenes and NH2 • radical are obtained to be exothermic, proceeding through a hydrogen-bonded prereactive complex. The resemblance between the structures of the reactants and the transition states indicates that the transition states appear earlier for the considered exothermic reactions. The calculated ΔH0 298 and ΔETS are in good correlation with the spherical excesses φi, showing upward trend with increasing curvature on silicon sites. The reactions of Si20H20 and Si20F20 with NH2 • and NF2 • radicals were also investigated. The reaction between Si20H20 and NF2 • radical also proceeds through a hydrogen-bonded prereactive complex. However, this reaction is endothermic, and as compared with the reaction of NH2 •+ Si20H20, the prereactive complex has stronger hydrogen bonding which leads to higher activation barrier. The reaction of Si20F20 and NF2 • radical as well as NH2 • radical is obtained to be endothermic, having significantly higher barriers via “late” transition states, in comparison to the reaction of NH2 •+ Si20H20.








Similar content being viewed by others

References
Fthenakis ZG, Havenith RWA, Menon M, Fowler PW (2007) Surface physics, nanoscale physics, low-dimensional systems-structural and electronic properties of the fullerene isomers of Si38: a systematic theoretical study. Phys Rev B 75:155435-1–155435-8
Ona O, Bazterra VE, Caputo MC, Facelli JC, Fuentealba P, Ferraro MB (2006) Modified genetic algorithms to model cluster structures in medium-sized silicon clusters: Si18−Si60. Phys Rev A 73:053203-1–05320311
Zhao J, Ma L, Wen B (2007) Lowest-energy endohedral fullerene structure of Si60 from a genetic algorithm and density-functional theory. J Phys Condens Matter 19:226208-1–226208-6
Belomion G, Therrien J, Smith A, Rao S, Twesten R, Chaieb S, Nayfeh MH, Wagner L, Mitas L (2002) Observation of a magic discrete family of ultrabright Si nanoparticles. Appl Phys Lett 80:841–843
Kumar V, Kawazoe Y (2003) Hydrogenated silicon fullerenes: effects of H on the stability of metal-encapsulated silicon clusters. Phys. Rev. Lett. 90:055502-1-4
Galashev AE (2008) Thermal instability of silicon fullerenes stabilized with hydrogen: computer simulation. Semiconductors 42:596–603
Zdetsis AD (2007) High-symmetry high-stability silicon fullerenes: a first-principles study. Phys Rev B 76:075402-1–075402-5
Karttunen AJ, Linnolahti M, Pakkanen TA (2007) Icosahedral polysilane nanostructures. J Phys Chem C 111:2545–2547
Earley CW (2000) Ab initio investigation of strain in group 14 polyhedrane clusters (M n H n : n = 4, 6, 8, 10, 12, 16, 20, 24). J Phys Chem A 104:6622–6627
Wang L, Li D, Yang D (2006) Fully exohydrogenated Si60 fullerene cage. Mol Simulat 32:663–666
Pichierri F, Kumar V, Kawazoe Y (2004) Exohedral functionalization of the icosahedral cluster Si20H20: a density functional theory study. Chem Phys Lett 383:544–548
Anafcheh M, Ghafouri R (2014) Exploring electronic properties of Si20-n H20−P n heterofullerenes (N = 1, 2, 5, and 10) based on NMR and NBO analysis: a DFT study. Phosphorus Sulfur Silicon Relat Elem 189:60–73
Anafcheh M, Ghafouri R, Hadipour NL (2012) 1H and 29Si NMR investigation of SinHn polysilanes with n < 60: a DFT study. Phys E 44:2099–2104
Anafcheh M, Ghafouri R (2014) Evaluation of on-cage phosphorus doping of hydrogenated silicon fullerenes: a computational study. Struct Chem 25:37–42
Anafcheh M, Ghafouri R (2014) (SiH)48X12 heterofullerenes with the group III and V dopants: a DFT prediction of geometry, stability, and electronic structure. J Clust Sci 25:505–515
Ghafouri R, Anafcheh M, Zahedi M (2014) Fully and partially exohydrogenated Si80 fullerene cage. Struct Chem 25:575–581
Ghafouri R, Ektefa F (2016) Exploring the mechanism of reactions of SiX3 and CX3 radicals with Si20X20 fullerenes (X = H, F): a density functional study. J Clust Sci 27:1719–1728
Demissy M, Lesclaux R (1980) A flash photolysis-laser resonance absorption study. J Am Chem Soc 102:2897–2902
Sun W, Yang L, Yu L, Saeys M (2009) Ab initio reaction path analysis for the initial hydrogen abstraction from organic acids by hydroxyl radicals. J Phys Chem A 113:7852–7860
Li QS, Lu RH (2002) Direction dynamics study of the hydrogen abstraction reaction CH2O + NH2 → CHO + NH3. J Phys Chem A 106:9446–9450
Mebel AM, Lin MC (1999) Prediction of absolute rate constants for the reactions of NH2 with alkanes from ab initio G2M/TST calculations. J Phys Chem A 103:2088–2096
Yu Y-X, Li S-M, Xu Z-F, Li Z-S, Sun C-C (1998) An ab initio study on the reaction NH2 + CH4 →NH3 + CH3. Chem Phys Lett 296:131–136
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120:215–241
Hariharan PC, Pople JA (1974) Accuracy of AH n equilibrium geometries by single determinant molecular orbital theory. Mol Phys 27:209–214
Zhang Y, Wu A, Xu X, Yan Y (2007) Geometric dependence of the B3LYP-predicted magnetic shieldings and chemical shifts. J Phys Chem A 111:9431–9437
Ghafouri R, Ektefa F (2015) Functionalization of carbon ad-dimer defective single-walled carbon nanotubes through 1,3-dipolar cycloaddition: a DFT study. Struct Chem 26:507–515
Anafcheh M, Ghafouri R (2014) Mono-and multiply-functionalized fullerene derivatives through 1, 3-dipolar cycloadditions: a DFT study. Phys E 56:351–356
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Gordon MS, Schmidt MW (2005) Advances in electronic structure theory: GAMESS a decade later. In: Dykstra CE, Frenking G, Kim KS, Scuseria GE (eds) Theory and applications of computational chemistry: the first forty years. Elsevier, Amsterdam
Pichierri F, Kumar V (2009) Geometries and electronic structures of phosphorous-doped silicon fullerenes: a DFT study. J Mol Struc-THEOCHEM 900:71–76
Zhang H, Liu P, Liu J-Y, Li Z-S (2013) Theoretical study and rate constant calculations for the reactions of SiHX3 with CF3 and CH3 radicals (X = F, Cl). J Mol Model 19:1515–1525
Jeffrey GA (1997) An introduction to hydrogen bonding. Oxford University Press, Oxford
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Para RD, Bulusu S, Zeng XC (2003) Cooperative effects in one-dimensional chains of three-center hydrogen bonding interactions. J Chem Phys 118:3499–3509
Hammond GS (1955) A correlation of reaction rates. J Am Chem Soc 77:334–338
Neretin IS, Lyssenko KA, Antipin MY, Slovokhotov YL, Boltalina OV, Troshin PA, Lukonin AY, Sidorov LN, Taylor R (2000) C60F18, a flattened fullerene: alias a hexa-substituted benzene. Angew Chem Int Ed 39:3273–3276
Ghafouri R, Anafcheh M (2013) Fully and partially exohydrogenated Si80 fullerene cage. J Fluor Chem 145:88–94
Gutsev GL (1993) A theoretical investigation of the electronic and geometrical structure of silicon fluorides SiF n and their anions SiF n −,n=1−6. Russ Chem Bull 42:36–45
Wang H, Wu L (2011) A density functional investigation of fluorinated silicon clusters. Chin J Chem 29:735–740
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Anafcheh, M., Naderi, F. & Ghafouri, R. Hydrogen-abstraction reactions of fully hydrogenated silicon fullerene cages with the amino radical: a density functional study. Struct Chem 29, 607–614 (2018). https://doi.org/10.1007/s11224-017-1057-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-1057-1