
Abstract
In the present research work, four new heterocyclic Schiff base ligands (1–4) were synthesized by the condensation of 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)phenol with salicylaldehyde derivatives in 1:1 molar ratio. The synthesized Schiff base ligands were allowed for complexation with Co(II), Ni(II), Cu(II), Zn(II) metal ions. The structure of the newly synthesized compounds (1–20) was elucidated with the help of various spectral and physicochemical techniques. Spectroscopic data confirm the tridentate nature of ligands which coordinate to the metal via deprotonated oxygen, azomethine nitrogen and thiol sulphur. Conductivity data showed the non-electrolytic nature of complexes. Furthermore, the synthesized compounds were evaluated for their in-vitro antimicrobial activity against four pathogenic bacterial strains and two pathogenic fungal strains. The observed results of microbial activity reveals that compound 3 and its complexes (13–16) were found most potent against the pathogenic strains. In addition, the anticancer activity of all the synthesized compounds was evaluated against human carcinoma cell lines i.e. HCT-116, DU145 and A549 using MTT assay. Among the tested compounds 12, 19, and 20 were found to show promising potency against the cancer cell lines. To rationalize the preferred modes of interaction of most potent compounds with the active site of human EGFR protein (PDB id: 5XGM), molecular docking studies were performed.
Graphical abstract

Similar content being viewed by others
Introduction
Cancer emerges as a serious health problem in all populations and ranks as the second most leading cause of death all over the world. The International Agency for Research on Cancer (IARC) in 2020 estimated 19.3 million new cancer cases and almost 10.0 million cancer deaths worldwide [1]. The projected death toll due to cancer is increasing at an alarming rate. It is estimated that 29.4 million people will have cancer in 2040 (WHO, 2020) [2]. Recent studies have shown that cancer patients are more likely to be infected with COVID-19 and serious side effects such as ARDS, liver injury, sepsis, renal insufficiency and myocardial injury are commonly observed in cancer patients with COVID‐19 [3]. The current therapeutic strategies used in the treatment of cancer involve chemotherapy, radiotherapy, surgery and immunotherapy either alone or in combination, but the constitutional treatment of cancer is based on chemotherapy which incorporates the use of various chemical entities of natural and synthetic origin [4]. The serendipitous discovery of cis-platin drug has established the importance of metal complexes in cancer cure [5]. But the clinical application of cis-platin is limited due to significant side effects such as bone marrow suppression, hearing problems, vomiting and kidney damage etc. [6, 7]. Therefore the development of novel metal-based drugs with no toxicity or very less toxicity for the treatment of cancer is nowadays attracting the attention of biochemists.
The coordination chemistry of Schiff base ligands still evokes much current interest and possesses a variety of applications [8,9,10,11,12,13,14]. Furthermore, it is also reported that the activity of Schiff base ligands gets increased when administered as metal complexes [15]. Schiff base complexes possess remarkable properties which make these compounds effective as stereospecific catalysts towards hydrolysis, oxidation, reduction and other transformations of inorganic and organic chemistry [16,17,18,19,20]. Heterocyclic ligands containing nitrogen as donor atom, especially 1,2,4-triazole have received much attention due to their huge applications in analytical chemistry [21,22,23,24]. 1,2,4-triazole possesses a wide variety of pharmacological activity including antimicrobial, anti-cancer, anti-diabetic, anti-tubercular, anxiolytics, anticonvulsants, etc. [25,26,27,28,29,30]. Schiff base ligands having triazole ring can be considered as “privileged structures” and are widely used due to their novel structural features. Transition metal complexes have effectively established them as good antimicrobial and anticancer agents [31, 32]. Therefore, the formation of transition metal chelates of triazole derived Schiff base ligands as new metallodrugs for the treatment of various diseases is found to have great potential.
Keeping in view the immense biological activity exhibited by 1,2,4-triazole motif and the potential chemistry of transition metals, we find it lively to join the chemistry of both moieties in a single molecular framework. In the recent study, we report the synthesis of some new Schiff base ligands derived from the condensation of 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)phenol with salicylaldehyde derivatives. Using these triazole containing Schiff base ligands Co(II), Ni(II), Cu(II), Zn(II) complexes were synthesized. Characterization of the synthesized compounds was done by 1H NMR, 13C NMR, FTIR, HRMS, XRD, ESR, TGA and SEM. The work was further extended to investigate the antimicrobial and anticancer activities of the synthesized compounds. Molecular docking studies on the most potent compounds were carried out to check the binding conformations.
Experimental
Materials
The entire chemicals employed in this research work were procured from Sigma Aldrich. The chemicals viz., 3-ethoxysalicylaldehyde (97%), 5-bromosalicylaldehyde (98%), 5-nitrosalicyladehyde (98%), 3,5-dibromosalicylaldehyde (98%), 4-hydroxybenzhydrazide (≥ 98%), Hydrazine hydrate (50–60%) and metal acetates solids i.e. Co(CH3COO)2·4H2O (98%), Ni(OCOCH3)2·4H2O (98%), Cu(CH3COO)2·H2O (≥ 98%), Zn(CH3COO)2·2H2O (≥ 98%) were used as such in the present study. The organic solvents were of analytical grade and used after drying with standard procedures [33]. The progress of the reaction and the purity of synthesized compounds were monitored regularly by TLC (thin layer chromatography).
Instrumentation
FTIR spectra were recorded on Perkin Elmer BX II spectrometer by using KBr matrix in the wavenumber range of 4000–400 cm−1. 1H and 13C NMR spectra of the compounds were recorded on Bruker Avance III (400 MHz) spectrometer by using DMSO as a solvent. Thermo Scientific FLASH 2000 CHN elemental analyser was used for calculating the percentages of C, H, and N. Melting points were measured by the capillary method using Thermo scientific apparatus and were reported uncorrected. Molar conductance was measured in DMF by utilising Systronics conductivity type bridge model-306. The mass spectra were obtained using SCIEX Triple TOF 5600 instrument by using methanol and DMSO as a solvent. The absorption spectra were recorded in DMF on a Varian cary-5000 spectrometer at room temperature. The X-ray analysis was carried out using a Rigaku table top X-ray diffractometer with a scan rate of 2 min−1 in the 2θ range of 10°–80°. TGA was recorded using SDT Q600 instrument at a heating rate of 0.1 to 100 °C/min. The electron spin resonance (ESR) studies of copper complexes were carried out on JES-FA200 spectrometer using tetracyanoethylene (TCNE) as an internal standard. Scanning electron microscopy (SEM) with energy dispersive X-ray analysis (EDAX) was carried out on a JEOL 7610F plus instrument and the micrographs were recorded in a dynamic nitrogen atmosphere.
Synthesis
Synthesis of 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)phenol precursor
The precursor was prepared as per the earlier reported method (Sahoo et al.) [34]. The identity of the synthesized compound was established after comparing its spectral data with the literature reported values.
Synthesis of Schiff base ligands (1–4)
To a methanolic solution of 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)phenol (0.624 g, 3 mmol) was added 3-ethoxysalicylaldehyde (0.498 g, 3 mmol)/5-bromosalicylaldehyde (0.603 g, 3 mmol)/5-nitrosalicylaldehyde (0.624 g, 3 mmol)/3,5-dibromosalicylaldehyde (0.839 g, 3 mmol) (Scheme 1). The reaction mixture was refluxed for 4–5 h at room temp. The progress of the reaction was monitored regularly by TLC. The coloured products so obtained were filtered, dried and recrystallized with the mixture of methanol and chloroform (1:1, v/v) to obtain pure products.

Synthesis of Schiff base ligands (1–4) and their transition metal complexes (5–20) (i) methanol, reflux, 4–5 h. (ii) methanol, metal acetates, stirring, 2–3 h
2-ethoxy-6-(((3-(4-hydroxyphenyl)-5-mercapto-4H-1,2,4-triazol-4-yl)imino)methyl)phenol, HL1 (1) Pale yellow, Yield: 81%, M.p.: 94–96 °C, Conductivity (Ohm−1 cm2 mol−1) in DMF: 14.09, Anal. calcd for C17H16N4O3S (%): C: 57.29, H: 4.52, N: 15.72, Anal. found (%): C: 57.02, H: 4.32, N: 15.16. HRMS m/z: calcd for C17H17N4O3S [M + H]+: 357.1016, found: 357.1021. 1H NMR (400 MHz, DMSO) δ: 14.04 (s, 1H, S–H), 10.10 (s, 1H, O–Hb), 9.93 (s, 1H, O–Ha), 9.67 (s, 1H, CH=N), 7.70 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 7.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 7.17 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 8.1 Hz, 2H), 4.17 (q, J = 8 Hz, 2H, CH2), 1.52 (t, J = 8.0 Hz, 3H, CH3). 13C NMR (100 MHz, DMSO) δ: 163.71, 162.38, 160.02, 149.28, 149.03, 147.81, 130.46, 120.04, 119.74, 119.05, 117.20, 116.63, 115.93, 64.83, 15.02. IR (KBr, cm−1): 3288 (O–Hb), 3121 (O-Ha), 2362 (S–H), 1603 (C=N).
4-bromo-2-(((3-(4-hydroxyphenyl)-5-mercapto-4H-1,2,4-triazol-4-yl)imino)methyl)phenol, HL 2 (2) Orange, Yield: 79%, M.p.: 97–99 °C, Conductivity (Ohm−1 cm2 mol−1) in DMF: 13.53. Anal. calcd for C15H11BrN4O2S (%): C: 46.05, H: 2.83, N: 14.32, Anal. found (%): C: 46.01, H: 2.62, N: 14.29. HRMS m/z: calcd for C15H12BrN4O2S [M + H]+: 390.9859, found: 390.9889. 1H NMR (400 MHz, DMSO) δ: 14.04 (s, 1H, S–H), 10.83 (s, 1H, O–Hb), 10.11 (s, 1H, O–Ha), 9.96 (s, 1H, CH=N), 7.89 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 8.7 Hz, 2H), 7.59 (dd, J = 8.8, 2.4 Hz, 1H), 6.99 (s, 1H), 6.89 (d, J = 8.7 Hz, 2H). 13C NMR (100 MHz, DMSO) δ: 162.31, 161.03, 160.07, 158.13, 149.49, 136.99, 130.59, 129.20, 121.01, 119.55, 116.53, 115.92, 111.33. IR (KBr, cm−1): 3275 (O-Hb), 3166 (O–Ha), 2363 (S–H), 1615 (C=N).
2-(((3-(4-hydroxyphenyl)-5-mercapto-4H-1,2,4-triazol-4-yl)imino)methyl)-4-nitrophenol, HL3 (3) Yellow, Yield: 87%, M.p.: 100–102 °C, Conductivity (Ohm−1 cm2 mol−1) in DMF: 13.97. Anal. calcd for C15H11N5O4S (%): C: 50.42, H: 3.10, N: 19.60, Anal. found (%): C: 50.29, H: 3.09, N: 19.07. HRMS m/z: calcd for C15H12N5O4S [M + H]+: 358.0605, found: 358.0629. 1H NMR (400 MHz, DMSO) δ: 14.08 (s, 1H, S–H), 12.09 (s, 1H, O–Hb), 10.21 (s, 1H, O–Ha), 10.12 (s, 1H, CH=N), 8.64 (d, J = 2.9 Hz, 1H), 8.30 (dd, J = 9.1, 2.9 Hz, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.18 (d, J = 9.2 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO) δ: 164.07, 162.27, 160.11, 159.38, 149.67, 140.47, 130.70, 129.51, 123.08, 119.60, 117.91, 116.47, 115.91. IR (KBr, cm−1): 3297 (O–Hb), 3135 (O–Ha), 2364 (S–H), 1610 (C=N).
2,4-dibromo-6-(((3-(4-Hydroxyphenyl)-5-mercapto-4H-1,2,4-triazol-4-yl)imino)methyl)phenol, HL4 (4) Yellow, Yield: 82%, M.p.: 93–95 °C, Conductivity (Ohm−1 cm2 mol−1) in DMF: 12.98. Anal. calcd for C15H10Br2N4O2S (%): C: 38.32, H: 2.14, N: 11.92, Anal. found (%): C: 38.20, H: 2.08, N: 11.86. HRMS m/z: calcd for C15H11Br2N4O2S [M + H]+: 468.8964, found: 468.8997. 1H NMR (400 MHz, DMSO) δ: 14.14 (s, 1H, S–H), 12.28 (s, 1H, O–Hb), 10.15 (s, 1H, O–Ha), 10.00 (s, 1H, CH=N), 8.02 (d, J = 2.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.66 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO) δ: 163.01, 162.57, 160.19, 154.87, 149.47, 138.86, 130.98, 130.59, 122.28, 116.23, 116.03, 113.76, 111.88. IR (KBr, cm−1): 3241 (O–Hb), 3130 (O–Ha), 2363 (S–H), 1609 (C=N).
Synthesis of transition metal complexes (5–20)
The metal complexes (5–20) were prepared by combining hot methanolic solution (20 mL) of synthesized Schiff base ligands (1–4) with a methanolic solution (20 mL) of metal acetates in 1:1 molar ratio. The reaction mixture was stirred vigorously for 2–3 h at room temperature. The different coloured products so obtained were filtered and finally dried over vacuum to obtain pure complexes (Scheme 1).
Co(L1)(H2O)(CH3COO) (5) Light red, Yield: 70%, M.p.: 160–163 °C, Conductivity (Ohm−1 cm2 mol−1) in DMF: 13.05. Anal. calcd for C19H20CoN4O6S (%): C: 46.44, H: 4.10, N: 11.40, Anal. found (%): C: 46.40, H: 4.01, N: 11.29. HRMS m/z: calcd for C19H21CoN4O6S [M + H]+: 492.0508, found: 492.0514. IR (KBr, cm−1): 3422 (O–Ha, M–H2O), 2347 (S–H), 1745 (M–CH3COO), 1593 (C=N), 525 (M–N), 475 (M–O), 435 (M–S).
Ni(L1)(H2O)(CH3COO) (6) Brown, Yield: 72%, M.p.: 170–172 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 12.76. Anal. calcd for C19H20NiN4O6S (%): C: 46.46, H: 4.10, N: 11.41, Anal. found (%): C: 46.34, H: 3.79, N: 11.11. HRMS m/z: calcd for C19H21NiN4O6S [M + H]+: 491.0530, found: 491.0520. IR (KBr, cm−1): 3393 (O–Ha, M–H2O), 2345 (S–H), 1736 (M–CH3COO), 1593 (C=N), 539 (M–N), 482 (M–O), 440 (M–S).
Cu(L1)(H2O)(CH3COO) (7) Dark brown, Yield: 69%, M.p.: 165–168 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.5. Anal. calcd for C19H20CuN4O6S (%): C: 46.01, H: 4.06, N: 11.30, Anal. found (%): C: 45.98, H: 3.86, N: 11.30. HRMS m/z: calcd for C19H21CuN4O6S [M + H]+: 496.0472, found: 496.0436. IR (KBr, cm−1): 3422 (O–Ha, M–H2O), 2346 (S–H), 1743 (M–CH3COO), 1595 (C=N), 533 (M–N), 471 (M–O), 431 (M–S).
Zn(L1)(H2O)(CH3COO) (8) Pale yellow, Yield: 75%, M.p.: 164–166 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 13.79, Anal. calcd for C19H20ZnN4O6S (%): C: 45.84, H: 4.05, N: 11.25, Anal. found (%): C: 45.22, H: 3.83, N: 11.21. HRMS m/z: calcd for C19H21ZnN4O6S [M + H]+: 497.0468, found: 497.0316. 1H NMR (400 MHz, DMSO) δ: 13.05 (s, 1H, S–H), 9.89 (s, 1H, O–Ha), 8.09 (s, 1H, CH=N), 7.79 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 8.6, 7.1 Hz, 1H), 6.88 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 7.4 Hz, 1H), 3.93 (q, J = 7.5 Hz, 2H, CH2), 1.85 (s, 3H, CH3COO), 1.21 (t, J = 8 Hz, 3H, CH3). 13C NMR (100 MHz, DMSO) δ: 172.72, 164.34, 162.65, 161.31, 151.34, 149.41, 148.51, 130.72, 122.91, 120.91, 119.25, 117.69, 116.79, 116.09, 65.04, 22.21, 15.34. IR (KBr, cm−1): 3421 (O–Ha, M–H2O), 2344 (S–H), 1743 (M–CH3COO), 1596 (C=N), 538 (M–N), 485 (M–O), 436 (M–S).
Co(L2)(H2O)(CH3COO) (9) Orange red, Yield: 73%, M.p.: 180–182 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 12.02, Anal. calcd for C17H15BrCoN4O5S (%): C: 38.80, H: 2.87, N: 10.65, Anal. found (%): C: 38.67, H: 2.65, N: 10.65, HRMS m/z: calcd for C17H16BrCoN4O5S [M + H]+: 525.9351, found: 525.8321, IR (KBr, cm−1): 3400 (O–Ha, M–H2O), 2349 (S–H), 1730 (M–CH3COO), 1602 (C=N), 530 (M–N), 458 (M–O), 430 (M–S).
Ni(L2)(H2O)(CH3COO) (10) Dark red, Yield: 71%, M.p.: 165–168 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.7, Anal. calcd for C17H15BrNiN4O5S (%): C: 38.82, H: 2.87, N: 10.65, Anal. found (%): C: 38.81, H: 2.09, N: 10.41. HRMS m/z: calcd for C17H16BrNiN4O5S [M + H]+: 524.9373, found: 524.9300. IR (KBr, cm−1): 3400 (O–Ha, M–H2O), 2349 (S–H), 1746 (M–CH3COO), 1604 (C=N), 533 (M–N), 464 (M–O), 431 (M–S).
Cu(L2)(H2O)(CH3COO) (11) Brown red, Yield: 70%, M.p.: 174–176˚C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 13.16, Anal. calcd for C17H15BrCuN4O5S (%): C: 38.46, H: 2.85, N: 10.55, Anal. found (%): C: 38.01, H: 2.69, N: 10.49. HRMS m/z: calcd for C17H16BrCuN4O5S [M + H]+: 529.9315, found: 529.9293. IR (KBr, cm−1): 3377 (O–Ha, M–H2O), 2346 (S–H), 1740 (M–CH3COO), 1603 (C=N), 529 (M–N), 462 (M–O), 438 (M–S).
Zn(L2)(H2O)(CH3COO) (12) Yellow, Yield: 76%, M.p.: 182–184 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 12.61. Anal. calcd for C17H15BrZnN4O5S (%): C: 38.33, H: 2.84, N: 10.52, Anal. found (%): C: 38.02, H: 2.35, N: 10.47. HRMS m/z: calcd for C17H16BrZnN4O5S [M + H]+: 530.9311, found: 530.9233. 1H NMR (400 MHz, DMSO) δ: 13.11 (s, 1H, S–H), 9.94 (s, 1H, O–Ha), 8.07 (s, 1H, CH=N), 7.85 (d, J = 3.4 Hz, 1H), 7.59 (d, J = 8.6 Hz, 2H), 7.28 (dd, J = 8.2, 3.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 2H), 6.63 (d, J = 8.1 Hz, 1H), 1.81 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 173.08, 163.61, 162.04, 160.22, 159.44, 149.77, 137.34, 130.70, 129.94, 122.32, 119.87, 116.98, 116.24, 112.72, 22.99. IR (KBr, cm−1): 3412 (O–Ha, M–H2O), 2349 (S–H), 1749 (M–CH3COO), 1607 (C=N), 533 (M–N), 488 (M–O), 447 (M–S).
Co(L3)(H2O)(CH3COO) (13) Dark green, Yield: 71%, M.p.: 160–162 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 13.14. Anal. calcd for C17H15CoN5O7S (%): C: 41.47, H: 3.07, N: 14.22, Anal. found (%): C: 41.38, H: 2.59, N: 14.11. HRMS m/z: calcd for C17H16CoN5O7S [M + H]+: 493.0097, found: 493.0079. IR (KBr, cm−1): 3411 (O–Ha, M–H2O), 2343 (S–H), 1735 (M–CH3COO), 1605 (C=N), 539 (M–N), 456 (M–O), 427 (M–S).
Ni(L3)(H2O)(CH3COO) (14) Green, Yield: 74%, M.p.: 163–165 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 13.56. Anal. calcd for C17H15NiN5O7S (%): C: 41.49, H: 3.07, N: 14.23, Anal. found (%): C: 41.38, H: 2.89, N: 14.14. HRMS m/z: calcd for C17H16NiN5O7S [M + H]+: 492.0118, found: 492.0092. IR (KBr, cm−1): 3404 (O–Ha, M–H2O), 2350 (S–H), 1751 (M–CH3COO), 1604 (C=N), 524 (M–N), 463 (M–O), 431 (M–S).
Cu(L3)(H2O)(CH3COO) (15) Dark brown, Yield: 66%, M.p.: 178–180 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.07, Anal. calcd for C17H15CuN5O7S (%): C: 41.09, H: 3.04, N: 14.09, Anal. found (%): C: 41.09, H: 2.49, N: 14.07. HRMS m/z: calcd for C17H16CuN5O7S [M + H]+: 497.0061, found: 497.0048. IR (KBr, cm−1): 3402 (O–Ha, M–H2O), 2351 (S–H), 1753 (M–CH3COO), 1604 (C=N), 511 (M–N), 467 (M–O), 434 (M–S).
Zn(L3)(H2O)(CH3COO) (16) Light yellow, Yield: 72%, M.p.: 171–173 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.22. Anal. calcd for C17H15ZnN5O7S (%): C: 40.94, H: 3.03, N: 14.04, Anal. found (%): C: 40.93, H: 2.67, N: 14.01. HRMS m/z: calcd for C17H16ZnN5O7S [M + H]+: 498.0056, found: 498.0053. 1H NMR (400 MHz, DMSO) δ: 13.22 (s, 1H, S–H), 10.09 (s, 1H, O–Ha), 8.38 (s, 1H, CH=N), 7.97 (d, J = 8.5 Hz, 1H), 7.84 (dd, J = 8.2, 4.1 Hz, 1H), 7.43 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.9 Hz, 2H), 6.60 (d, J = 8.1 Hz, 1H), 1.82 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 175.38, 167.13, 165.39, 163.66, 161.40, 149.88, 141.60, 137.53, 130.81, 129.83, 120.06, 118.63, 116.80, 116.28, 22.78. IR (KBr, cm−1): 3410 (O–Ha, M–H2O), 2344 (S–H), 1736 (M-CH3COO), 1607 (C=N), 516 (M–N), 496 (M–O), 429 (M–S).
Co(L4)(H2O)(CH3COO) (17) Green, Yield: 73%, M.p.: 177–179 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.7. Anal. calcd for C17H14Br2CoN4O5S (%): C: 33.74, H: 2.33, N: 9.26, Anal. found (%): C: 33.57, H: 2.09, N: 9.01. HRMS m/z: calcd for C17H15Br2CoN4O5S [M + H]+: 602.8384, found: 602.8380. IR (KBr, cm−1): 3368 (O–Ha, M–H2O), 2342 (S–H), 1742 (M–CH3COO), 1596 (C=N), 537 (M–N), 480 (M–O), 441 (M–S).
Ni(L4)(H2O)(CH3COO) (18) Brown, Yield: 76%, M.p.: 163–165 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 13.09. Anal. calcd for C17H14Br2NiN4O5S (%): C: 33.76, H: 2.33, N: 9.26, Anal. found (%): C: 33.54, H: 2.12, N: 9.27. HRMS m/z: calcd for C17H15Br2NiN4O5S [M + H]+: 602.8478, found: 602.8464. IR (KBr, cm−1): 3399 (O–Ha, M–H2O), 2348 (S–H), 1759 (M–CH3COO), 1596 (C=N), 536 (M–N), 457 (M–O), 428 (M–S).
Cu(L4)(H2O)(CH3COO) (19) Brown red, Yield: 73%, M.p.: 162–164 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 14.08. Anal. calcd for C17H14Br2CuN4O5S (%): C: 33.49, H: 2.31, N: 9.19, Anal. found (%): C: 33.02, H: 2.02, N: 9.01. HRMS m/z: calcd for C17H15Br2CuN4O5S [M + H]+: 607.8420, found: 607.8357. IR (KBr, cm−1): 3432 (O–Ha, M–H2O), 2346 (S–H), 1745 (M–CH3COO), 1596 (C=N), 533 (M–N), 469 (M–O), 432 (M–S).
Zn(L4)(H2O)(CH3COO) (20) Orange yellow, Yield: 78%, M.p.: 167–169 °C, Conductivity: (Ohm−1 cm2 mol−1) in DMF: 12.05. Anal. calcd for C17H14Br2ZnN4O5S (%): C: 33.39, H: 2.31, N: 9.16, Anal. found (%): C: 33.21, H: 2.12, N: 9.11. HRMS m/z: calcd for C17H15Br2ZnN4O5S [M + H]+: 608.8416, found: 608.7999. 1H NMR (400 MHz, DMSO) δ: 13.13 (s, 1H, S–H), 10.00 (s, 1H, O–Ha), 8.18 (s, 1H, CH=N), 7.76 (d, J = 2.8 Hz, 1H), 7.64 (d, J = 2.5 Hz, 1H), 7.58 (d, J = 8 Hz, 2H), 6.89 (d, J = 8.2 Hz, 2H), 1.84 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 173.75, 163.84, 163.21, 160.76, 157.10, 149.57, 138.86, 130.98, 130.59, 122.93, 116.23, 116.03, 113.99, 112.54, 24.28. IR (KBr, cm−1): 3422 (O–Ha, M–H2O), 2346 (S–H), 1739 (M–CH3COO), 1598 (C=N), 533 (M–N), 458 (M–O), 430 (M–S).
Pharmacology
Antimicrobial activity assay
The in-vitro antimicrobial activity of all the synthesized compounds (1–20) was assessed against four bacterial strains and two fungal strains. Norfloxacin and Fluconazole were used as positive control for bacteria and fungi [35]. DMSO was used as a negative control. The synthesized compounds were assessed against two Gram-positive bacteria Bacillus subtilis (NCIM 2063) and Staphylococcus aureus (MTCC 2901), two Gram-negative bacteria Pseudomonas aeruginosa (MTCC 424) and Escherichia coli (MTCC 732). The antifungal activity was assessed against two fungal strains namely Aspergillus niger (MTCC 9933) and Candida albicans (MTCC 227). The pathogens were collected from the Department of Bio and Nano Technology, GJUS&T, Hisar.
Compounds concentration
The stock solutions of concentration 1000 µg/mL were prepared by dissolving 5 mg of the sample compounds in 5 mL of DMSO. 1 mL of the above prepared stock solution was mixed with 9 mL of DMSO to get a stock solution of concentration 100 µg/mL [36].
Subculture and preservation of pathogens
The bacteria were subcultured on the NB (nutrient broth) and the fungi were subcultured on the PDB (potato dextrose broth) which were prepared by dissolving NB (1.3 g) and PDB (2.4 g) in 100 mL of distilled water. The resulting mixture was autoclaved at 15 psi for 30 min. The prepared cultures were incubated at 37 °C for 24 h in case of antibacterial culture and at 27 °C for seven days in case of antifungal culture [35].
Determination of MIC values
The serial dilution method was employed for carrying out the antimicrobial activity [37,38,39,40]. In this method, 1 mL of stock solution was added to the test tube containing 1 mL of broth and diluted serially up to 3.125 µg/mL concentrations. After dilution, the subcultured bacteria and fungi were impregnated in each test tube. Then, the test tubes were kept in an incubator for a fixed time to develop the culture. Pathogens growth was checked visually and spectrophotometrically. The MICs (minimum inhibitory concentration) value for tested compounds was determined. The obtained values were compared to those of reference drugs. The overall process was repeated twice to obtain more accurate and precise results.
Anticancer activity assay
The in-vitro anticancer activity of the synthesized compounds (1–20) was assessed on the basis of measurement of inhibition of cell lines in 96-well microtitre plates by cell mediated reduction of tetrazolium salt to water insoluble formazan crystals using Paclitaxel as a standard. The cytotoxic evaluation was performed against a panel of three cell lines namely: HCT-116 (ATCC No. CCL-247) derived from human colon cancer cell line, DU145 (ATCC No. HTB-81) derived from human prostate cancer cell line, A549 (ATCC No. CCL-185) derived from human alveolar adenocarcinoma epithelial cell line, using the MTT assay [41]. To study the effects of synthesized compounds on the viability of human cancer cell lines, the absorbance was noted at 595 nm using a micro plate reader (InfiniteM200, Tecan, Mannedorf, Switzerland). The effect of test samples on the procreation of cells was expressed in the form of % inhibition and is calculated as
From the dose–response curves, IC50 values of the compounds were reported as average of three independent experiments. The IC50 value is expressed in the form of mean ± standard deviation.
Molecular docking
The crystal structure of the human epidermal growth factor receptor (EGFR) was retrieved from the RCSB protein data bank (https://www.rcsb.org) and was encoded with PDB code 5XGM [42]. The complexes bound to the receptor, such as non-essential water molecules and any inhibitors were removed before the docking run. Hex 8.0.0 which is an interactive molecular graphics program (http://www.loria.fr) and is used to perform rigid docking to compute possible interactions of 5XGM protein with potent compounds. During docking the following parameters were used: correlation type − shape + electrostatics, FFT mode—3D, post processing- mm energies, grid dimension − 0.6, receptor range − 180, ligand range − 180, twist range − 360, distance range − 40. After docking, the detailed protein–ligand visualization and comparison of the docked sites of target proteins and ligands were done by Chimera 1.14 and Ligplot software [43, 44].
Results and discussion
Chemistry
Schiff base ligands (1–4) were synthesized by the condensation of 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)phenol with salicylaldehyde derivatives. The solid products so obtained have different colours owing to the presence of a chromophoric group in the molecule. Moreover, the treatment of synthesized compounds with corresponding metal acetates leads to formation of new complexes (5–20). Various spectral (1H, 13C NMR, FTIR and HRMS) and physicochemical techniques (XRD, ESR, TGA and SEM) were utilised for the characterization of synthesized compounds. The synthesized complexes (5–20) are coloured solids which are soluble in polar aprotic solvents like DMF, DMSO. The melting point of synthesized complexes was higher than those of Schiff base ligands, which means the complexes are more stable as compared to ligands. The molar conductivity values of synthesized compounds lie between 12 - 15 (Ohm−1 cm2 mol−1), which indicate that the complexes are non-electrolytic in nature.
FTIR spectra
The FTIR spectra of synthesized compounds (1–20) were recorded over the wavenumber range of 4000–400 cm−1. Comparison between infrared absorption frequencies of ligands and their respective metal complexes helps in understanding the binding modes of the ligand with the metal. The most explicit IR bands recorded for the synthesized compounds with the probable assignment are given in Table 1. The band appearing at 1615–1603 cm−1 in the IR spectra of ligands is due to ν(C=N) group which shifted to lower frequency region 1607–1593 cm−1 on complexation with metal atom, these shifting in spectra justified that lone pair of nitrogen is involved in bonding with the metal centre [45,46,47]. A band at 3297–3241 cm−1 is observed due to ν(O–Hb) group which gets disappeared in complexes due to deprotonation of phenolic O–H on complexation with the metal atom. The bands appearing in ligands in the range of 2364–2362 cm−1 are due to ν(S–H) group [48] which gets shifted to slightly lower values in complexes, due to the participation of sulphur in bonding with the metal atom. Thiol sulphur coordinates to metal without deprotonation. Some new bands appears at 496–456 cm−1 [due to ν(M–O)] and 447–427 cm−1 [due to ν(M-S)] [49]. The coordinated acetate molecule in complexes was ascertained by the presence of a band in the range of 1759–1730 cm−1 [50]. A new broad band near 3432–3368 cm−1 in complexes is due to the coordinated water molecule and ν(O–Ha) group. Furthermore, some new bands in complexes were observed in the range 539–511 cm−1 which are due to ν(M–N) [51]. The above results imply that ligands behave in a tridentate manner and the metal complexes have pentacoordinated geometry.
1H NMR spectra
1H NMR spectra of the synthesized Schiff base ligands (1–4) and their corresponding zinc (II) complexes were recorded in DMSO-d6 at 400 MHz. The spectra of compound (2) and its corresponding zinc complex (12) are depicted in Fig. 1. The 1H NMR spectra of synthesized ligands showed one sharp singlet in the range of δ 12.28–10.10 ppm (due to O–Hb proton) [52] which got disappeared in complexes due to involvement of oxygen in complexation. Another singlet for ligands was observed in the range of δ 14.14–14.04 ppm (due to S–H proton) [52], which shift to slightly lower values on complexation with metal atom. The ligands showed another singlet in the range of δ 10.21–9.93 ppm (due to another O-Ha proton), which remained very less changed in complexes. The azomethine (H–C=N) peak of ligands appeared in the range of δ 10.12–9.67 ppm. In case of complexes, it undergoes upfield shifting up to 8.07 ppm, which clearly indicates the confinement between metal ion and imine nitrogen. The upfield shifting is due to chelation [53]. The aromatic protons in ligands appeared at δ 8.64–6.88 ppm which remained unaffected or very less affected in complexes with a slight change in chemical shift values. In the spectra of complexes, a new singlet of three protons appeared at δ 1.84–1.81 ppm, which further confirms the presence of acetate group in complexes. The observed NMR data were found to be in good correlation with the expected values.

1H NMR spectra of compounds A HL2 (2) and B [Zn(L2)(H2O)(CH3COO)] (12)
13C NMR spectra
In the 13C NMR spectra of ligands, (1–4) the peak for azomethine carbon atom appeared at δ 159.38–149.28 ppm which showed downfield shifting on complexation, suggesting that nitrogen atom is involved in coordination with the metal ion [50]. In compound (1) extra peaks at δ 64.83 ppm and δ 15.02 ppm were observed due to the presence of an ethoxy group in the aliphatic region. Two peaks at δ 164.07–162.31 ppm and δ 162.57–161.03 ppm were observed due to carbon atom attached to oxygen and sulphur which was shifted downfield in complexes, thus indicating the involvement of oxygen and sulphur in complexation. But the peaks appearing at δ 160.19–160.02 ppm in ligands remains unchanged on complexation, which showed that the O-Ha group attached with this carbon is not involved in complexation. Two new peaks arising at δ 175.38–172.72 ppm and δ 24.28–22.21 ppm, due to the presence of a coordinated acetate group in complexes. The 13C NMR data confirm the authenticity of the purposed structures.
Mass spectra
Mass spectrometry plays a valuable role in determining the molecular weight and stoichiometry of the newly synthesized compounds. The molecular ion peak of ligand (3) appeared at m/z 358.0629 which was same as the estimated value. Similarly, for ligand (4) the molecular ion peak appeared at m/z 468.8997, while the signals at m/z 470.8977 and 472.8955 were assigned to (M + H + Br79) and (M + H + Br81) isotopes. In case of complex (14) molecular ion peak appeared at m/z 492.0092. In case of complex (18) molecular ion peak appeared at m/z 602.8464 and the other signals observed at m/z 604.8502 and 606.8515 were due to (M + H + Br79) and (M + H + Br81) isotopes [54]. The mass spectral data results were found in good agreement with theoretical values and support the formation of [M(L1−4)(H2O)(CH3COO)] type complexes in 1:1 stoichiometry.
Electronic spectra
The electronic transitions involved in the ligands (1–4) and their corresponding transition metal complexes (5–20) were recorded in DMF over the range 200–800 nm. The synthesized ligands showed absorption bands around 265–296 nm due to intra-ligand π → π*transitions. Moreover, the absorption bands appearing in the range of 319–370 nm are due to the occurrence of n → π* transitions. These bands shift to a slightly higher wavelength on complexation. For all the complexes n → π* transitions are red shifted, due to coordination of azomethine nitrogen to the central metal ion. For the Ni(II) complexes, electronic spectrum shows bands in the range of 757–798, 607–630, 345–388 nm which are typical for a pentacoordinated square pyramidal Ni(II) complex and could be assigned to d-d transitions. For Cu(II) centres the visible spectra exhibits broad band in the range of λmax at 628–655 nm which are specifically consistent for five-coordinate geometry and are correlated to d-d transition of Cu(II) ions. Zinc(II) complexes do not show any d-d transitions. In case of complexes, some new bands appeared in the range of 416–447 nm, which were attributed to ligand to metal charge transfer [55].
Molar conductivity studies
The molar conductivity studies of synthesized compounds (1–20) were undertaken using a digital conductivity meter at room temperature and using DMF as a solvent. The DMF solution of synthesized complexes (10–3 M) exhibited lower molar conductance values (12–15 Ω−1 cm2 mol−1), which supports the non-electrolytic nature of complexes [56].
X-ray diffraction studies
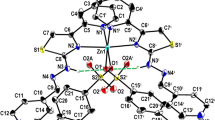
Powder X-ray diffraction method is one of the primary techniques used by solid-state chemists to explore the physicochemical parameters of synthesized materials. The prepared ligand (2) and their metal complexes (9–11) were analysed by the use of this technique over the 2θ range 10°–80° at a wavelength of 1.5406Ȧ. The obtained X-ray diffraction patterns are shown in Fig. 2. Schiff base ligand (2) showed the peaks at 15.42°, 16.46°, 18.14°, 20.64°, 24.18°, 25.92°, 27.28°, 29.92°, 35.98°, 41.34°. The two very important characteristic peaks appearing in Schiff base ligand at 2θ = 18.14° and 20.64° confirms the formation of Schiff base imine [57]. The observed diffraction pattern confirms that the compound is semi-crystalline in nature. The peaks for cobalt complex (9) were observed at 12.18°, 15.12°, 18.04°, 22.78°, 29.38°, 32.46°, 35.22°, 38.48°, 41.88°. The peaks for nickel complex (10) existed at 17.08°, 18.18°, 19.82°, 21.44°, 22.28°, 23.4°, 24.02°, 26.76°, 28.28°, 32.16°, 33.5°, 35.72°, 36.14°, 40.3°. Copper complex (11) showed the peaks at 12.74°, 16.42°, 19.64°, 21.7°, 23.28°, 25.82°, 26.96°, 27.92°, 29.38°, 30.9°, 32.3°, 35.2°, 37.6°, 39.32°, respectively. The diffraction pattern of the ligand is completely different from metal complexes, thus indicating the quantum confinement of ligands by metal ions [58]. The XRD data observed for complexes confirm the dominant amorphous nature with very less crystallinity.

XRD diagrams of A [HL2] (2), B [Co(L2)(H2O)(CH3COO)] (9), C [Ni(L2)(H2O)(CH3COO)] (10) and D [Cu(L2)(H2O)(CH3COO)] (11)
Electron spin resonance studies
The ESR spectra of copper complexes give information about the geometry and bonding sites. The solid-state X-band ESR spectrum of copper complex (7) was recorded in powder form at room temperature (300 K) and the micrograph obtained is presented in Fig. 3. The values of g║ and g┴ in case of Cu(II) complex lies at 2.15 and 2.09 which holds in good agreement with the condition g║ > g┴ > 2.0023. Since complexes are pentacoordinated, therefore two basic configurations with five coordinated geometry are square pyramidal and trigonal bipyramidal. The two configurations are characterised by the ground state dx2─y 2 and dz2, respectively. For systems with g║ > g┴ the geometry is square pyramidal, while systems with g║ < g┴˔ the geometry is trigonal bipyramidal [59, 60]. From the observed results it can be concluded that there exists a square pyramidal geometry around the Cu(II) metal ion with unpaired electron lying mainly in the dx2─ y2 ground state orbital. The gaverage and G values were computed from the given equations

The X-band ESR spectrum of complex [Cu(L1)(H2O)(CH3COO)] (7)
From the above calculations, the value of gaverage and G was found to be 2.11 and 1.68. According to Hathway and Billing [61, 62], if the value of G > 4.0 then we can consider negligible exchange interaction between Cu(II) centres in the solid state and if the value of G < 4.0 there will be significant exchange interactions. Here, the value of G was found to be 1.68, which indicates that exchange interactions are operative. By using the g-vlaue, magnetic moment can be calculated using the following equation.
The calculated value of magnetic moment for complex 7 was found to be 1.82.
Thermal studies
Thermal analysis of copper complex (15) was carried out over the temperature range 0 to 900 °C. The percentage weight loss was recorded in the temperature range of 22–790 °C, which gives information about the stability and volatility of the complex. The thermogram showed a total 55.10% weight loss at 790 °C temperature which was observed in three steps: (1) about 7% weight loss in the range of 75–100 °C which was due to trace amount of moisture present in sample (2) 20% weight loss in the range of 150–272 °C was due to loss of coordinated water molecule (3) about 18% weight loss in the range of 272–459 °C which might be due to complete decomposition of ligand around metal ion. Finally, the complex was converted into metal oxide. The endothermic band observed in the range of 150–272 °C, confirmed the presence of water molecule in the complex [63].

SEM and EDAX
The SEM images of the ligand (3) and its zinc (II) complex (16) was viewed at different magnifications and the micrographs obtained are presented in Fig. 4. The micrograph of ligand exhibited a rough surface with numerous territorial patches. On the other hand, the micrograph of zinc complex indicated an ice rock like shape which may be due to the tightening of voids on the outer surface by coordination of ligand with the metal ion. The SEM-micrographs indicated that ligand has completely different surface morphology from complex, which is another evidence for coordination of ligand to metal ion.

A SEM image of [HL3] (3), B EDAX image of [HL3] (3), C SEM image of [Zn(L3)(H2O)(CH3COO)] (16) and D EDAX image of [Zn(L3)(H2O)(CH3COO)] (16)
Moreover, Energy dispersive X-ray analysis (EDAX) of ligand and its Zn(II) complex was also performed to check the elemental composition of prepared materials. EDAX spectra shown in Fig. 4 clearly show that complexation successfully occurred.
Antimicrobial activity
To investigate the antimicrobial action of synthesized compounds (1–20), four bacterial and two fungal strains were used. The serial dilution method was used for screening the antimicrobial activity. Norfloxacin and Fluconazole were used as reference drugs and the MIC values were calculated in µmol/mL. The obtained activity data are presented in Table 2 and Fig. 5.

Graph showing antimicrobial activity of the compounds (1–20) and standard drugs
By comparing the activity of ligands with their respective metal complexes, it was found that the metal complexes exhibit more potency as compared to ligands. This is due to the chelation of metal which increases the hydrophobic and lipophilic character as discussed in tweedy chelation theory [64]. The results of the activity data are summarized below:
Among the synthesized ligands (1–4), HL3 (MIC = 0.0175–0.0350 µmol/mL) was the most active compound. The higher activity of HL3 is due to the presence of strong electron withdrawing group (nitro group), while due to the presence of electron donating group (ethoxy group) HL1 showed least potency. The observed results indicate that compounds having electron withdrawing groups attached were found to be more active than compounds having electron donating groups attached [65]. The order of activity of ligands is HL3 > HL4 > HL2 > HL1.
Among the transition metal complexes, zinc complexes showed good activity as compared to other metal complexes. The general order of the activity of complexes follows the trend [Zn(L1−4)(H2O)(CH3COO)] > [Cu(L1−4)(H2O)(CH3COO)] > [Ni(L1−4)(H2O)(CH3COO)] > [Co(L1−4)(H2O)(CH3COO)] [66].
Compounds 3, 13, 14, 15 and 16 were found to be active against all bacterial strains except S. aureus with MIC values in the range of (0.0251–0.0350 µmol/mL), as compared to standard drug Norfloxacin (0.0195–0.0391 µmol/mL). The compounds 3, 4, 13, 14, 15, 16, 19 and 20 were active against fungal strains with MIC values in the range of (0.0125–0.0266 µmol/mL) as compared to standard drug Fluconazole (MIC = 0.0204–0.0408 µmol/mL). Also, it was found that compounds were more active against A. niger as compared to C. albicans.
Anticancer assay
The synthesized compounds (1–20) were screened for in-vitro anticancer activity against three cancer cell lines i.e. HCT-116 (human colon cancer), DU145 (prostate cancer) and A549 (lung cancer) by using MTT colorimetric assay. The cytotoxicity of the synthesized compounds was represented in terms of median growth inhibitory concentration (IC50) and values were compared to standard anticancer drug Paclitaxel. The obtained results are presented in Table 3.
Based on the cytotoxicity data, it may be concluded that compound 12 displayed the highest potency towards all cancer cell lines with IC50 value of 8.66 (HCT-116), 12.66 (DU145) and 9.75 (A549) µM. Compound 20 also showed good inhibition and its IC50 values are 6.89 (HCT-116), 10.45 (DU145) and 8.30 (A549) μM. However, these compounds do not completely touch the potential of the known anticancer drug Paclitaxel. Compounds 10, 11, 15, 16 and 19 displayed moderate activity, while the rest of compounds show very less inhibition towards the cancer cell lines.
Structure–activity relationship
The SAR of compounds (1–20) was also studied to check the effect of substituents on biological activity. The synthesized compounds having different substituents attached were found to be active against the three cancer cell lines (except compounds 3–6 and 8), but particularly the compounds 12 and 20 were found to be more active against the cancer cell lines. Compound 1 having ethoxy substitution on the phenyl ring was very less active. On the other hand, its complexation with different metals (compounds 5–8) is ineffective for the enhancement of anticancer activity. Compound 2 having bromo substitution on the phenyl ring itself shows very less activity, but its complexation with nickel (compound 10) and copper metal (compound 11) metal makes them moderately active. The complexation of compound 2 with cobalt metal (compound 9) improved its activity to some extent. However, the complexation of compound 2 with zinc metal (compound 12) results in a significant increase of activity, thus making it the most outstanding compound of the series. Compound 3 having nitro substitution on the phenyl ring was negligibly active, but its complexation with copper (compound 15) and zinc metal (compound 16) resulted in significant improvement of its anticancer activity up to a moderate level. However, its complexation with cobalt (compound 13) and nickel metal (compound 14) resulted in no remarkable increase in anticancer activity. The anticancer activity of compound 4 was almost negligible, but its complexation with copper (compound 19) and zinc metal (compound 20) resulted in a significant increase in anticancer activity. However, its complexation with cobalt (compound 17) and nickel (compound 18) has no effects on its anticancer activity. After accessing the biological potential, it was observed that the activity of metal complexes gets enhanced on complexation. The higher activities of complexes may be due to an increase in their lipophilicity on complexation [66].

Molecular docking studies
Among the synthesized compounds 12, 19, and 20 showed good inhibition against tested cancer cell lines that are closely associated with Human Epidermal Growth Factor Receptor (EGFR) which is a cell surface protein molecule and found to be associated with various types of cancer. Many drugs have been developed as a targeted approach to inhibit over-expressed EGFR, but these drugs are not able to inhibit its over-expression due to drug resistance. Drug resistance is a major challenge in targeted therapy of EGFR-mutated non-small cell lung cancers (NSCLCs). However, the efficacy of the tyrosine kinase inhibitors used in targeted therapy is often limited due to the emergence of drug-resistant mutations in the targeted kinases [42, 67, 68]. Resistance to osimertinib, a third generation EGFR tyrosine kinase inhibitor used to treat NSCLCs with specific mutations, caused by EGFR at position 797 is currently a new challenge in the targeted therapy of EGFR-mutated NSCLCs. Therefore, mutated EGFR was chosen for molecular docking studies to support the observed in-vitro anticancer activity and to add more insight into the possible mechanism of action.
To understand the probable binding modes and estimate the binding energies, three compounds (12, 19 and 20) were docked into EGFR-mutated NSCLCs binding cavity. The docking studies were performed using Hex 8.0.0 and the results are shown in Table 4. Further, the most suitable docking pose of the protein receptor with preferred target ligands was selected and then analysed using Chimera 1.14 and Ligplot software. The obtained docking poses are shown in Fig. 6A–C. The binding energy value of compound 12 is −268.27 kcal/mol, and it shows good interaction with eight amino acid residues of the protein structure i.e. Leu718A, Gly719A, Val726A, Ala743A, Met793A, Gly796A, Cys797A, and Leu844A. Most interesting feature revealed by the docking of this compound is its interaction with Cys797A site, which is responsible for the mutated and drug-resistant behaviour of EFGR, thereby making this compound of utmost importance. The binding energy value of compound 20 is −262.34 kcal/mol and it shows interaction with three amino acid residues of the protein structure i.e. Pro772A, Lys852A, and Asp1014A. Similarly, compound 19 shows appreciable binding energy (−248.39 kcal/mol), but the values are slightly lower as compared to compounds 12 and 20. Compound 19 shows interaction with amino acid residues i.e. Gln820A, Gln849A via covalent bonding, while it shows non-covalent bonding interactions with His773A, Val819A, Lys823A, His850A, Ser969A amino acid residues. The results of docking studies were found to be in line with the results of in-vitro anticancer studies. Based on docking results, it may be concluded that compound 20 is having highest anticancer potential.

Interaction view of 5XGM protein with compounds 12 (A), 19 (B) and 20 (C)
Conclusion
In the present study, four new Schiff base ligands (1–4) and their transition metal (II) complexes (5–20) were synthesized and characterized by various spectral and physicochemical techniques. The obtained spectroscopic data confirm that the synthesized ligands coordinate to metal in a tridentate manner via azomethine nitrogen, deprotonated oxygen and thiol sulphur. The antimicrobial assessment data showed that metal complexes exhibit more potency as compared to ligands. Compound 3 and its metal complexes (13–16) showed reliable activity against the tested pathogenic strains. Furthermore, the results of anticancer activity of the compounds were encouraging and compounds 12, 19 and 20 showed significant anticancer activity against the three tested cancer cell lines (HCT-116, DU145, and A549). The docking results correlate very well with the IC50 data and enabled the importance of interactions for depicting the binding energies of the potent compounds. Based on the results in the current manuscript, the compounds 12, 19 and 20 may be considered as good cytotoxic drugs and this class of compounds could be a better model to develop new leads in the treatment of cancer. The procured results also showed that structural modifications in the synthesized compounds may provide products with different pharmacological activities.
References
H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray, CA: Cancer J. Clin. 71, 209 (2021)
World Health Organisation (WHO) (2020).
F. Yang, S. Shi, J. Zhu, J. Shi, K. Dai, X. Chen, J. Med. Virol. 92, 2067 (2020)
N. Hoti, D. Zhu, Z. Song, Z. Wu, S. Tabassum, M. Wu, J. Pharmacol. Exp. Ther. 311, 22 (2004)
Z.H. Siddik, Oncogene 22, 7265 (2003)
J. Reedijk, Eur. J. Inorg. Chem. 2009, 1303 (2009)
L. Kelland, Nat. Rev. Cancer. 7, 573 (2007)
A.M. Abu-Dief, I.M. Mohamed, Beni-Seuf Univ. J. Appl. Sci. 4, 119 (2015)
L.H. Abdel‐Rahman, A.M. Abu‐Dief, M. Basha, A.A. Hassan Abdel‐Mawgoud, Appl. Organomet. Chem. 31, e3750 (2017).
L.H. Abdel‐Rahman, M.S.S. Adam, A.M. Abu‐Dief, H. Moustafa, M.T. Basha, A.S. Aboraia, B.S. Al‐Farhan, H.E.S. Ahmed, Appl. Organomet. Chem. 32, e4527 (2018).
L.H. Abdel-Rahman, A.M. Abu-Dief, A.A.H. Abdel-Mawgoud, J. King Saud Univ. Sci. 31, 52 (2019)
F.S. Aljohani, A.M. Abu-Dief, R.M. El-Khatib, H.A. Al-Abdulkarim, A. Alharbi, A. Mahran, M.E. Khalifa, N.M. El-Metwaly, J. Mol. Struct. 1246, 131139 (2021).
L.H. Abdel-Rahman, A.M. Abu-Dief, H. Moustafa, A.A.H. Abdel-Mawgoud, Arab. J. Chem. 13, 649 (2020)
E.M.M. Ibrahim, L.H. Abdel-Rahman, A.M. Abu-Dief, A. Elshafaie, S.K. Hamdan, A.M. Ahmed, Mater. Res. Bull. 107, 492 (2018)
R. Antony, S.T. David, K. Saravanan, K. Karuppasamy, S. Balakumar, Spectrochim. Acta A Mol. Biomol. Spectrosc. 103, 423 (2013)
H. Chakraborty, N. Paul, M.L. Rahman, Transit. Met. Chem. 19, 524 (1994)
A.A. Alshaheri, M.I.M. Tahir, M.B.A. Rahman, T. Begum, T.A. Saleh, J. Mol. Liq. 240, 486 (2017)
V. Arun, N. Sridevi, P.P. Robinson, S. Manju, K.M. Yusuff, J. Mol. Catal. A Chem. 304, 191 (2009)
S. Iyer, G.M. Kulkarni, C. Ramesh, Tetrahedron 60, 2163 (2004)
G. Desimani, G. Dasi, G. Paita, P. Quadrelle, P. Righille, Tetrahedron 51, 4131 (1995)
T.T. Calam, Int. J. Environ. Anal. Chem. 99, 1298 (2019)
M. Finsgar, Corros. Sci. 77, 350 (2013)
R.M. Izatt, G.C. LindH, R.L. Bruening, P. Huszthy, C.W. McDaniel, J.S. Bradshaw, J.J. Christensen, Anal. Chem. 60, 1694 (1988)
R.V. Gadag, M.R. Gajendragad, Talanta 25, 418 (1978)
E. Stingaci, M. Zveaghinteva, S. Pogrebnoi, L. Lupascu, V. Valica, L. Uncu, A. Smetanscaia, M. Drumea, A. Petrou, A. Ciric, J. Glamoclija, Bioorg. Med. Chem. Lett. 30, 127368 (2020).
H.A. El-Sherief, B.G. Youssif, S.N.A. Bukhari, A.H. Abdelazeem, M. Abdel-Aziz, H.M. Abdel-Rahman, Eur. J. Med. Chem. 156, 774 (2018)
R.S. Gani, K. Timanagouda, S. Madhushree, S.D. Joshi, M.B. Hiremath, S.B.H. Mujawar, A.K. Kudva, J. King Saud Univ. Sci. 32, 3388 (2020)
K.N. Venugopala, M. Kandeel, M. Pillay, P.K. Deb, H.H. Abdallah, M.F. Mahomoodally, D. Chopra, Antibiotics 9, 559 (2020)
Y. Pruglo, ScienceRise: Pharm Sci. 3, 19 (2018)
B. Kaproń, J.J. Łuszczki, A. Płazińska, A. Siwek, T. Karcz, A. Gryboś, G. Nowak, A. Makuch-Kocka, K. Walczak, E. Langner, K. Szalast, Eur. J. Pharm. Sci. 129, 42 (2019)
A.Z. El‐Sonbati, M.A. Diab, S.M. Morgan, A.M. Eldesoky, M.Z. Balboula, Appl. Organomet. Chem. 32, e4207 (2018).
B. Abulhasanov, U. Yıldız, S. Akkoç, B. Coban, Appl. Organomet. Chem. 34, e5866 (2020).
A.I. Vogel, fifth ed. (Longmans, Edison, Wesley, London, 1999).
P.K. Sahoo, R. Sharma, P. Pattanayak, Med. Chem. Res. 19, 127 (2010)
L. Deswal, V. Verma, D. Kumar, A. Kumar, M. Bhatia, Y. Deswal, A. Kumar, Future Med. Chem. 13, 975 (2021)
A. Ahlawat, P. Khatkar, V. Singh, S. Asija, Res. Chem. Intermed. 44, 4415 (2018)
L.H. Abdel‐Rahman, A.M. Abu‐Dief, M.R. Shehata, F.M. Atlam, A.A.H. Abdel‐Mawgoud, Appl. Organomet. Chem. 33, e4699 (2019).
A.M. Abu‐Dief, H.M. El‐Sagher, M.R. Shehata, Appl. Organomet. Chem. 33(8), e4943 (2019).
A.M. Abu-Dief, L.H. Abdel-Rahman, A.A. Abdelhamid, A.A. Marzouk, M.R. Shehata, M.A. Bakheet, O.A. Almaghrabi, A. Nafady, Spectrochim. Acta A Mol. Biomol. Spectrosc. 228, 117700 (2020).
H.A. Al-Abdulkarim, R.M. El-khatib, F.S. Aljohani, A. Mahran, A. Alharbi, G.A. Mersal, N.M. El-Metwaly, A.M. Abu-Dief, J. Mol. Liq. 339, 116797 (2021).
T. Mosmann, J. Immunol. Methods 65, 55 (1983)
L.L. Kong, R. Ma, M.Y. Yao, X.E. Yan, S.J. Zhu, P. Zhao, C.H. Yun, Biochem. Biophys. Res. Commun. 488, 266 (2017)
E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E. Ferrin, J. Comput. Chem. 25, 1605 (2004)
R.A. Laskowski, M.B. Swindells, J. Chem. Inf. Model. 51, 2778 (2011)
L.H. Abdel-Rahman, R.M. El-Khatib, L.A. Nassr, A.M. Abu-Dief, M. Ismael, A.A. Seleem, Spectrochim. Acta A Mol. Biomol. Spectrosc. 117, 366 (2014)
A.M. Abu-Dief, L.A. Nassr, J. Iran. Chem. Soc. 12, 943 (2015)
L.H. Abdel-Rahman, A.M. Abu-Dief, R.M., El-Khatib, S.M. Abdel-Fatah, Bioorg. Chem. 69, 140 (2016).
N. Mehrotra, R.M. Tripathi, F. Zafar, M.P. Singh, IEEE Trans. Nanobiosci. 16, 280 (2017)
A.A. Olanrewaju, F.S. Fabiyi, C.U. Ibeji, E.G. Kolawole, R. Gupta, J. Mol. Struct. 1211, 128057 (2020)
M. Yadav, S. Sharma, J. Devi, J. Chem. Sci. 133, 1 (2021)
V.V. Dhayabaran, T.D. Prakash, R. Renganathan, E. Friehs, D.W. Bahnemann, J. Fluoresc. 27, 135 (2017)
M.M. Slaihim, F.S.R. Al-Suede, M. Khairuddean, M.B.K. Ahamed, A.M.S.A. Majid, J. Mol. Struct. 1196, 78 (2019)
V. Gomathi, R. Selvameena, Asian J. Chem. 25, 2083 (2013)
J. Devi, M. Yadav, D. Kumar, L.S. Naik, D.K. Jindal, Appl. Organomet. Chem. 33, e4693 (2019)
S.S.A. Fathima, M.M.S. Meeran, E.R. Nagarajan, J. Mol. Liq. 279, 177–189.
W.J. Geary, Coord. Chem. Rev. 7, 81 (1971)
M. Murugaiyan, S.P. Mani, M.A. Sithique, New J. Chem. 43, 9540 (2019)
H.F. Abd El‐Halim, G.G. Mohamed, M.N. Anwar, Appl. Organomet. Chem. 32, e3899 (2018)
S. Chandra, A. Kumar, Spectrochim. Acta A Mol. Biomol. Spectrosc. 68, 1410 (2007)
B. Hathaway, D.E. Billing, Coord. Chem. Rev. 5, 143 (1970)
B.J. Hathaway, A.A.G. Tomlinson, Coord. Chem. Rev. 5, 1 (1970)
N. Kavitha, P.V.A. Lakshmi, J. Saudi Chem. Soc. 21, S457 (2015)
J. Porkodi, N. Raman, Appl. Organomet. Chem. 32, e4030 (2018)
B.B. Beyene, A.M. Mihirteu, A.T. Ayana, A.W. Yibeltal, Results Chem. 2, 100073 (2020)
N. Beyazit, D. Çakmak, C. Demetgül, Tetrahedron 73, 2774 (2017)
J. Devi, S. Sharma, S. Kumar, D.K. Jindal, P.P. Dutta, D. Kumar, Res. Chem. Intermed. 47, 2433 (2021)
D. Ercan, H.G. Choi, C.H. Yun, M. Capelletti, T. Xie, M.J. Eck, N.S. Gray, P.A. Jänne, Clin. Cancer Res. 21, 3913 (2015)
C. Hu, A. Wang, H. Wu, Z. Qi, X. Li, X.E. Yan, C. Chen, K. Yu, F. Zou, W. Wang, Oncotarget 8, 18359 (2017)
Acknowledgements
The author Mr. Yogesh Deswal (SRF) expresses their thanks to CSIR, New Delhi, for providing the financial support in the form of JRF-SRF fellowship with Reference No.: 09/752(0087)/2018-EMR-I dated 31/12/18. The authors are also highly thankful to Department of Chemistry and Dr. APJ Abdul Kalam Central Instrumentation Laboratory (CIL) of GJUS&T, Hisar, Haryana, for providing the needed research facilities to pursue this research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflicts in publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deswal, Y., Asija, S., Kumar, D. et al. Transition metal complexes of triazole-based bioactive ligands: synthesis, spectral characterization, antimicrobial, anticancer and molecular docking studies. Res Chem Intermed 48, 703–729 (2022). https://doi.org/10.1007/s11164-021-04621-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-021-04621-5