
Abstract
Diabetes mellitus is a metabolic disorder denoted by chronic hyperglycemia that drives maladaptive structural changes and functional damage to the vasculature. Attenuation of this pathological remodeling of blood vessels remains an unmet target owing to paucity of information on the metabolic signatures of this process. Ca2+/calmodulin-dependent kinase II (CaMKII) is expressed in the vasculature and is implicated in the control of blood vessels homeostasis. Recently, CaMKII has attracted a special attention in view of its chronic upregulated activity in diabetic tissues, yet its role in the diabetic vasculature remains under investigation.
This review highlights the physiological and pathological actions of CaMKII in the diabetic vasculature, with focus on the control of the dialogue between endothelial (EC) and vascular smooth muscle cells (VSMC). Activation of CaMKII enhances EC and VSMC proliferation and migration, and increases the production of extracellular matrix which leads to maladaptive remodeling of vessels. This is manifested by activation of genes/proteins implicated in the control of the cell cycle, cytoskeleton organization, proliferation, migration, and inflammation. Endothelial dysfunction is paralleled by impaired nitric oxide signaling, which is also influenced by CaMKII signaling (activation/oxidation). The efficiency of CaMKII inhibitors is currently being tested in animal models, with a focus on the genetic pathways involved in the regulation of CaMKII expression (microRNAs and single nucleotide polymorphisms). Interestingly, studies highlight an interaction between the anti-diabetic drugs and CaMKII expression/activity which requires further investigation. Together, the studies reviewed herein may guide pharmacological approaches to improve health-related outcomes in patients with diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Vascular smooth muscle cells and endothelial cells: the major constituents of the arterial wall
The structure of the normal artery consists of three main layers: the intima, the media, and the adventitia [1]. The media is mainly composed of vascular smooth muscle cells (VSMCs), whilst the intima constitutes the innermost monolayer of endothelial cells (ECs). The occasional existence of VSMCs in the intima of children has been reported, while an infiltration of VSMCs into the intima may occur with aging or following vessel injury [2,3,4,5]. The adventitia harbors a mixture of fibroblasts and VSMCs along with components of the extracellular matrix (e.g. collagen and proteoglycans) [6].
In blood vessels, VSMCs play a major role in vessel contraction and the control of its diameter, and thus the distribution of blood flow. VSMCs usually proliferate at a low rate and possess poor synthetic activity [7], but under specific conditions proliferation and phenotypic changes contribute to the development of atherosclerosis, arteriosclerosis, and hypertension [8]. In response to various factors, VSMCs can reversibly switch from a “contractile” to a “synthetic” phenotype, and proliferate [9]. For example, the platelet-derived growth factor (PDGF), which is released during vessel injury, is pivotal for the proliferation of VSMCs [10, 11].
ECs are restricted to the intima, and they are structurally and functionally coupled to the VSMCs and they regulate vascular function and structure. The interaction between ECs and VSMCs starts early as ECs signal to VSMCs to cause differentiation at embryonic stages by inducing the expression of smooth muscle cell specific markers (SM-Myosin, SM22alpha, and calponin) [12,13,14]. ECs release vasoactive substances that control the relaxation, contraction, and growth of VSMCs, especially nitric oxide (NO), prostaglandins, and endothelium-derived hyperpolarizing factor to specifically induce vasodilation [3, 15, 16]. This crosstalk between ECs and VSMCs appears impaired during remodeling of blood vessels. Remodeling may be manifested by either pathological lesions, changes (increase/decrease) in the thickness of the media or the lumen of the vessels, functionally reflected by changes in blood flow and blood pressure dynamics [17, 18].
The maladaptive changes in the vascular wall have been the major focus for therapeutic interventions in various conditions i.e. hypertension, stenosis/restenosis, inflammation and diabetes [17,18,19,20,21]. In fact, whilst various diabetes-related factors could contribute to vasculopathies, hyperglycemia remains a major cause of the pathological remodeling of the micro- and macro-vasculature [22], thus contributing to vascular pathology in diabetic subjects [14]. Endothelial impairment contributes to the development of atherosclerosis and vascular disease [23, 24], with coronary and peripheral arterial disease highly prevalent in diabetic patients [25,26,27]. Chronic hyperglycemia in diabetes results in oxidative stress and activation of protein kinase C (PKC) and polyol pathways [23, 28, 29] that might contribute to proliferation of vessels and altered signal transduction [30]. Further, hyperglycemia encourages attachment of inflammatory cells to the endothelium of the vasculature [23, 31].
There are several signaling networks that are involved in the pathological remodeling of blood vessels under hyperglycemic conditions, one of these that appears to be important is the Calcium/Calmodulin-dependent protein kinase II (CaMKII) pathway. This pathway is also implicated in diabetic cardiomyopathy [32,33,34,35]. Thus CaMKII has recently emerged as a possible therapeutic target in various conditions (reviewed in [36]). In the subsequent sections, we will examine the role of CaMKII in diabetes-induced remodeling of the EC-VSMC interaction and blood vessel homeostasis, with a focus on anti-diabetic therapies and their effect on CaMKII signaling.
2 Characterization of the four CaMKII isoforms and their function
CaMKII, a calcium (Ca2+)-sensing enzyme, is a serine-threonine kinase with a dodecameric structure [37]. Each monomer consists of three domains, an N- terminal catalytic domain that incorporates the kinase activity, an autoregulatory domain that mediates the function of the catalytic domain, and a C-terminal end that drives multimeric assembly [38]. It is a member of a large family of diverse CaM kinases including CaMKI, II and IV. CaMKII is distinguished by its four isoforms α, β, γ, and δ that are encoded by separate genes [39]. CaMKII α and β are predominantly expressed in the brain and also in ECs, whilst CaMKII δ and γ are found in a broader range of tissues, particularly in the heart [40, 41]. The gene coding for the δ variant is the main isoform expressed in cultured VSMCs with δ2 being the major CaMKII splice variant [42], but differentiated VSMCs express both CaMKII γ and δ [43].
An increase in intracellular Ca2+ triggers the activation of all CaMKII isoforms and coordinates ion channel and proteins involved in Ca2+ homeostasis [44, 45]. In neurons, CaMKII is critically involved in complex cognitive and behavioral responses, including synaptic plasticity, learning, and long-term memory [46, 47]. At the cardiac level, CaMKII is crucial for excitation–contraction coupling and excitation–transcription coupling that occurs in cardiomyocytes. Not only is CaMKII critical for normal function of the myocardium, but it also plays a central role in signaling pathways mediating pathological remodeling of the heart [48]. For example, oxidized CaMKII enhances proinflammatory cytokines by upregulating nuclear factor- kB (NFkB) expression [49].
Whilst most studies have been carried out on neuronal cells and cardiomyocytes, some evidence suggests that CaMKII also plays an essential role in the regulation of glycogenolysis/gluconeogenesis [50], and the immune system [51].
3 Implication of CaMKII in vascular physiology
A growing body of evidence suggests that CaMKII exerts a central effect on the function of vascular cell types, specifically the ECs and VSMCs [45, 52]. It has been implicated in the regulation of EC proliferation, migration and inflammation [53]. CaMKII may also be important in the regulation of endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) enzymes [54]. When these enzymes are activated, they cause nitric oxide (NO) release locally which induces relaxation in VSMCs and thus vasodilatation [55]. Following stimulation by receptor-dependent and -independent agonists, there is an increase in the intracellular cytosolic concentrations of free Ca2+. When Ca2+ levels rise, Ca2+/calmodulin complexes bind to CaMKII, triggering its activation, which enhances NO production in ECs. CaMKII has been shown to increase eNOS activity by direct phosphorylation of the enzyme, which enhances its catalytic activity and results in increased production of NO. The phosphorylation promotes eNOS coupling with its cofactor, tetrahydrobiopterin (BH4). BH4 is a critical cofactor for the production of NO, it preserves eNOS dimerization and improves endothelial function [56, 57]. This activation has been shown to play a critical role in regulating vascular tone and blood pressure [54]. CaMKII has also been shown to modulate iNOS expression and activity. Inhibiting CaMKII activity causes an accumulation of a CaMKII/iNOS complex in an aggresome-like structure with consequent decrease in iNOS activity [58].
3.1 CaMKII in VSMCs
CaMKII is abundantly expressed in VSMCs [38] suggesting a role in vasoconstriction [45]. A study by Humphries et al., was the first to explore the role of CaMKII in initiating cellular activity linked to vasodilatation. It revealed that exchange protein directly activated by cAMP (Epac)-induced spontaneous transient outward currents (STOCs) activity in contractile vascular smooth muscle occurs via the activation of CaMKII and thus induces relaxation in VSMC by increasing the sensitive release channels of peripheral sarcoplasmic reticulum ryanodine receptor (RyR) [59].
CaMKII has been shown to be involved in the regulation of L-type Ca2+ channels, potassium channels [60], as well as VSMCs migration [61] and proliferation [62]. Numerous factors promote VSMCs migration including PDGF stimulation, reactive oxygen species (ROS), and matrix metalloproteinase-9 (MMP-9) [32, 63,64,65]. PDGF and other growth factors cause VSMC migration that is associated with a rapid activation of CaMKII [64]. A cytoplasmic increase in Ca2+ causes enhanced CaMKII activity during VSMCs migration [66]. ROS-activated CaMKII stimulates VSMCs migration which is evident following vascular damage [67]. CaMKII is one of the key downstream targets of ROS signaling. ROS oxidize and activate CaMKII, leading to its autophosphorylation at threonine 287 and its sustained activation. Activated CaMKII subsequently catalyzes phosphorylation of proteins and transcription factors involved in VSMCs migration. CaMKII is also activated by methionine 281/282 oxidation (ox-CaMKII), which maintain CaMKII in an active state [68]. Carriers of a mutant CaMKII (by knock-in replacement of methionine 281 & 282 with valine) exhibited reduced oxidation-dependent activation of CaMKII and decreased VSMCs migration [69, 70].
Under normal circumstances, VSMCs remain in a non-proliferative state (G0), however, they enter the cell cycle when provoked by injury. Several mediators/regulators of the cell cycle appear to be influenced by CaMKII and include PDGF, cyclin-dependent kinase 2 (Cdk2), and cyclin E. CaMKII activation promotes production of Cdk2 and cyclin E, which drives transition from G1 to S phase. The cell cycle down regulator p21 reduces Cdk2 and cyclin E activity and in CaMKIIδ−/− mice with carotid artery injury, p21 is overexpressed compared with control mice, which exhibits marked inhibition of VSMCs proliferation [62, 71].
The cAMP responsive element binding protein (CREB) downregulates VSMC growth; CREB is itself inhibited by CaMKIIδ2 (through Ser142 phosphorylation) enhancing VSMC growth [72]. Passaged primary cultures of de-differentiated VSMCs predominantly express CaMKIIδ2 that promote transition from G1 to G2/M phase of the cell cycle [43].
Another important role of CaMKII in VSMCs proliferation is exerted by the Raf/MEK/ERK pathway. Activated CaMKII forms a protein complex with the extracellular-signal-regulated kinase (ERK), but does not directly phosphorylate it. CaMKII rather activates Raf1, which indirectly causes ERK phosphorylation (via MEK) and this complex translocates to the nucleus, promoting proliferation [73]. Together, these findings suggest an important role for CaMKII in VSMCs proliferation in vitro and in vivo. Figure 1 summarizes the dynamics of CaMKII in ECs and VSMCs.

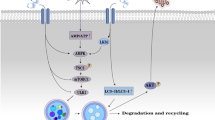
Control and function of CaMKII in ECs and VSMCs. Schematic drawing summarizing the major regulators of CaMKII function in ECs and VSMCs. The lines/effectors of same color depict a signaling cascade and the role of CaMK on the effectors in EC and VSMC. Abbreviations: EC: endothelial cell; VSMC: vascular smooth muscle cell; CaMKII: Ca2+/calmodulin-dependent kinase II; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; BH4: tetrahydrobiopterin; LTCC: L-type calcium channel; Na+: Sodium; K+: potassium; PDGF: platelet-derived growth factor; ERK: extracellular-signal-regulated kinase; CREB: cAMP responsive element binding protein; Cdk2: cyclin-dependent kinase 2; ROS: reactive oxygen species; Ox-M: Ox-Methionine. Figure created with Biorender.com
4 Role of CaMKII in vascular injury in diabetes mellitus
4.1 CaMKII-dependent regulation of ECs structure and function in diabetic conditions
Although CaMKII is primarily involved in the pathogenesis of direct vascular damage e.g. that caused by pressure injury (hypertension), it is not yet clear whether it is as involved when such damage occurs secondary to other insults (e.g. diabetes) [74].
In diabetic environment vaso-responsiveness is significantly impaired. For example, in vitro, both the vasodilation to NO and the vasoconstriction to endothelin-1 are significantly attenuated [75,76,77]. These are further impacted by functional deterioration of the peripheral sympathetic nervous system [75, 78], increased activity of PKC, expression of pro-inflammatory transcription factor nuclear factor kappa B (NF-κΒ), and generation of oxygen-derived free radicals [75, 79, 80].
In contrast, induced NO release from lipopolysaccharide which upregulates iNOS, is markedly increased in diabetic rats compared to their control counterparts [35]. This increase is followed by an increase in intracellular Ca2+ and activation of CaMKII via phosphorylation. A role for CaMKII in endothelial injury has been shown using chronic CaMKII inhibition (with KN-93) which halted the progression of endothelial dysfunction in diabetic rats [81]. This could be due to the effect of CaMKII on the subcellular localization of filamin, an actin-binding protein implicated in the rearrangement of the cell cytoskeleton and the control of cell size and shape, as well as the regulation of gene expression [82]. CaMKII promotes the formation of tight junctions (TJ) in ECs in an Adenosine Monophosphate-Activated Protein Kinase (AMPK) and claudin-1 dependent manner [83]. The inhibition of CaMKII using KN93 causes a spontaneous polymerization of claudin-1, which promotes the development of TJs [83]. Interestingly, CaMKII also influences thrombin/fibronectin-induced EC migration, which points to CaMKII as a target for pathological remodeling of blood vessels [84]. The reorganization of these structural proteins could be impaired in diabetic conditions. These findings suggest that CaMKII may be a driver of endothelial dysfunction in hyperglycemic conditions, independent of NO signaling. It is worth noting that CaMKII inhibition enhances the contraction and relaxation of myocardial tissue in diabetic rats, which suggests that it may have an action on cardiac myocytes as well [85].
Based on these findings, potential therapeutic strategies revolving around the inhibition of CaMKII may be developed for the treatment of diabetes-associated vascular damage.
4.2 CaMKII-dependent regulation of VSMCs in diabetic conditions
In contrast to ECs, VSMCs are organized in multiple layers across the wall of blood vessels and, as indicated earlier, their remodeling in diabetes translates into many pathological processes including hypertension, atherosclerosis, and plaque rupture. The interplay of CaMKII with ROS in diabetic VSMCs may contribute to vasculopathies [86]. It has been shown that hydrogen peroxide (H2O2) and/or glucose induced phosphorylation of ERK1/2 in VSMCs is under the control of CaMKII, and this promotes growth, proliferation, and hypertrophy of VSMCs [87]. Additionally, either the inhibition, or the silencing of CaMKII (in H2O2 stimulated cells) attenuates phosphorylation of the insulin-like growth factor-1 receptor (IGF-1R), suggesting a role for CaMK in insulin metabolism and implies a diabetogenic role for this enzyme [87, 88]. On the other hand, the pharmacological challenge of VSMCs (from diabetic rats) with lipopolysaccharide showed increased expression of iNOS, activated CaMKII, but a weaker interaction between iNOS and CaMKIIδ2 compared with non-diabetic animals [35]. These studies suggest that CaMKII activity and its interaction with iNOS are altered in VSMCs under hyperglycemic conditions. In ECs, CaMKIIδ contributes to the production of ROS via phosphorylation of NADPH oxidase [63]. Following vascular insult, the expression of CaMKIIδ2 is promoted [43, 89], resulting in the activation of the MAPK pathway [43, 89, 90], increased proliferation and migration, which ultimately manifests as neo-intimal hyperplasia [43, 89]. Thus, these studies suggest that the maladaptive remodeling of blood vessels in hyperglycemic conditions may result from an aberrant activity of isoforms of CaMKII. Other studies have shown that CaMKIIδ involvement in diabetic injuries is tissue- and cell-type specific: this is evident in CaMKIIδ-deficient mice (crossed with leptin receptor-mutant mice) that do not develop hyperglycemia, but show improved glucose transport into skeletal muscle and reduced production of hepatic glucose. However, these mice exhibited diabetic nephropathy without diabetic retinopathy, most probably due to differences in glucose metabolism between the kidneys and the retina [91]. The authors proposed that CaMKIIδ may phosphorylate the insulin receptor to block its activity and inhibit the expression of GLUT4, as highlighted by other studies [92, 93]. While pharmacological inhibition (KN93) of CaMKII attenuates the activity of CaMKII in diabetic vessels, there is urgency to define the molecular cues behind the CaMKII isoforms-induced diabetic vasculopathy, particularly CaMKIIγ and δ that are predominantly expressed in VSMCs, to better guide therapeutic interventions focused on either ECs, or VSMCs, or probably both.
5 Impact of CaMKII on macrophages and progenitor cells: possible therapeutic targets in diabetes?
The reduced ambient NO levels in patients with diabetes may contribute to enhanced expression of leukocyte adhesion molecules, chemokines and cytokines. These actions promote monocyte migration into the intima and the formation of foam cells from macrophage [75]. The accumulation of macrophages in the vascular wall causes plaque instability and rupture by reducing VSMCs proliferation. Within the unstable plaque, CaMKII expression is increased in macrophages and actively regulates chemokine production [94]. Knockout models of CaMKII in cardiomyocytes showed a reduction in chemokines which implies that this may also occur in the vasculature [95].
Endothelial progenitor cells (EPCs) are also implicated in the repair process of the damaged blood vessel. EPCs are damaged in type 2 diabetes (T2D), with few studies suggesting that EPC dysfunction precedes the onset of the disease itself [96]. This defect in EPCs leads to a lower recruitment of progenitor cells to the site of injury, which may manifest as nephropathy [97], retinopathy [98], and cardiomyopathy [99] in diabetic patients. Taking retinopathy as an example, this disease becomes clinically noticeable when the microenvironment in the eye is altered by proangiogenic factors, thus favoring neovascularization. In contrast to other areas in the body, this neovascularization promotes endothelial progenitor cell recruitment, further exacerbating the degree of retinopathy [98]. In this context, CaMKII contributes to pathological retinal angiogenesis, and appears to be a key signaling step in angiogenic activity in human retinal ECs. In fact, the signaling of several angiogenic molecules including fibroblast growth factor, hepatocyte growth factor, insulin-like growth factor-1, and the vascular endothelial growth factor, converge towards CaMKII in the retina. Whilst these factors induce cell proliferation and migration, pharmacological inhibition of CaMKII (KN93) abolished these effects [100]. These data crystallize CaMKII as a novel therapeutic target for diabetes-induced retinopathies.
CaMKII is also involved in the proliferation of hematopoietic stem and progenitor cells in response to bone marrow injury [101]. Here, CaMKII knockout mice exhibited faster regeneration of peripheral blood and bone marrow injury, and they were more resilient to radiation-induced insults compared with CaMKII intact mice. This crystallized CaMKII as a negative regulator of cell proliferation and highlighted a possible role for CaMKII inhibition in enhancing cell expansion and recovery. However, this is difficult to reconcile with other findings where CaMKII was found to be proliferative and stimulated migration in retinal ECs, suggesting tissue specificity in CaMKII actions [98].
The role of the oxidation of CaMKIIδ in arteriogenesis and the infiltration of macrophages in the perivascular space has also been studied [65]. Outward remodeling of arteries appears to be controlled by the oxidation of CaMKIIδ and the secretion of MMP-9 by macrophage. Studies have identified CaMKIIδ as a mediator of ROS-signaling in the vasculature, and highlighted a role for CaMKII in diabetic vasculopathy due to the activation of ROS in this setting. Further, endogenous CaMKIIγ serves as a functional brake on CaMKIIδ-mediated signaling, which otherwise promotes VSMCs proliferation and vascular wall remodeling, hinting at different repair functions amongst the various CaMKII isoforms [102]. In addition, CaMKIV, which is mainly expressed in the brain and hematopoietic stem cells (HSC), may be essential for cell survival [103]. Taken together, these studies underscore a role for CaMKII in the repair and maintenance of the injured blood vessels.
6 CaMKII miRNA targets in the vasculature and its genetic polymorphism in diabetes
MicroRNAs (miRNAs) are a class of small non-coding RNAs that modulate post-transcriptional protein expression and have been suggested to have a key regulatory role in diverse biologic processes [104]. They emerged as regulators of several pathophysiological conditions including vasculoproliferative diseases. The expression of CaMKIIδ in VSMCs may be regulated by miR-30 family members (miR-30), miR-143, and miR-145 following vascular injury [105]. It appears that miR-30 inhibits CaMKIIδ-dependent VSMCs function and neointimal VSMCs hyperplasia induced by vascular injury. Interestingly, the miR-143/145 cluster overexpressed in VSMCs obtained from rat aorta with a contractile phenotype, also inhibit CaMKIIδ protein expression [105, 106]. Both miR-143 and miR-145 also regulate the insulin signaling pathway and glucose uptake in VSMCs. Deletion of the miR-143/145 cluster results in potentiated insulin signaling and insulin-induced glucose uptake [106]. Thus miR-143/145 may be endogenous inhibitors of CaMKII, but enhance insulin-induced glucose uptake, providing perhaps a novel therapeutic target for diabetes. Several single nucleotide polymorphisms (SNPs) have been identified in the genes encoding CAMK2 isoforms. The SNPs have been found to be associated with a range of phenotypes, including T2D, Parkinson's disease, Alzheimer's disease, atrial fibrillation, chronic bronchitis, human height, and liver-related alkaline phosphatase disease. A summary of the relationship between the phenotypes and the associated SNPs in CAMK2 isoforms is provided in Table 1. These associations suggest that genetic variation in CAMK2 isoforms may contribute to the development or modulation of the clinical manifestations of a number of the above mentioned disorders.
7 Pharmacologic and genetic control of CaMKII
The use of pharmacologic inhibitors of CaMKII activity to date has been mainly limited to empirical research with little or no translation to interventions, at least in humans. There are different approaches to suppress the signaling of CaMKII and these include direct pharmacological inhibitors using small molecules (e.g. KN-93, AS105 and GS-680), peptides, and RNA interference (RNAi).
KN-93, one of the first CaMKII inhibitors, is an allosteric inhibitor preferentially binding to CaMKII in the inactive state [32, 123]. This mechanism of inhibition is thought to be less efficient at inhibiting non-active CaMKII, including autonomously activated CaMKII (from Thr287 auto-phosphorylation). Unfortunately KN-93 is poorly selective, with a half maximum inhibitory concentration (IC50) in the 1 to 4 µM range [124]. Nevertheless, it has been useful as a research tool, and it has emerged lately as a focus for pharmacological inhibition of CaMKII in clinical settings [125]. A novel CaMKII inhibitor, AS105, was developed by Allosteros Therapeutics as an ATP-competitive pyrimidine-based molecule. AS105 displayed a remarkable in vitro IC50 of 8 nM and effectively mitigated Ca2+ dysregulation in adult cardiomyocytes from CaMKIIδC-overexpressing mice. Importantly, AS105 inhibition of CaMKII had no adverse effects on baseline Ca2+ handling, consistent with results from CaMKII knockout mice [126]. These findings surface the potential of AS105 as a specific and effective CaMKII inhibitor, holding a promising therapeutic applications in the treatment of cardiac conditions.
GS-680 is another novel selective and ATP-competitive CaMKII inhibitor that has been developed by Gilead Sciences, with an IC of 2.3 nM against CaMKIIδ. Biopsies from patients undergoing surgeries revealed that GS-680 demonstrated effectiveness in preventing CaMKII-dependent proarrhythmic activity and in reducing sarcoplasmic reticulum Ca2+ leak in human atrial trabeculae [127].
Peptide inhibitors have been developed and studied as potential inhibitors of CaMKII. Autocamtide-2-related inhibitory peptide (AIP) [128] and autocamtide-3 derived inhibitory peptide (AC3-I) [129] are designed based on the structure/sequence of the auto-regulatory domain of CaMKII. These inhibitors mimic the pseudosubstrate domain, with a key mutation at T287 to a phosphorylation-resistant alanine, effectively sequestering the catalytic domain. In contrast with the complete CaMKII structure, these peptides lack the CaM binding domain, enabling them to remain bound even in the presence of Ca2+/CaM [130]. Consequently, these inhibitors have been employed both pharmacologically and genetically to effectively impede CaMKII's pathological signaling. CaMKIIN including CaMKIINα and CaMKIIβ were subsequently identified. These peptides selectively inhibit CaMKII activity with an IC50 of 50 nM [131]. CaMKIIN molecules exhibit a higher degree of selectivity by directly binding with the kinase domain in the active conformation. Peptide inhibitors are genetically encoded, they can be specifically delivered to selective regions by using locally administered viral vectors and guided to specific intracellular locations with suitable targeting sequences [132].
On the other hand, targeted gene silencing is a more selective tool to suppress CaMKII expression and activity. Here single-stranded antisense oligonucleotides, double-stranded siRNAs, or miRNAs [133] are used to knock-down CaMKII expression. Although theoretically downregulating CaMKII with RNA based therapy looks promising, current agents are delivered systemically either by intravenous or intramuscular injections, which adds to the complexity of selectively targeting CaMKII in specific tissues. Recently, CRISPR-Cas9 targeting CaMKIIδ in cardiomyocytes from human induced pluripotent stem cell effectively suppressed the oxidation of methionine on CaMKII, thus rendering these cells more tolerant to ischemia-reperfusion injuries [134]. Moreover, ablating the autophosphorylation site of CaMKIIδ using CRISPR-Cas9 in mice protected their hearts from transaortic constriction induced heart failure [135]. Together, these studies offer hope for direct targeting of CaMKII domains to better control its dynamics in diabetes settings. To our advantage, this gene editing approach is permanent and may provide a unique therapeutic tool to control CaMKII activity in patients with diabetes [136]. Together, these studies offer hope for direct targeting of CaMKII domains to better control its dynamics in diabetes settings [137].
8 Intersection of the anti-diabetes therapy and CaMKII signaling pathways
Upregulation and constitutive over-activity of CaMKII are features of several pathologies in diabetes mellitus including heart failure and possibly chronic kidney disease (CKD). It is therefore helpful to evaluate whether the anti-diabetes therapy can modulate CaMKII signaling.
It is well known that excess calorie intake and a sedentary lifestyle are associated with the development of T2D. The literature contains a few studies linking lifestyle interventions to the expression/activity of CaMKII. One study examined the impact of exercise and endurance training on angiogenesis-related genes in cardiac tissue of diabetic rats. It was found that six weeks of moderate-intensity exercise training reduced the expression of CaMKII and allowed more effective control of glucose homeostasis by improving glucose uptake in adipose and muscle tissue [138]. Another study reported that a chronically high salt intake induced an increase in intracellular ROS and high CaMKII activity which led to impaired endothelial function [139]. Studies in obese individuals show that adipocytes have higher intracellular mitochondrial Ca2+ levels and reduced glucose uptake [140]. Inhibition or deletion of CaMKII in the liver of obese mice protects against insulin resistance and this is similar in adipose tissue [141, 142]. Interestingly, in a study of diabetic mice (leptin receptor mutation) crossed with CaMKII knockout mice, there was no development of hyperglycemia, but normal glucose uptake into muscle, reduced hepatic glucose synthesis, and the absence of retinopathy. However, despite normoglycemia, these mice still developed diabetic nephropathy, suggesting that hyperglycemia per se is a symptom, and not a direct cause, of diabetic nephropathy. However, CaMKII may be directly involved in nephropathy progression as it has been shown that CaMKII upregulation contributes to renal fibrosis [143] and enhanced matrix production in CKD [144].
Many drugs may directly or indirectly interfere with or affect CaMKII signaling. Insulin stimulates growth and proliferation of many cells involving CaMKII signaling pathway and CaMKII signaling may be further involved in signals downregulation after insulin stimulation [93]. CaMKII appears to modify insulin-induced ERK1/2 activation and cell proliferation, and after stimulation, CaMKII mediates the down-regulation of glucose uptake. Thus, CaMKII may play a central role in long term insulin response and modulation of this pathway may enhance insulin action. Somewhat counterintuitively, the biguanide metformin, has been shown to enhance CaMKII dependent protein synthesis in mouse liver [145]. Nevertheless, there is paucity of data on the effects of biguanides on CaMKII activation. Biguanides and sulphonylureas are widely used in the treatment of T2D, and whilst there is little direct evidence on the effect of these drugs on CaMKII pathways, there is some tangential evidence that they may change the CaMKII signaling pathway. The sulphonylurea, glyburide, activates CaMKII and enhances anti-inflammatory responses which improves wound healing in a high fat diet obese mouse model [146], but the significance of these findings remains unclear.
Thiazolidinediones or Glitazones are a class of drugs commonly used to treat T2D by improving insulin sensitivity in peripheral tissues [147]. Several studies have investigated the impact of glitazone treatment on CaMKII in the heart. Glitazones (and/or curcumin) were shown to reduce the expression of CaMKII in diabetic cardiomyopathy in streptozotocin induced diabetic rats [148], but little evidence on human exist to support these findings.
EMPA-REG was an international, prospective, placebo-controlled clinical study investigating the cardiovascular outcome of empagliflozin, a sodium-glucose co-transporter-2 inhibitor (SGLT2i), in patients with heart failure and diabetes. This study showed that this treatment significantly reduced mortality [149], and similar results have been seen in diabetic and non-diabetic nephropathic patients [150, 151]. Thus, it is of considerable interest that SGLT2i appears to downregulate CaMKII activity [149, 152], and therefore myocyte CaMKII pathways may be a target for SGLT2i in diabetes [153].
Oxidation of CaMKII causing constitutive activation may be central to the toxic effects of mineralocorticoids e.g. aldosterone [154, 155]. Thus, steroidal mineralocorticoid receptor antagonists like spironolactone and eplerenone, and the non-steroidal mineralocorticoid antagonists (MRA), finerenone, may have some activity reducing CaMKII activity. The newer MRA have shown very good clinical results in patients with diabetes mellitus and CKD, especially more recently in the ARTS-DN/FIGARO-DKD/FIDELIO-DKD studies with improved renal and cardiac outcomes [156]. Whether or not this clinical improvement is due to MRA’s effects on the CaMKII pathways is not presently known.
The angiotensin receptor antagonist valsartan has been shown to be associated with inhibition of CaMKII in heart failure [157]. It is established that β-blockers are one of the standard therapeutic approaches for the treatment of chronic heart failure. While CaMKII activity is also increased by beta adrenergic stimulation (reviewed in [158]), β-blockers do not appear to downregulate CaMKII in experimental models [159].
Lipid pathways may also intersect with these signaling pathways and LDL appears to activate CaMKII [160], thus statins and other lipid lowering drugs might be expected to reduce CaMKII activation. The fact that we cannot show evidence for this suggests that this effect may be an epiphenomenon [161, 162].
Antioxidants in general might be expected to downregulate CaMKII activation, which makes it clinically important to counteract the CaMKII-induced upregulation of ROS [163]. Whilst many antioxidants have been shown to downregulate CaMKII pathways (e.g. resveratrol downregulates CaMKII in neurons), disappointingly untargeted antioxidant therapy has not been shown to therapeutically and materially affect outcomes, especially in diabetes [164]. On the other hand, studies have shown that intake of some phenolic compounds improves endothelial function and reduces CaMKII activity. A single ingestion of coffee polyphenols improved peripheral endothelial function after glucose loading in healthy subjects [165]. Furthermore, the 3′,4′-dihydroxyflavonol (DiOHF), a synthetic flavonol, was shown to alleviate diabetes-induced vascular dysfunction [166] and to modulate the p38 MAPK and JNK signaling cascades through inhibition of CaMKII with a potency (IC50 0.25 μM) that is superior to that of the well-established CaMKII inhibitor, KN-93 (IC50 3.3 μM) [167].
9 Conclusion and future perspectives
There is a need to further characterize and define the role of CaMKII in regulating the interplay between ECs and VSMCs in diabetes. Chronic activation of CaMKII has been shown to be associated with inflammation, increased proliferation, migration and hypertrophy leading to abnormal vascular remodeling (Fig. 2). Whilst the current pharmacotherapy of diabetes is effective in abating hyperglycemia, the interaction of certain treatments with CaMKII should not be ignored. In vivo studies implicating animal models with targeted deletions of the CaMKII isoforms in the settings of diabetes are imperative to assess the changes in ECs and VSMCs under these conditions. These models could also serve as valuable tools to address the dynamics of EC/VSMC in the micro vasculatures (i.e. liver, retina, …etc) and the macrovasculature (i.e. coronary arteries). While targeting CaMKII signaling may offer a promising therapeutic approach for preventing or treating the observed complications in diabetes, delivering the different modalities of the discussed CaMKII inhibitors (drugs, siRNA, gene guided therapy-CRISPR Cas9, …etc) in a tissue-specific manner remains a major challenge. In fact, the advent of nanoparticles technology for drug delivery combined to genetic tools (i.e. siRNA or CRISPR Cas9) may provide a suitable solution to this complication. This opens the door for novel delivery methods of CaMKII inhibitors in patients with diabetes in a more effective and targeted manner. Nonetheless, the emergence of the mRNA-based vaccines may also reveal promising mostly by implicating the use of either RNAi for CaMKII isoforms, or expression cassettes for inhibitory peptides for CaMKII isoforms. In addition, a genetic screening of patients with diabetes for the various SNPs of the CaMKII isoforms may give hope for a better therapeutic approach to avoid vasculature-related complications. More research is needed to materialize these concepts, and a careful monitoring of the maladaptive remodeling of blood vessels is vitally important for patients with diabetes.

Role of CaMKII in healthy vessel and in diabetes-induced vascular injury. Abbreviations: CaMKII: Ca2+/calmodulin-dependent kinase II; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; LTCC: L-type calcium channel; NF-κΒ: nuclear factor kappa B; PKC: protein kinase C; ROS: reactive oxygen species. Figure created with Biorender.com
Abbreviations
- CaMKII:
-
Ca2+/calmodulin-dependent kinase II
- Calcium:
-
Ca2+
- ECs:
-
Endothelial cells
- VSMCs:
-
Vascular smooth muscle cells
- NO:
-
Nitric oxide
- PDGF:
-
Platelet-derived growth factor
- PKC:
-
Protein kinase C
- BH4:
-
Tetrahydrobiopterin
- eNOS:
-
Endothelial nitric oxide synthase
- iNOS:
-
Inducible nitric oxide synthase
- ROS:
-
Reactive oxygen species
- Cdk2:
-
Cyclin-dependent kinase 2
- CREB:
-
cAMP responsive element binding protein
- ERK:
-
Extracellular-signal-regulated kinase
- NF-κΒ:
-
Nuclear factor kappa B
- AMPK:
-
Adenosine Monophosphate-Activated Protein Kinase
- H2O2 :
-
hydrogen peroxide
- IGF-1R:
-
Insulin-like growth factor-1 receptor
- TJ:
-
Tight junctions
- T2D:
-
Type 2 diabetes
- EPCs:
-
Endothelial progenitor cells
- MMP-9:
-
Matrix metalloproteinase-9
- miRNAs:
-
MicroRNAs
- SNPs:
-
Single nucleotide polymorphisms
- RNAi:
-
RNA interference
- CKD:
-
Chronic kidney disease
- MRA:
-
Mineralocorticoid antagonists
- SGLT2i:
-
Sodium-glucose co-transporter-2 inhibitor
- DiOHF:
-
3′,4′-dihydroxyflavonol
- Epac:
-
Exchange protein directly activated by cAMP
- STOCs:
-
Spontaneous transient outward currents
- RyR:
-
Ryanodine receptor
- AIP:
-
Autocamide-2-related inhibitory peptide
- AC3-I:
-
Autocamide-3 derived inhibitory peptide
References
Stenmark KR, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47.
Schwartz SM, deBlois D, O’Brien ER.The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77(3):445–65.
Wang M, et al. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50(1):219–27.
Miller SJ, et al. Development of progressive aortic vasculopathy in a rat model of aging. Am J Physiol Heart Circ Physiol. 2007;293(5):H2634-43.
Monk BA, George SJ. The Effect of Ageing on Vascular Smooth Muscle Cell Behaviour–A Mini-Review. Gerontology. 2015;61(5):416–26.
Ross R, Glomset JA. The pathogenesis of atherosclerosis (second of two parts). N Engl J Med. 1976;295(8):420–5.
Kaiser N, et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes. 1993;42(1):80–9.
Schwartz SM, Campbell GR, Campbell JH. Replication of smooth muscle cells in vascular disease. Circ Res. 1986;58(4):427–44.
Nguyen HT, Medford RM, Nadal-Ginard B. Reversibility of muscle differentiation in the absence of commitment: analysis of a myogenic cell line temperature-sensitive for commitment. Cell. 1983;34(1):281–93.
Takagi Y, et al. Effects of protein kinase inhibitors on growth factor-stimulated DNA synthesis in cultured rat vascular smooth muscle cells. Atherosclerosis. 1988;74(3):227–30.
Yang X, et al. Curcumin inhibits platelet-derived growth factor-stimulated vascular smooth muscle cell function and injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2006;26(1):85–90.
Ferreira LS, et al. Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ Res. 2007;101(3):286–94.
Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141(3):805–14.
Brownrigg JR, Schaper NC, Hinchliffe RJ. Diagnosis and assessment of peripheral arterial disease in the diabetic foot. Diabet Med. 2015;32(6):738–47.
Messina EJ, et al. Role of endothelium-derived prostaglandins in hypoxia-elicited arteriolar dilation in rat skeletal muscle. Circ Res. 1992;71(4):790–6.
Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–6.
Renna NF, De Las Heras N, Miatello RM.Pathophysiology of vascular remodeling in hypertension. Int J Hypertens. 2013;808353.
Lyle AN, Taylor WR. The pathophysiological basis of vascular disease. Lab Invest. 2019;99(3):284–9.
Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75(4):640–8.
Hu D, et al. Vascular Smooth Muscle Cells Contribute to Atherosclerosis Immunity. Front Immunol. 2019;10:1101.
Mao L, et al. Role of advanced glycation end products on vascular smooth muscle cells under diabetic atherosclerosis. Front Endocrinol (Lausanne). 2022;13.
Keats EC, Khan ZA. Vascular stem cells in diabetic complications: evidence for a role in the pathogenesis and the therapeutic promise. Cardiovasc Diabetol. 2012;11:37.
Cutiongco MFA, et al. Functional differences between healthy and diabetic endothelial cells on topographical cues. Biomaterials. 2018;153:70–84.
Polovina MM, Potpara TS. Endothelial dysfunction in metabolic and vascular disorders. Postgrad Med. 2014;126(2):38–53.
Onat D, et al. Human vascular endothelial cells: a model system for studying vascular inflammation in diabetes and atherosclerosis. Curr Diab Rep. 2011;11(3):193–202.
Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond). 2005;109(2):143–59.
Gu K, Cowie CC, Harris MI.Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971-1993. Diabetes Care. 1998;21(7):1138–45.
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–25.
Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115(10):1285–95.
Kuboki K, et al. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation. 2000;101(6):676–81.
Fowler M.Microvascular and macrovascular complications of diabetesClinical 435. Diabetes. 2011;29(116–122):436.
Erickson JR, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502(7471):372–6.
Hegyi B, et al. CaMKII Serine 280 O-GlcNAcylation Links Diabetic Hyperglycemia to Proarrhythmia. Circ Res. 2021;129(1):98–113.
Nishio S, et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. 2012;52(5):1103–11.
Di Pietro N, et al. Increased iNOS activity in vascular smooth muscle cells from diabetic rats: potential role of Ca(2+)/calmodulin-dependent protein kinase II delta 2 (CaMKIIdelta(2)). Atherosclerosis. 2013;226(1):88–94.
Zhang M, Lam CK. CaMKII: Therapeutic target in vascular diseases warranted for Clinical and Translational Discovery? Clin Trans Discov. 2022;2(3).
Rosenberg OS, et al. Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. FEBS J. 2006;273(4):682–94.
Ebenebe OV, Heather A, Erickson JR. CaMKII in Vascular Signalling: "Friend or Foe"? Heart Lung Circ. 2018;27(5):560–7.
Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31.
Tobimatsu T, Fujisawa H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem. 1989;264(30):17907–12.
Toussaint F, et al. CaMKII regulates intracellular Ca(2)(+) dynamics in native endothelial cells. Cell Calcium. 2015;58(3):275–85.
Schworer CM, et al.Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268(19):14443–9.
House SJ, et al. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292(6):C2276-87.
Beckendorf J, van den Hoogenhof MMG, Backs J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res Cardiol. 2018;113(4):29.
Toussaint F, et al. Vascular CaMKII: heart and brain in your arteries. Am J Physiol Cell Physiol. 2016;311(3):C462-78.
Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510.
Ataei N, Sabzghabaee AM, Movahedian A. Calcium/Calmodulin-dependent Protein Kinase II is a Ubiquitous Molecule in Human Long-term Memory Synaptic Plasticity: A Systematic Review. Int J Prev Med. 2015;6:88.
Rusciano MR, et al.CaMKII Activity in the Inflammatory Response of Cardiac Diseases. Int J Mol Sci. 2019;20(18).
Singh MV, et al. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52(5):1135–44.
Ozcan L, et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15(5):739–51.
Bui JD, et al. A role for CaMKII in T cell memory. Cell. 2000;100(4):457–67.
Roberts-Craig FT, et al.CaMKII Splice Variants in Vascular Smooth Muscle Cells: The Next Step or Redundancy? Int J Mol Sci. 2022;23(14).
Zhao X, et al. FMRP regulates endothelial cell proliferation and angiogenesis via the miR-181a-CaM-CaMKII pathway. Cell Biol Int. 2018;42(10):1432–44.
Förstermann U, Sessa WC.Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–37, 837a–837d.
Tousoulis D, et al. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. 2012;10(1):4–18.
Yang Y-M, et al. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiology-Heart Circ Physiol. 2009;297(5):H1829–36.
Murthy S, et al. Endothelial CaMKII as a regulator of eNOS activity and NO-mediated vasoreactivity. PLoS ONE. 2017;12(10).
Jones RJ, et al. iNOS regulation by calcium/calmodulin-dependent protein kinase II in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292(6):H2634-42.
Humphries ESA, et al. Calcium/calmodulin-dependent kinase 2 mediates Epac-induced spontaneous transient outward currents in rat vascular smooth muscle. J Physiol. 2017;595(18):6147–64.
Prasad AM, et al. Differential control of calcium homeostasis and vascular reactivity by Ca2+/calmodulin-dependent kinase II. Hypertension. 2013;62(2):434–41.
Li H, et al. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol. 2010;298(2):H688-98.
Li W, et al. The multifunctional Ca2+/calmodulin-dependent kinase II delta (CaMKIIdelta) controls neointima formation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J Biol Chem. 2011;286(10):7990–9.
Pandey D, et al. Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol Pharmacol. 2011;80(3):407–15.
Pauly RR, et al. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation. 1995;91(4):1107–15.
Scott JA, et al. The multifunctional Ca(2)(+)/calmodulin-dependent kinase IIdelta (CaMKIIdelta) regulates arteriogenesis in a mouse model of flow-mediated remodeling. PLoS ONE. 2013;8(8).
Nguyen EK, et al. CaMKII (Ca(2+)/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation. Arterioscler Thromb Vasc Biol. 2018;38(6):1333–45.
Trebak M, et al. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal. 2010;12(5):657–74.
Erickson JR, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–74.
Zhu LJ, et al. Oxidative activation of the Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) regulates vascular smooth muscle migration and apoptosis. Vascul Pharmacol. 2014;60(2):75–83.
Scott JA, et al. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol Heart Circ Physiol. 2012;302(10):H1953-64.
Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4(1):104–11.
Liu Y, et al. CaMKIIδ-dependent inhibition of cAMP-response element-binding protein activity in vascular smooth muscle. J Biol Chem. 2013;288(47):33519–29.
Cipolletta E, et al. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology. 2010;151(6):2747–59.
Kobayashi T, et al. Involvement of CaM kinase II in the impairment of endothelial function and eNOS activity in aortas of Type 2 diabetic rats. Clin Sci (Lond). 2012;123(6):375–86.
Creager MA, et al. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108(12):1527–32.
Williams SB, et al. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27(3):567–74.
Nugent AG, et al. Impaired vasoconstriction to endothelin 1 in patients with NIDDM. Diabetes. 1996;45(1):105–7.
McDaid EA, et al. Peripheral autonomic impairment in patients newly diagnosed with type II diabetes. Diabetes Care. 1994;17(12):1422–7.
Hattori Y, et al. High-glucose-induced nuclear factor kappaB activation in vascular smooth muscle cells. Cardiovasc Res. 2000;46(1):188–97.
Inoguchi T, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–45.
Yousif MH, et al. Role of Ca2+/calmodulin-dependent protein kinase II in development of vascular dysfunction in diabetic rats with hypertension. Cell Biochem Funct. 2008;26(2):256–63.
Cai H, Liu D, Garcia JG. CaM Kinase II-dependent pathophysiological signalling in endothelial cells. Cardiovasc Res. 2008;77(1):30–4.
Shiomi R, et al. CaMKII regulates the strength of the epithelial barrier. Sci Rep. 2015;5:13262.
Meoli DF, White RJ. Thrombin induces fibronectin-specific migration of pulmonary microvascular endothelial cells: requirement of calcium/calmodulin-dependent protein kinase II. Am J Physiol Lung Cell Mol Physiol. 2009;297(4):L706-14.
Daniels LJ, et al. Inhibition of calcium/calmodulin-dependent kinase II restores contraction and relaxation in isolated cardiac muscle from type 2 diabetic rats. Cardiovasc Diabetol. 2018;17(1):89.
Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: a focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016;9:244–53.
Bouallegue A, Pandey NR, Srivastava AK. CaMKII knockdown attenuates H2O2-induced phosphorylation of ERK1/2, PKB/Akt, and IGF-1R in vascular smooth muscle cells. Free Rad Biol Med. 2009;47(6):858–66.
Fernández AM, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15(15):1926–34.
House SJ, Singer HA. CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28(3):441–7.
Abraham ST, et al. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circ Res. 1997;81(4):575–84.
Chen J, et al. CaM kinase II-δ is required for diabetic hyperglycemia and retinopathy but not nephropathy. Diabetes. 2021;70(2):616–26.
Ojuka EO, Goyaram V, Smith JA. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am J Physiology-Endocrinol Metabol. 2012;303(3):E322–31.
Illario M, et al. Calcium-calmodulin-dependent kinase II (CaMKII) mediates insulin-stimulated proliferation and glucose uptake. Cell Signal. 2009;21(5):786–92.
Maione AS, et al. Cellular subtype expression and activation of CaMKII regulate the fate of atherosclerotic plaque. Atherosclerosis. 2017;256:53–61.
Weinreuter M, et al. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med. 2014;6(10):1231–45.
Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3(1):46–56.
Baigent C, Burbury K, Wheeler D. Premature cardiovascular disease in chronic renal failure. Lancet. 2000;356(9224):147–52.
Liu X, et al. Endothelial progenitor cells (EPCs) mobilized and activated by neurotrophic factors may contribute to pathologic neovascularization in diabetic retinopathy. Am J Pathol. 2010;176(1):504–15.
Fein FS. Diabetic cardiomyopathy. Diabetes Care. 1990;13(11):1169–79.
Ashraf S, et al.CAMKII as a therapeutic target for growth factor–induced retinal and choroidal neovascularization. JCI insight. 2019;4(6).
Racioppi L, et al. Calcium/calmodulin-dependent kinase kinase 2 regulates hematopoietic stem and progenitor cell regeneration. Cell Death Dis. 2017;8(10).
Saddouk FZ, et al. Ca2+/calmodulin-dependent protein kinase II-gamma (CaMKIIgamma) negatively regulates vascular smooth muscle cell proliferation and vascular remodeling. FASEB J. 2016;30(3):1051–64.
Kitsos CM, et al. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J Biol Chem. 2005;280(39):33101–8.
Kutter C, Svoboda P. miRNA, siRNA, piRNA: Knowns of the unknown. RNA Biol. 2008;5(4):181–8.
Liu YF, et al. MicroRNA-30 inhibits neointimal hyperplasia by targeting Ca(2+)/calmodulin-dependent protein kinase IIdelta (CaMKIIdelta). Sci Rep. 2016;6:26166.
Lan S, Albinsson S. Regulation of IRS-1, insulin signaling and glucose uptake by miR-143/145 in vascular smooth muscle cells. Biochem Biophys Res Commun. 2020;529(1):119–25.
Xue A, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9(1):2941.
Vujkovic M, et al.Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Gen. 2020;52(7):680–691.
Wang Z, et al. Identification of novel functional CpG-SNPs associated with type 2 diabetes and coronary artery disease. Mol Genet Genomics. 2020;295(3):607–19.
Sinnott-Armstrong N, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53(2):185–94.
Sakaue S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–24.
Mahajan A, et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet. 2022;54(5):560–72.
Shrine N, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet. 2019;51(3):481–93.
Jian X-Q, et al. Association of ADAM10 and CAMK2A Polymorphisms with Conduct Disorder: Evidence from Family-Based Studies. J Abnorm Child Psychol. 2011;39(6):773–82.
Bufill E, et al. Reelin Signaling Pathway Genotypes and Alzheimer Disease in a Spanish Population. Alzheimer Dis Associated Disorders. 2015;29(2):169.
Nalls MA, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Smeland OB, et al. Genome-wide Association Analysis of Parkinson’s Disease and Schizophrenia Reveals Shared Genetic Architecture and Identifies Novel Risk Loci. Biol Psychiatry. 2021;89(3):227–35.
Chang D, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017;49(10):1511–6.
Cárcel-Márquez J, et al. A Polygenic Risk Score Based on a Cardioembolic Stroke Multitrait Analysis Improves a Clinical Prediction Model for This Stroke Subtype. Front Cardiovascular Med. 2022;9.
Nielsen JB, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50(9):1234–9.
Yengo L, et al. A saturated map of common genetic variants associated with human height. Nature. 2022;610(7933):704–12.
Kichaev G, et al. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Human Gen. 2019;104(1):65–75.
Sumi M, et al. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181(3):968–75.
Pellicena P, Schulman H.CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5.
Mavunkel B, et al. Pyrimidine-based inhibitors of CaMKIIdelta. Bioorg Med Chem Lett. 2008;18(7):2404–8.
Neef S, et al. Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J Mol Cell Cardiol. 2018;115:73–81.
Lebek S, et al. The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J Mol Cell Cardiol. 2018;118:159–68.
Ishida A, et al. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212(3):806–12.
Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca (2+)-calmodulin-dependent protein kinase in human epithelial cells. J Physiol. 1995;488(1):37–55.
Nassal D, Gratz D, Hund TJ. Challenges and opportunities for therapeutic targeting of calmodulin kinase II in heart. Front Pharmacol. 2020;11:35.
Chang B, Mukherji S, Soderling T. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience. 2001;102(4):767–77.
Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5:21.
Laina A, et al. RNA Therapeutics in Cardiovascular Precision Medicine. Front Physiol. 2018;9:953.
Lebek S, et al. Ablation of CaMKIIδ oxidation by CRISPR-Cas9 base editing as a therapy for cardiac disease. Science. 2023;379(6628):179–85.
Lebek S, et al.Elimination of CaMKIIδ Autophosphorylation by CRISPR-Cas9 Base Editing Improves Survival and Cardiac Function in Heart Failure in Mice. Circulation. 2023.
Karri DR, et al. Long-term maintenance of dystrophin expression and resistance to injury of skeletal muscle in gene edited DMD mice. Mol Ther Nucleic Acids. 2022;28:154–67.
Reyes Gaido OE, et al. CaMKII as a therapeutic target in cardiovascular disease. Annu Rev Pharmacol Toxicol. 2023;63:249–72.
Khajehlandi M, et al.Endurance Training Regulates Expression of Some Angiogenesis-Related Genes in Cardiac Tissue of Experimentally Induced Diabetic Rats. Biomolecules. 2021;11(4).
Bkaily G, et al. High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells. Biomedicines. 2021;9(8):883.
Wright LE, et al. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am J Physiol Endocrinol Metab. 2017;313(6):E641–50.
Dai W, et al. Adipocyte CAMK2 deficiency improves obesity-associated glucose intolerance. Mol Metab. 2021;53.
Ozcan L, et al. Treatment of Obese Insulin-Resistant Mice With an Allosteric MAPKAPK2/3 Inhibitor Lowers Blood Glucose and Improves Insulin Sensitivity. Diabetes. 2015;64(10):3396–405.
Zhou J, et al. NMDA receptor-mediated CaMKII/ERK activation contributes to renal fibrosis. BMC Nephrol. 2020;21(1):392.
Srivastava A, et al. RIPK3-MLKL signaling activates mitochondrial CaMKII and drives intrarenal extracellular matrix production during CKD. Matrix Biol. 2022;112:72–89.
Senesi P, et al. Metformin Treatment Prevents Sedentariness Related Damages in Mice. J Diabetes Res. 2016;2016:8274689.
Lin YW, et al. Glyburide and retinoic acid synergize to promote wound healing by anti-inflammation and RIP140 degradation. Sci Rep. 2018;8(1):834.
Saltiel AR, Olefsky JM. Thiazolidinediones in the Treatment of Insulin Resistance and Type II Diabetes. Diabetes. 1996;45(12):1661–9.
Gbr AA, et al. Cardioprotective effect of pioglitazone and curcumin against diabetic cardiomyopathy in type 1 diabetes mellitus: impact on CaMKII/NF-kappaB/TGF-beta1 and PPAR-gamma signaling pathway. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(2):349–60.
Mustroph J, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018;5(4):642–8.
Perkovic V, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–306.
Heerspink HJ, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–46.
Trum M, et al. CaMKII and GLUT1 in heart failure and the role of gliflozins. Biochim Biophys Acta Mol Basis Dis. 2020;1866(6).
Kadosaka T, et al.Empagliflozin attenuates arrhythmogenesis in diabetic cardiomyopathy by normalizing intracellular Ca(2+) handling in ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol. 2023.
He BJ, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17(12):1610–8.
Bienvenu LA, et al. Cardiomyocyte Mineralocorticoid Receptor Activation Impairs Acute Cardiac Functional Recovery After Ischemic Insult. Hypertension. 2015;66(5):970–7.
Akhavan Sepahi M, et al.Administration of finerenone in chronic kidney disease. J Nephropathol. 2022.
Wu Y, et al. Cardiac Protection of Valsartan on Juvenile Rats with Heart Failure by Inhibiting Activity of CaMKII via Attenuating Phosphorylation. Biomed Res Int. 2017;2017:4150158.
Erickson JR, et al. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev. 2011;91(3):889–915.
Dewenter M, et al. Calcium/Calmodulin-Dependent Protein Kinase II Activity Persists During Chronic beta-Adrenoceptor Blockade in Experimental and Human Heart Failure. Circ Heart Fail. 2017;10(5).
Ma Y, et al. Atherogenic L5 LDL induces cardiomyocyte apoptosis and inhibits K(ATP) channels through CaMKII activation. Lipids Health Dis. 2020;19(1):189.
Adameova A, Ravingerova T. 13 Distinct role of Ca<sup>2+</sup>–calmodulin-dependent protein kinase II in excitation–contraction coupling under certain conditions. Heart. 2011;97(1):e4–e4.
Szobi A, et al. Pleiotropic Effects of Simvastatin on Some Calcium Regulatory and Myofibrillar Proteins in Ischemic/Reperfused Heart: Causality of Statins Cardioprotection? Curr Pharm Des. 2016;22(42):6451–8.
Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol. 2015;89(Pt B):160–7.
Johansen JS, et al. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5.
Ochiai R, et al. Coffee polyphenols improve peripheral endothelial function after glucose loading in healthy male adults. Nutr Res. 2014;34(2):155–9.
Leo CH, Woodman OL. Flavonols in the Prevention of Diabetes-induced Vascular Dysfunction. J Cardiovasc Pharmacol. 2015;65(6):532–44.
Lim NR, et al. Cardioprotective 3′, 4′-dihydroxyflavonol attenuation of JNK and p38MAPK signalling involves CaMKII inhibition. Biochem J. 2013;456(2):149–61.
Funding
This work was supported by Khalifa University of Science and Technology to MN (FSU-2021-024).
Author information
Authors and Affiliations
Contributions
All authors made a significant contribution to the work reported. They took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interests
The authors declare no conflict of interest and have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chacar, S., Abdi, A., Almansoori, K. et al. Role of CaMKII in diabetes induced vascular injury and its interaction with anti-diabetes therapy. Rev Endocr Metab Disord 25, 369–382 (2024). https://doi.org/10.1007/s11154-023-09855-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-023-09855-9