
Abstract
Ti(1-x)MoxO2-carbon composites are promising new supports for Pt-based electrocatalysts in polymer electrolyte membrane fuel cells offering exciting catalytic properties and enhanced stability against electrocorrosion. Pt and the mixed oxide form a couple liable for strong metal-support interaction (SMSI) phenomenon, generally manifesting itself in decoration of the metal particles by ultrathin layers of the support material upon annealing under reductive conditions. The aim of this work is to evaluate the SMSI phenomenon as a potential strategy for tailoring the properties of the electrocatalyst. A 20 wt% Pt/50 wt% Ti0.8Mo0.2O2-50 wt% C electrocatalyst prepared on Black Pearls 2000 carbon functionalized with HNO3 and glucose was reduced at 250 °C in H2 in order to induce SMSI. The electrocatalytic properties and the stability of the reduced and the original catalysts were analyzed by cyclic voltammetry and COads stripping voltammetry. Structural investigations as well as X-ray photoelectron spectroscopy (XPS) measurements were performed in order to obtain information about the details of the interaction between the oxide and the Pt particles. The electrochemical experiments pointed out a small loss of the electrochemically active surface area of Pt in the reduced catalyst along with enhanced stability with respect to the original one, while structural studies suggested only a minimal decrease of the Pt dispersion. At the same time, hydrogen exposure experiments combined with XPS demonstrated the presence of Mo species directly adsorbed on the Pt surface. Thus, the properties of the reduced catalyst can be traced to decoration of the surface of Pt by Mo-containing species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Composites of titania-based mixed oxides and carbon have recently been identified as promising novel supports for Pt electrocatalysts in polymer electrolyte membrane fuel cells [1,2,3,4,5]. These composites are multifunctional materials, which offer solutions for the issues of the traditional Pt/C electrocatalysts used in this class of fuel cells. While the carbonaceous backbone ensures the appropriate surface area and the good electrical conductivity of the catalyst, the TiO2 component helps to stabilize the nanometer-sized Pt particles against dissolution and/or sintering driven by oxidation of carbon during potential jumps (start-stop cycles) in the acidic milieu of the membrane-electrode assembly. At the same time, dopant metal cations (e.g. W, Mo, Nb or Sn) incorporated into the titania not only may improve its conductivity, but also can provide valuable co-catalytic function such as unique tolerance against CO poisoning at the anode side of the cell (W, Mo, Sn dopants) [1,2,3,4,5,6] or promotion of the otherwise sluggish oxygen reduction reaction at the cathode side (Nb, Sn dopants) [7,8,9,10] via the bifunctional mechanism [11]. Importantly, the enhanced stability and catalytic performance offered by the composite support can allow the decrease of the Pt content of the electrocatalysts, which is a key point in determining the price of the polymer electrolyte membrane fuel cells.
For preparation of the composites, a sol–gel synthesis strategy for different dopants and carbon materials was developed in our recent works [1, 2, 4, 5]. The structural and electrocatalytic features of the composite supports and the related catalysts were identified by widespread physicochemical and electrochemical investigations. These studies demonstrated that high long-term stability of the catalysts can only be achieved if the incorporation of the dopant into the titania is complete, i.e. formation of segregated dopant oxide phases is avoided [12]. It was established that the rutile polymorph of TiO2 is preferred as the dopant incorporation is much better than in the case of anatase. The superior performance (involving the CO-tolerance) of the composite-supported catalysts was demonstrated also in fuel cell test experiments [2, 13]. A recent model investigation suggested that the active sites responsible for the beneficial properties of the mixed oxide – carbon composite supported electrocatalysts are located at the perimeter of the Pt particles where atomic closeness with the surface dopant species belonging to the oxide is ensured [14].
A characteristic feature of the platinum group catalysts supported on TiO2 is their affinity towards the phenomenon of strong metal-support interaction (SMSI), which is interpreted as decoration of the metal particles by an ultrathin layer of the partially reduced support oxide [15]. After many years of debate, today it is generally accepted that the driving force for the effect is minimizing the surface energy of the system and it occurs via migration of interstitial Ti ions to the surface of the Pt particles [16]. The importance of the diffusion of the Ti species in the process was also confirmed by studies involving thick Pt-group metal overlayers [17]. Because of the involved long-range material transfer, the SMSI effect is thermally activated and can only be initiated by an appropriate heat treatment. The appearance of the partially reduced oxide species on the surface of the metal particles can very significantly alter their catalytic properties, thus it is widely believed that the SMSI effect can be used for tailoring the performance, especially the selectivity of a catalyst [18]. For example, in selective hydrogenation/dehydrogenation reactions such as selective hydrogenation of carbonyl groups [19] or propane dehydrogenation [20] catalysts showing the SMSI effect may be highly beneficial. Even the CO oxidation features can be enhanced by the bifunctional effect due to the atomic closeness of the metal and the oxide, although complete encapsulation is not desirable in this case [21]. According to literature examples, both electronic (charge transfer from the oxide towards the metal) and bifunctional considerations explain the enhanced oxygen reduction activity of Pt/TiO2 [22], Pt/WO3 [23, 24], Pt/Ti0.7Mo0.3O2 [25] or Pt/SnO2 [26] electrocatalysts showing strong metal-support interaction.
Since SMSI in its classical manifestation involves covering the surface of the metal particles by an ultrathin oxide layer, it is plausible to assume that catalysts in the SMSI state are tolerant against degradation processes such as sintering. Indeed, strong metal-support interaction is identified as a promising way for enhancing the stability of catalysts [27,28,29]. Importantly, the stabilization effect of SMSI can be observed also in electrocatalytic systems [7].
Considering that in the Pt/Ti(1-x)MoxO2-C composite supported electrocatalyst the metal and the oxide represent a couple liable for strong metal-support interaction, a short study was conducted to explore the effect of a reductive treatment (intended for inducting SMSI) on the features of the system. Accordingly, the comparison of the structural as well as the catalytic properties of an original and a reduced Pt/Ti0.8Mo0.2O2-C sample derived from X-ray diffraction, electron microscopy, electron spectroscopy measurements and electrochemical investigations with special emphasis on stability will be presented in this report.
Experimental
Preparation of the 20 wt% Pt/Ti0.8Mo0.2O2-C composite supported electocatalysts
For the purposes of this study a Pt electrocatalyst deposited on a Ti0.8Mo0.2O2-C composite support with 50:50 mixed oxide/carbon weight ratio was prepared. The carbonaceous part was based on Black Pearls 2000 commercial carbon (BP: Cabot, Boston, MA, USA), which was previously pre-treated in nitrogen at 1000 °C and then functionalized with HNO3 and glucose in order to ensure a better dispersion of the oxide phase [30,31,32]. Details of the two-step functionalization treatment of carbon are given in Ref. [4]. The composite material was synthesized using the optimized preparation route described in our previous study [5]. Shortly, during the sol–gel-based multistep synthesis, the first step is the deposition of TiO2-rutile nuclei on the carbon backbone by adding transparent acidic TiO2 colloidal solution, derived from Ti(O-i-Pr)4, to sonicated functionalized carbon–water mixture under acidity control with cc. HNO3 and aging this slurry at room temperature for 4 days. The aging procedure is finished by heating the synthesis mixture to 65 °C for 8 h [5]. The next steps are the introduction and incorporation of Mo into the TiO2-rutile particles by adding the precursor compound [(NH4)6Mo7O24 × 4H2O] to the TiO2-C dispersion, drying the material and annealing the system under inert atmosphere at 600 °C for 8 h. Our previous investigations pointed out that this treatment results in almost exclusive incorporation of Mo into the rutile crystallites while very little segregated Mo-oxides remain on the surface if the Mo content is below the solubility limit (20–30 atomic% with respect to the Ti content) [2, 5, 12, 33].
At the last step of the synthesis of the electrocatalyst, the support was loaded with 20 wt% Pt, using H2PtCl6 as a precursor compound, by the reduction-precipitation method described with more details in our previous studies [3, 12].
After catalyst preparation, a certain fraction of the material was annealed in hydrogen flow at 250 °C for 2 h in order to induce the metal-support interaction. The classic SMSI phenomenon can be induced in the Pt/TiO2 system by reductive treatments at 500–600 °C [34, 35], however, we suspect that in case of a Ti(1-x)MoxO2-C-type composite decoration of Pt starts at much lower temperatures. Thus the 250 °C annealing temperature was selected as a trade-off where sintering of Pt is still small. This reduced fraction will be denoted below as Pt/50C-250H, in contrast to the non-annealed original sample denoted as Pt/50C.
Phase composition and morphology
X-ray diffraction patterns of the original and the reduced electrocatalyst samples were recorded by a Philips model PW 3710 based PW 1050 Bragg–Brentano parafocusing goniometer using CuKα radiation, graphite monochromator and proportional counter.
Morphology of the catalysts was investigated by Transmission Electron Microscopy (TEM) by means of a FEI Titan Themis 200 kV Cs—corrected TEM with 0.09 nm HRTEM resolution. Size distribution of the Pt particles was determined by measuring the diameters of at least 800 randomly selected metal particles in five micrographs of each sample taken from non-aggregated areas using the ImageJ software.
Electrochemical characterization
The activity and the stability of the reduced and the original catalysts were analyzed by cyclic voltammetry and COads stripping voltammetry measurements done before and after the 500-cycle stability test, as well as by the long-term stability test involving 10,000 polarization cycles, which were carried out in a standard three-electrode electrochemical cell using a 0.5 M H2SO4 solution as electrolyte.
The working electrode was prepared by drying drops of an ink containing the suspension of the electrocatalyst in isopropanol, ultrapure water and Nafion onto a polished glassy carbon material (ink composition: 5 mg catalyst in 4 ml H2O + 1 ml isopropanol + 20 µl Nafion solution (D520 Nafion Dispersion—Alcohol based 1000 EW at 5 wt%, DuPont™ Nafion®), electrode surface area: 0.0707 cm2, Pt loading of the electrode: 10 µg cm−2). A platinum wire was installed as counter electrode and a hydrogen electrode was used as reference. All the potentials are given in the RHE scale. The details of the catalyst ink composition and electrocatalytic measurements were described in Ref. [5].
The electrochemically active surface area (ECSA) of the electrocatalysts was determined from the charge needed for oxidation of the underpotentially deposited hydrogen [36] using conventional baseline correction as described in our previous studies [5, 33]. The change of the electrochemically active Pt surface area (ΔECSA) value is defined by Eq. 1 [5]:
where ECSAN/ECSA1 is the change of the electrochemically active surface area of Pt upon the N-cycle stability test presented as ECSAN (N: 500, 2500, 5000 and 10,000) value normalized to ECSA1 measured in the first cycle on the same sample.
Electrochemical performance of the Pt/50C and Pt/50C-250H electrocatalysts was compared with that of commercial 20 wt% Pt/C (Quintech).
Surface chemical characterization
The surface analysis of the electrocatalysts was performed by X-ray photoelectron spectroscopy (XPS) using an Omicron EA 125 electron spectrometer operated in the constant analyser energy mode (pass energy 30 eV, resolution around 1 eV). A non-monochromatic Mg Kα X-ray source (1253.6 eV) was used as excitation source. The reported binding energies are accurate within ± 0.15 eV. The catalyst powder was suspended in isopropanol and drops of this suspension were dried onto the standard stainless steel sample plates. Spectra were collected from both the Pt/50C original and the Pt/50C-250H reduced catalysts.
In our previous work [14] it was established that the nature of the Mo-Pt interaction in the electrocatalysts can be sensitively monitored by analyzing the chemical state changes of Mo after low temperature in situ H2 exposure in the electron spectrometer. In order to compare the surface state of the original and the 250 °C reduced catalysts, a similar route was followed here: a step-wise in situ treatment was performed in 100 mbar H2 for 1 h (at room temperature and 100 °C) in the high pressure/high temperature treatment chamber of the spectrometer. Spectra of the samples were recorded after each hydrogen exposure steps.
For each experiment, we collected high-resolution spectra from the regions of interest (Mo 3d, Ti 2p, Pt 4f, C 1s, O 1s). The spectra were processed with the software Casa XPS [37] by fitting the data with a combination of Gaussian–Lorentzian peaks with Linear and Shirley background. The details of the spectral fitting procedure were described in our previous works [14]. In the case of the quantitative analysis, we made the evaluation with the software XPS MultiQuant [38, 39] assuming a homogeneous distribution of the components.
Results and discussion
Initial characterization of the electrocatalysts
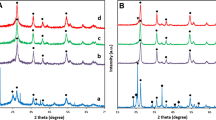
Electrocatalysts prepared in our research program are routinely checked by X-ray diffraction prior to any further investigation. Diffractograms of the Pt/50C and the Pt/50C-250H samples are shown in Fig. 1. The measurements confirmed the success of the synthesis: diffraction patterns indicated the exclusive presence of rutile crystallites with slightly altered cell parameters due to Mo incorporation, as described e.g. in Refs. [2, 5]. The 250 °C reductive treatment did not result in any noticeable structural change in the composite material. Broad reflections from Pt demonstrated the presence of well-dispersed platinum particles. Nevertheless, the appearance of a sharper Pt-related contribution in addition to the original broad peaks after the reductive heat treatment suggested the coexistence of small and somewhat larger particles, indicating the initial phase of Pt sintering.

Influence of the pre-treatment in hydrogen at 250 °C for 2 h on the XRD patterns of the Pt/Ti0.8Mo0.2O2-C electrocatalyst. Black filled circle- Rutile, *-Pt
Transmission electron micrographs and particle size distributions derived from them (Fig. 2) confirmed the conclusion drawn from the XRD experiments. Both in the original state and after reductive heat treatment 2–3 nm sized particles were the dominant structural elements. However, several larger particles were also formed during the reduction process, which gives a tail region to the distribution curve towards the larger particle sizes. Probably these larger particles are responsible for the sharper Pt contribution in the XRD pattern.

TEM images and corresponding histograms of particle size distribution for the Pt/50C (A, B) and Pt/50C-250H (C, D) catalysts
The apparent surface composition of the original and the 250 °C reduced electrocatalysts was determined by XPS and is summarized in Table 1.
The results are in good agreement with previous results obtained on similar electrocatalysts and with the observations of the XRD analysis. The high apparent Pt content (38–39 weight% instead of the nominal 20 weight%) is the expected consequence of the high dispersion and the surface location of the Pt species; bulk-sensitive measurements indeed indicated a Pt content close to 20 weight% in these systems [5]. The deviation of the apparent oxide/carbon weight ratio from the nominal 50:50 value is again a frequently observed feature of such mixed oxide–carbon composites and is explained by the somewhat imperfect dispersion of the oxide (i.e. the coexistence of well-dispersed oxide crystallites with several larger ones, which results in inevitable underestimation of the oxide content by XPS) [5, 14]. Nevertheless, the Mo/Ti atomic ratio is very close to the nominal 1:4 value, which suggests the lack of non-incorporated surface Mo oxide species, in agreement with the XRD results. While the variation of the apparent Pt content is in the range of the experimental uncertainties, its slight decrease is probably the indication of the minimal sintering as suggested by the XRD and TEM observations.
The structural investigations therefore confirmed that the 250 °C reduction treatment did not result in significant Pt sintering. During comparison of the electrocatalytic properties of the original and reduced samples, we do not expect noticeable particle size effects as the influence of the less abundant larger Pt particles on the catalytic behavior is marginal.
Electrochemical behavior of the electrocatalysts
Fig. 3 summarizes the cyclic voltammograms of the original and the 250 °C hydrogen reduced electrocatalysts obtained before and after the short 500-cycle stability test (Fig. 3, panel A) and long-term stability test involving 10,000 polarization cycles (Fig. 3, panel B). The voltammograms indicate the characteristics of the Ti(1-x)MoxO2-C composite supported Pt electrocatalysts described in our previous works [5, 33]. In the cyclic voltammograms of Fig. 3 the region between 50 mV and 350 mV contains the typical adsorption/desorption peaks of underpotentially deposited hydrogen on the Pt surface. Features in the region between 380 mV and 530 mV arise from the reduction/oxidation peaks of Mo species [40,41,42,43,44,45].

Influence of the pre-treatment in hydrogen on the electrochemical performance of the Pt electrocatalysts. Cyclic voltammograms of the Pt/50C-250H (a) and Pt/50C (b) catalysts obtained during (A: left panel) 500-cycle stability test and (B: right panel) long-term stability test involving 10,000 polarization cycles. Recorded in Ar purged 0.5 M H2SO4 solution between 50 and 1000 mV potential limits with 100 mV s−1 sweep rate
From these voltammograms it appears that the electrochemical features are only slightly affected by the 250 °C hydrogen treatment, although, as shown in Table 2, some decrease of the electrochemically active surface area (ECSA) is evident. As can be seen from Fig. 3 (panel A, curve a) the short (500-cycle) stability test resulted in only a minimal decrease of the current density associated with desorption of underpotentially deposited hydrogen, suggesting a very small decrease of the ECSA for the 250 °C reduced (Pt/50C-250H) catalyst. Despite a slight decrease of the Pt ECSA, the electrochemical double-layer region remained practically unchanged, indicating good stability of the Ti0.8Mo0.2O2-C composite support material. In addition, it may be noted that a broad and very stable redox peak pair appeared in the CV around 0.6 V in the hydrogen treated Pt/50C-250H catalyst; its origin deserves further investigations.
As shown in Fig. 3 (panel A, curve b), in the case of the Pt/50C catalyst, the changes in the shape of the voltammograms, observed during 500-cycle stability test, affect not only the peaks associated with the hydrogen adsorption/desorption on platinum at 50–350 mV, but also the Mo redox peaks.
As far as the extended stability investigation is concerned, (Fig. 3, panel B, curve a) the Pt/50C-250H catalyst maintained its high stability during 10,000 polarization cycles and only minimal changes in the shape of the voltammograms were observed after 2500 cycles. This observation is in good agreement with the assumption that the rate of degradation decreases with the age of the electrodes [46]. Moreover, the insignificant loss of ECSA most likely indicates that the Pt nanoparticles have reached a size at which their stability has increased [47].
It should be noted that, despite the high stability of both samples, the changes observed during 10,000 polarization cycles in the voltammograms of the Pt/50C sample were more pronounced (see Fig. 3, panel B, curve b).
The relevant data obtained from the voltammograms depicted in Fig. 3 are summarized in Table 2.
In Fig. 4 ECSA change results after 500 cycles (panel A) and extended cycling (panel B) in the 50–1000 mV potential range are visualized; results obtained in Ref. [5] on the commercial 20 wt% Pt/C catalyst are given for comparison. As it was already pointed out there is not significant ECSA decrease in the hydrogen treated sample in the 500-cycle stability test (see Table 2). Moreover, as can be seen from Fig. 4 (panel A), during the short stability test, the Pt/50C-250H sample showed unusual behavior: a slight increase in the electrochemically active surface area of Pt compared to the ECSA value measured in the 1st cycle was observed until the 150th cycle; this small increase was then followed by a gradual slight decrease in ECSA. This behavior of the hydrogen-treated sample observed at the beginning of the stability test can be attributed to cleaning of the catalyst surface from residual impurities or deposited metals/metal oxides causing a site-blocking effect.

ECSA change during 500 CV cycles (A) and ECSA change during 10,000 CV cycles (B). ECSAN/ECSA1: comparison of the ECSA measured after N cycles normalized to ECSA measured in the 1st cycle of the Pt/50C and Pt/50C-250H catalysts as a function of the number of cycles (N); results obtained in Ref. [5] on the 20 wt% Pt/C are given for comparison. ΔECSAN (N: 500 and 10,000): the loss in ECSA upon N-cycle stability test was calculated using the Eq. 1
It should be noted that this change in ECSA values is within the reproducibility of the electrochemical measurements, demonstrating extremely high stability of the hydrogen treated catalyst.
As shown in Fig. 4, the Pt/50C electrocatalyst showed also improved stability in the short and long-term stability tests compared to the reference Pt/C, as already reported for the mixed oxide–carbon composite supported catalysts [5].
Thus, as follows from the results presented in Fig. 4 and Table 2, after 10,000 polarization cycles the best stability in this series of experiments was obtained on the hydrogen-treated Pt/50C-250H catalyst (ΔECSA = 22.1%). The behavior of the original Pt/50C catalyst was a little bit worse (32.2%), but still much better than the stability of the reference 20 wt% Pt/C (ΔECSA = 47.8% according to the results presented in our recent study [5]).
The qualitative behavior of the COads stripping voltammograms obtained before (Fig. 5A) and after 500 cycles of the stability test (Fig. 5B) is also similar for the two catalysts. In the initial state of the catalysts a broad main CO oxidation peak consisting of two overlapping peaks centered at around 745 mV is accompanied by a pre-peak region between 50 mV and 500 mV; current in this region arises from oxidation of weakly-bound CO species as well as oxidation of surface Mo species. The appearance of the pre-peak and the relatively low potential of the main COads-electrooxidation peak indicate good CO tolerance in both catalysts (the main peak in Pt/C catalysts is typically around 800 mV [5, 33]). Nevertheless, the pre-peak seems to be slightly more pronounced for the 250 °C hydrogen treated sample.

COads stripping voltammograms of the electrocatalysts recorded in 0.5 M H2SO4 before (A) and after 500 cycles (B) of the stability test. Sweep rate: 10 mV s−1. Pt/50C (grey) and Pt/50C-250H (black) catalysts without and with treatment in H2 at 250 °C for 2 h
As shown in Fig. 5B the short aging results in similar changes in both catalysts. In particular, the main CO oxidation peak splits into two components: a peak around 775 mV is accompanied by a shoulder around 705 mV. The splitting is most probably due to the co-existence of two environments for the Pt particles: CO oxidation on Pt species in atomic contact with the oxide contributes to the shoulder, while the peak arises from CO oxidation on Pt less associated with the oxide [5, 33 its references]. The co-existence of these different environments is probably another indication of the imperfect dispersion of the oxide, as already suggested by the XPS data described above.
It should be noted that on both fresh catalysts, a broad main COads-electrooxidation peak was observed, which consists of two overlapping peaks at about 725 mV and 765 mV (see Fig. 5A). Thus, some heterogeneity of the catalyst surface can be assumed in the original catalysts even before the stability tests (e.g., due to the formation of Pt nanostructures with high surface defect density and/or due to the presence of highly dispersed and agglomerated Pt nanoparticles in the catalysts [48, 49]).
Nevertheless, from the COads stripping measurements one can conclude that the hydrogen treatment caused no change in the CO-tolerant behavior of the composite-supported electrocatalyst.
The results of the electrochemical investigations suggest therefore that the 250 °C hydrogen treatment did not change significantly the CO-tolerant behavior of the Pt/50C catalyst, but it had a beneficial effect on the long-term stability.
Surface spectroscopic comparison of the Pt/50C and the Pt/50C-250H electrocatalysts
Since neither the XRD measurements nor the quantitative XPS data indicated significant sintering of the Pt particles during the 250 °C hydrogen treatment, one can suspect that the smaller ECSA for the Pt/50C-250H sample and its unusual behavior during the stability test can be explained by the partial blocking of the surface Pt sites by the support material, similarly to the case of classic SMSI in the Pt/TiO2 system. In the followings the results of a detailed electron spectroscopy analysis addressing this situation will be summarized.
It has to be noted that the general literature on strong metal-support interaction indicates that the encapsulation of the metal particle causes negligible changes in the apparent composition measured by XPS, as the relevant information depths (around 10 nm) are large not only with respect to the thickness of the ultrathin encapsulation layer but they significantly exceed the Pt particle size, too. However, previous experience shows that the formation of the SMSI state is accompanied by chemical state changes of the support material, thus qualitative investigations can still provide spectroscopic evidence for the encapsulation. For example, Ti ions in the ultrathin encapsulation layer in a TiO2-based system are expected to be in the Ti2+ ionic state (with a Ti 2p3/2 binding energy around 455–456 eV [50], as evidenced e.g. in Ref. [51]). Unfortunately, in case of Mo-containing mixed oxide systems an analogous partially reduced state for Mo cannot be identified, as the bare support material already contains a range of reduced Mo species [2, 33]. Nevertheless, in one of our previous works it was established by model experiments that ionic Mo bound to Pt can be separated based on its unusually strong reducibility and surface alloy forming tendency upon low temperature hydrogen exposure [14]. Therefore, the changes of the Mo 3d spectra of the original Pt/50C and the reduced Pt/50C-250H catalyst were compared during an in situ hydrogen exposure series.
Before discussing the results of the Mo spectra, it should be mentioned that Pt was predominantly metallic already in the initial, as received state of both catalysts and the small ionic Pt content was completely reduced upon the room temperature hydrogen exposure.
Fig. 6 summarizes the Mo 3d spectra of the original and the 250 °C reduced catalysts at different stages of the in situ hydrogen exposure series. Apart from the raw data, fitting components arising from the observed Mo states are also shown. The fitting procedure was described in our previous publications [2, 14, 33]. Shortly, the measured spectra were modeled by a combination of signals arising from different Mo chemical states: the Mo4+ state was represented by a complex line shape based on reference measurements on MoO2 with its lowest binding energy contribution around 229.5–230 eV, while other Mo states were described by 3d5/2–3/2 spin–orbit doublets with intensity and doublet separation parameters derived from data on MoO3 (Mo6+), appearing around 3d5/2 binding energies of 232.0–232.5 eV (Mo6+), 231.0–231.5 eV (Mo5+) and, if needed, below 229.5 eV (strongly reduced Mo species) [52,53,54]. Binding energies, widths and intensities of the leading components of these line shapes were treated as variable parameters (between reasonable limits) during fitting. The fitting results (binding energies of the leading components after each treatment step) are summarized in Table 3.

Mo 3d spectra of the Pt/50C-250H (curves denoted as (a)) and the Pt/50C (curves denoted as (b)) electrocatalysts measured during the in situ hydrogen exposure series (initial state, room temperature and 100 °C hydrogen exposure). 3d5/2–3/2 binding energies of the different Mo chemical states are shown above the spectra (color codes: Mo6+: white, Mo5+: dark gray, Mo4+: gray, metallic Mo (alloyed with Pt): black)
The Mo 3d spectrum of the original (as received) state of the catalyst corresponds very well to that obtained on other Mo-doped TiO2-C (C: BP carbon) composite supported electrocatalysts [see e.g. 2, 14, 33]: the dominant Mo 3d5/2–3/2 doublet arising from Mo6+ ions is accompanied by a weaker doublet from Mo5+ ions and a weak contribution from the Mo4+ sates. The Mo spectrum of the 250 °C hydrogen treated sample in its initial state looks very similar to that of the original one.
After room temperature hydrogen exposure in the electron spectrometer (100 mbar, 1 h) the asymmetry of the Mo 3d doublet towards lower binding energies notably increased in case of both catalysts, which clearly indicates reduction of Mo. Detailed curve fitting revealed that in the case of the original Pt/50C catalyst the increasing asymmetry is due to the increase of the Mo4+ and Mo5+ contributions. At the same time, in the 250 °C reduced system a clearly established new component (black peak pair) at a 3d5/2 binding energy slightly below 229 eV was also identified. Further in situ H2 exposition at an elevated temperature of 100 °C followed the same trend: while in the Pt/50C catalyst the Mo4+ and Mo5+ signals increased on expense of the Mo6+ contribution, in the hydrogen-treated Pt/50C-250H catalyst the signal from the new state with binding energy below 229 eV also increased.
The general reduction of the surface or near-surface Mo species upon hydrogen exposure or annealing in hydrogen is similar to what was observed in case of the non-reduced electrocatalysts [14] and can be explained by spillover of hydrogen activated on the Pt particles. The appearance of the low binding energy component below 229 eV (clearly demonstrated even after the room temperature hydrogen exposure) is, however, a distinctive feature of the electrocatalyst ex situ reduced at 250 °C. Its binding energy corresponds to that observed for Mo forming a metallic surface alloy with Pt in [14] (note that the locations of the Mo0+ peaks on the bar indicate the expected positions of the signals from zero-valence Mo alloyed with Pt). Therefore the most probable interpretation for this component is that it arises from reduction products of Mo species bound to the Pt particles in the initial state of the catalyst. An alternative interpretation can be the formation of Mo-carbide, although the binding energy seems to be too high for that compound [55, 56] and the temperature seems to be too low for its formation.
Qualitative evaluation of the Mo 3d spectrum of the reduced catalyst, therefore, revealed that the system contained very easily reducible Mo species which behaved analogously to Mo deposited directly onto Pt. Accordingly, we believe Mo species migrated to the surface of the Pt particles during the 250 °C hydrogen treatment which exactly corresponds to what is expected during the strong metal-support interaction. As Ti remains almost exclusively in the fully oxidized state during the hydrogen treatment cycle (single Ti 2p3/2–1/2 doublet with 2p3/2 binding energy around 459.0 eV), XPS data did not indicate similar behavior for Ti, thus it seems that at this relatively low temperature the main component of the decoration layer on Pt is partially oxidized Mo.
In summary, comparison of the electrochemical behavior of the Pt/50C and the 250 °C hydrogen annealed Pt/50C-250H catalysts suggested a certain extent of Pt site blocking induced by the reductive pre-treatment. This site blocking was partially reversed during the initial stages of the potential cycling of the stability test. The electron spectroscopic investigations were consistent with this image and indicated the decoration of the Pt particles by oxidized Mo species. We believe that these surface oxides interfere with the coalescence of the Pt particles during aging, which explains the promising behavior of the hydrogen treated catalyst in the long-term electrochemical stability test. Another peculiarity of the hydrogen treated system is the easy reducibility and surface alloying tendency of the Mo species bound to the Pt particles. Even if these Mo species can be re-oxidized upon air exposure, their easy reducibility may facilitate the formation of alloy-type bimetallic catalysts under appropriate reductive conditions, which may open further perspectives in modulating the activity and selectivity of the system. It may be noted that recent investigations seem to point to the importance of alloying and oxidation-induced segregation in SMSI in purely TiO2- based catalysts also [35], in analogy with the observations of this work.
Conclusions
In this preliminary study the effect of reductive heat treatment on the properties of Pt electrocatalysts supported on a Ti(1-x)MoxO2-carbon composite was assessed by structural, electrochemical as well as surface spectroscopic investigations. The electrochemical experiments revealed a certain loss of the electrochemically active surface area of Pt in the catalyst annealed in hydrogen with respect to the original one, while XRD, TEM and XPS indicated only a marginal change in the dispersion of Pt. Furthermore, in situ hydrogen exposure measurements combined with XPS evidenced transfer of Mo species onto the surface of the Pt particles during the hydrogen annealing, thus the loss of the electrochemically active surface area can be interpreted as the result of partial blocking of Pt surface sites by a Mo-containing encapsulation layer. According to these results, the features of strong metal-support interaction appear in the Pt/Ti(1-x)MoxO2-C electrocatalysts already after a hydrogen treatment at the relatively low 250 °C temperature. An important feature of the hydrogen treated catalyst is its good long term stability, while the unique CO-tolerant property ensured by the composite support remained unchanged. These results confirm that utilization of the strong metal-support interaction phenomenon can be a valuable approach for improving the stability of the mixed oxide – carbon composite supported electrocatalysts.
References
Gubán D, Borbáth I, Pászti Z, Sajó I, Drotár E, Hegedűs M, Tompos A (2015) Preparation and characterization of novel Ti0.7W0.3O2–C composite materials for Pt-based anode electrocatalysts with enhanced CO tolerance. Appl Catal B: Environ 174:455–470. https://doi.org/10.1016/j.apcatb.2015.03.031
Gubán D, Tompos A, Bakos I, Vass Á, Pászti Z, Szabó EG, Sajó IE, Borbáth I (2017) Preparation of CO-tolerant anode electrocatalysts for polymer electrolyte membrane fuel cells. Int J Hydrogen Energy 42:13741–13753. https://doi.org/10.1016/j.ijhydene.2017.03.080
Vass Á, Borbáth I, Bakos I, Pászti Z, Sáfrán G, Tompos A (2019) Stability issues of CO tolerant Pt-based electrocatalysts for polymer electrolyte membrane fuel cells: comparison of Pt/Ti0.8Mo0.2O2–C with PtRu/C. React Kinet Mech Catal 126:679–699. https://doi.org/10.1007/s11144-018-1512-z
Borbáth I, Zelenka K, Vass Á, Pászti Z, Szijjartó GP, Sebestyén Z, Sáfrán G, Tompos A (2021) CO tolerant Pt electrocatalysts for PEM fuel cells with enhanced stability against electrocorrosion. Int J Hydrogen Energy 46:13534–13547. https://doi.org/10.1016/j.ijhydene.2020.08.002
Borbáth I, Tálas E, Pászti Z, Zelenka K, Ayyubov I, Salmanzade K, Sajó IE, Sáfrán G, Tompos A (2021) Investigation of Ti-Mo mixed oxide-carbon composite supported Pt electrocatalysts: effect of the type of carbonaceous materials. Appl Catal A Gen 620:118155. https://doi.org/10.1016/j.apcata.2021.118155
Li YB, Liu CT, Liu YY, Feng B, Li L, Pan HG, Kellogg W, Higgins D, Wu G (2015) Sn-doped TiO2 modified carbon to support Pt anode catalysts for direct methanol fuel cells. J Power Sources 286:354–361. https://doi.org/10.1016/j.jpowsour.2015.03.155
Shieh BJ, Tsai MC, Pan CJ, Su WN, Rick J, Chou HL, Lee JF, Hwang BJ (2017) Tuning metal support interactions enhances the activity and durability of TiO2-supported Pt nanocatalysts. Electrochim Acta 224:452–459. https://doi.org/10.1016/j.electacta.2016.12.020
Elezović NR, Babić BM, Lj G-K, Radmilović V, Krstajić NV, Vračar LJ (2010) Synthesis, characterization and electrocatalytical behavior of Nb-TiO2/Pt nanocatalyst for oxygen reduction reaction. J Power Sources 195:3961–3968. https://doi.org/10.1016/j.jpowsour.2010.01.035
Wang YJ, Wilkinson DP, Zhang J (2012) Synthesis of conductive rutile-phased Nb0.06Ti0.94O2 and its supported Pt electrocatalysts (Pt/Nb0.06Ti0.94O2) for the oxygen reduction reaction. Dalton Trans 41:1187–1194. https://doi.org/10.1039/C1DT11711D
Gao Y, Hou M, Shao Z, Zhang C, Qin X, Yi B (2014) Preparation and characterization of Ti0.7Sn0.3O2 as catalyst support for oxygen reduction reaction. J Energy Chem 23:331–337. https://doi.org/10.1016/S2095-4956(14)60155-8
Micoud F, Maillard F, Bonnefont A, Job N, Chatenet M (2010) The role of the support in COads monolayer electrooxidation on Pt nanoparticles: Pt/WOx vs. Pt/C Phys Chem Chem Phys 12:1182–1193. https://doi.org/10.1039/B915244J
Vass Á, Borbáth I, Pászti Z, Bakos I, Sajó IE, Németh P, Tompos A (2017) Effect of Mo incorporation on electrocatalytic performance of Ti-Mo mixed oxide-carbon composite supported Pt electrocatalysts. React Kinet Mech Cat 121:141–160. https://doi.org/10.1007/s11144-017-1155-5
Yazici MS, Dursun S, Borbáth I, Tompos A (2021) Reformate gas composition and pressure effect on CO tolerant Pt/Ti0.8Mo0.2O2–C electrocatalyst for PEM fuel cells. Int J Hydrogen Energy 46:13524–13533. https://doi.org/10.1016/j.ijhydene.2020.08.226
Diczházi D, Borbáth I, Bakos I, Szíjjáró GP, Tompos A, Pászti Z (2021) Design of Mo-doped mixed oxide–carbon composite supports for Pt-based electrocatalysts: the nature of the Mo-Pt interaction. Catal Today 366:31–40. https://doi.org/10.1016/j.cattod.2020.04.004
Fu Q, Wagner T (2007) Interaction of nanostructured metal overlayers with oxide surfaces. Surf Sci Rep 62:431–498. https://doi.org/10.1016/j.surfrep.2007.07.001
Fu Q, Wagner T, Olliges S, Carstanjen HD (2005) Metal−Oxide interfacial reactions: encapsulation of Pd on TiO2 (110). J Phys Chem B 109:944–951. https://doi.org/10.1021/jp046091u
Óvári L, Berkó A, Gubó R, Rácz Á, Kónya Z (2014) Effect of a gold cover layer on the encapsulation of rhodium by titanium oxides on titanium dioxide (110). J Phys Chem C 118:12340–12352. https://doi.org/10.1021/jp502748a
Pan CJ, Tsai MC, Su WN, Rick J, Akalework NG, Agegnehu AK, Cheng SY, Hwang BJ (2017) Tuning/exploiting strong metal-support interaction (SMSI) in heterogeneous catalysis. J Taiwan Inst Chem Eng 74:154–186. https://doi.org/10.1016/j.jtice.2017.02.012
Reyes P, Aguirre MC, Melián-Cabrera I, López Granados M, Fierro JLG (2002) Interfacial properties of an Ir/TiO2 system and their relevance in crotonaldehyde hydrogenation. J Catal 280:229–237. https://doi.org/10.1006/jcat.2002.3566
Chen JZ, Gao J, Probus PR, Liu W, Wu X, Wegener EC, Kropf AJ, Zemlyanov D, Zhang G, Yang X, Miller JT (2020) The effect of strong metal–support interaction (SMSI) on Pt–Ti/SiO2 and Pt–Nb/SiO2 catalysts for propane dehydrogenation. Catal Sci Technol 10:5973–5982. https://doi.org/10.1039/D0CY00897D
Bonanni S, Aït-Mansour K, Brune H, Harbich W (2011) Overcoming the strong metal−support interaction state: CO oxidation on TiO2(110)-supported Pt nanoclusters. ACS Catal 1:385–389. https://doi.org/10.1021/cs200001y
Huang SJ, Ganesan P, Popov BN (2011) Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell. Appl Catal B: Env 102:71–77. https://doi.org/10.1016/j.apcatb.2010.11.026
Shim J, Lee CR, Lee HK, Lee JS, Cairns AJ (2001) Electrochemical characteristics of Pt–WO3/C and Pt–TiO2/C electrocatalysts in a polymer electrolyte fuel cell. J Power Sources 102:172–177. https://doi.org/10.1016/S0378-7753(01)00817-5
Jaksic JM, Krstajic NV, Vracar LM, Neophytides SG, Labou D, Falaras P, Jaksic MM (2007) Spillover of primary oxides as a dynamic catalytic effect of interactive hypo-d-oxide supports. Electrochim Acta 53:349–361. https://doi.org/10.1016/j.electacta.2007.06.062
Ho VTT, Pan CJ, Rick J, Su WN, Hwang BJ (2011) Nanostructured Ti0.7Mo0.3O2 support enhances electron transfer to Pt: High-performance catalyst for oxygen reduction reaction. J Am Chem Soc 133:11716–11724. https://doi.org/10.1021/ja2039562
Takasaki F, Matsuie S, Takabatake Y, Noda Z, Hayashi A, Shiratori Y, Ito K, Sasaki K (2011) Carbon-free Pt electrocatalysts supported on SnO2 for polymer electrolyte fuel cells: electrocatalytic activity and durability. J El Chem Soc 158:B1270–B1275. https://doi.org/10.1149/1.3625918
Otor HO, Steiner JB, García-Sancho C, Alba-Rubio AC (2020) Encapsulation methods for control of catalyst deactivation: a review. ACS Catal 10:7630–7656. https://doi.org/10.1021/acscatal.0c01569
Cao XQ, Zhou J, Li S, Qin GW (2020) Ultra-stable metal nano-catalyst synthesis strategy: a perspective. Rare Met 39:113–130. https://doi.org/10.1007/s12598-019-01350-y
Jiménez-Morales I, Cavaliere S, Jones D, Roziére J (2018) Strong metal–support interaction improves activity and stability of Pt electrocatalysts on doped metal oxides. Phys Chem Chem Phys 20:8765–8772. https://doi.org/10.1039/C8CP00176F
Guha A, Lu W, Zawodzinski TA Jr, Schiraldi DA (2007) Surface modified carbons as platinum catalyst support for PEM fuel cells. Carbon 45:1506–1517. https://doi.org/10.1016/j.carbon.2007.03.023
Odetola C, Trevani L, Easton EB (2015) Enhanced activity and stability of Pt/TiO2/carbon fuel cell electrocatalyst prepared using a glucose modifier. J Power Sources 294:254–263. https://doi.org/10.1016/j.jpowsour.2015.06.066
Odetola C, Easton EB, Trevani L (2016) Investigation of TiO2/carbon electrocatalyst supports prepared using glucose as a modifier. Int J Hydrogen Energy 41:8199–8208. https://doi.org/10.1016/j.ijhydene.2015.10.035
Vass Á, Borbáth I, Bakos I, Pászti Z, Sajó IE, Tompos A (2018) Novel Pt electrocatalysts: multifunctional composite supports for enhanced corrosion resistance and improved CO tolerance. Top Catal 61:1300–1312. https://doi.org/10.1007/s11244-018-0988-0
Gao Y, Liang Y, Chambers SA (1996) Thermal stability and the role of oxygen vacancy defects in strong metal support interaction- Pt on Nb-doped TiO2(100). Surf Sci 365:638–648. https://doi.org/10.1016/0039-6028(96)00763-7
Beck A, Huang X, Artiglia L, Zabilskiy M, Wang X, Rzepka P, Palagin D, Willinger MG, Bokhoven JA (2020) The dynamics of overlayer formation on catalyst nanoparticles and strong metal-support interaction. Nature Comm 11:3220. https://doi.org/10.1038/s41467-020-17070-2
Bard AJ (1976) Electroanalytical chemistry : a series of advances. Volume 9. M. Dekker, New York, Basel
N. Fairley. (2006). CasaXPS: Spectrum processing software for XPS, AES and SIMS. Cheshire http://www.casaxps.com
Mohai M (2004) XPS MultiQuant: multimodel XPS quantification software. Surf Interface Anal 36:828–832. https://doi.org/10.1002/sia.1775
Mohai, M. (2011). XPS MultiQuant: Multi-model X-ray photoelectron spectroscopy quantification program. Version 7.00.92. http://aki.ttk.hu/XMQpages/XMQdownload.php
Grgur BN, Markovic NM, Ross PN (1999) The electro-oxidation of H2 and H2/CO mixtures on carbon-supported PtxMoy alloy catalysts. J Electrochem Soc 146:1613–1619. https://doi.org/10.1149/1.1391815
Mukerjee S, Lee SJ, Ticianelli EA, McBreen J, Grgur BN, Markovic NM, Ross PN, Giallombardo JR, De Castro ES (1999) Investigation of enhanced CO tolerance in proton exchange membrane fuel cells by carbon supported PtMo alloy catalyst. Electrochem Solid State Lett 2:12–15. https://doi.org/10.1149/1.1390718
Guillén-Villafuerte O, García G, Rodríguez JL, Pastor E, Guil-López R, Nieto E, Fierro JLG (2013) Preliminary studies of the electrochemical performance of Pt/X@MoO3/C (X= Mo2C, MoO2, Mo0) catalysts for the anode of a DMFC: Influence of the Pt loading and Mo-phase. Int J Hydrogen Energy 38:7811–7821. https://doi.org/10.1016/j.ijhydene.2013.04.083
Mukerjee S, Urian RC (2002) Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cells: electrochemical and in situ synchrotron spectroscopy. Electrochim Acta 47:3219–3231. https://doi.org/10.1016/S0013-4686(02)00242-6
Justin P, Rao GR (2011) Methanol oxidation on MoO3 promoted Pt/C electrocatalyst. Int J Hydrogen Energy 36:5875–5884. https://doi.org/10.1016/j.ijhydene.2011.01.122
Mikhailova AA, Pasynskii AA, Dobrokhotova ZV, Grinberg VA, Khazova OA (2008) CO oxidation at platinum–molybdenum electrodes. Russian J Electrochem 44(3):303–312. https://doi.org/10.1134/S1023193508030075
Sheng W, Chen S, Vescovo E, Shao-Horn Y (2012) Size influence on the oxygen reduction reaction activity and instability of supported Pt nanoparticles. J Electrochem Soc 159:B96–B103. https://doi.org/10.1149/2.009202jes
Martins PFBD, Ticianelli EA (2015) Electrocatalytic activity and stability of platinum nanoparticles supported on carbon-molybdenum oxides for the oxygen reduction reaction. ChemElectroChem 2(9):1298–1306. https://doi.org/10.1002/celc.201500196
Maillard F, Schreier S, Hanzlik M, Savinova ER, Weinkauf S, Stimming U (2005) Influence of particle agglomeration on the catalytic activity of carbon-supported Pt nanoparticles in CO monolayer oxidation. Phys Chem Chem Phys 7:385–393. https://doi.org/10.1039/B411377B
Maillard F, Peyrelade E, Soldo-Olivier Y, Chatenet M, Chaînet E, Faure R (2007) Is carbon-supported Pt-WOx composite a CO-tolerant material? Electrochim Acta 52:1958–1967. https://doi.org/10.1016/j.electacta.2006.08.024
Mayer JT, Diebold U, Madey TE, Garfunkel E (1995) Titanium and reduced titania overlayers on titanium dioxide (110). J Electron Spect Relat Phenom 73:1–11. https://doi.org/10.1016/0368-2048(94)02258-5
Gubó R, Óvári L, Kónya Z, Berkó A (2014) Growth of gold on a pinwheel TiO~1.2 encapsulation film prepared on rhodium nanocrystallites. Langmuir 30:14545–14554. https://doi.org/10.1021/la503756c
Baltrusaitis J, Mendoza-Sanchez B, Fernandez V, Veenstra R, Dukstiene N, Roberts A, Fairley N (2015) Generalized molybdenum oxide surface chemical state XPS determination via informed amorphous sample model. Appl Surf Sci 326:151–161. https://doi.org/10.1016/j.apsusc.2014.11.077
Scanlon OD, Watson GW, Payne DJ, Atkinson GR, Egdell RG, Law DSL (2010) Theoretical and experimental study of the electronic structures of MoO3 and MoO2. J Phys Chem C 114:4636–4645. https://doi.org/10.1021/jp9093172
Schroeder T, Zegenhagen J, Magg N, Immaraporn B, Freund HJ (2004) Formation of a faceted MoO2 epilayer on Mo(112) studied by XPS, UPS and STM. Surf Sci 552:85–97. https://doi.org/10.1016/j.susc.2004.01.010
Óvári L, Kiss J (2005) Angle-resolved XPS investigations of the interaction between O2 and Mo2C/Mo(100). Vacuum 80:204–207. https://doi.org/10.1016/j.vacuum.2005.08.008
Song HJ, Sung MC, Yoon H, Ju B, Kim DW (2019) Ultrafine α-phase molybdenum carbide decorated with platinum nanoparticles for efficient hydrogen production in acidic and alkaline media. Adv Sci 6:1802135. https://doi.org/10.1002/advs.201802135
Acknowledgements
The research within project No. VEKOP-2.3.2-16-2017-00013 was supported by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. Project No. 2017-2.3.7-TÉT-IN-2017-00049 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the TÉT-IN-2017 funding scheme. Project No. NNE130004 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the TR-NN-17 funding scheme. Project No. NNE 131270 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary financed under the M-ERA.NET-2018 funding scheme. We acknowledge that the purchase of THEMIS transmission electron microscope was financed by the Hungarian innovation project: VEKOP-2.3.3.15-2016-00002.
Funding
Open access funding provided by ELKH Research Centre for Natural Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Silva, C., Borbáth, I., Zelenka, K. et al. Effect of the reductive treatment on the state and electrocatalytic behavior of Pt in catalysts supported on Ti0.8Mo0.2O2-C composite. Reac Kinet Mech Cat 135, 29–47 (2022). https://doi.org/10.1007/s11144-021-02131-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-021-02131-4