
Abstract
Nuclear magnetic resonance (NMR) spectroscopy has contributed to structure-based drug development (SBDD) in a unique way compared to the other biophysical methods. The potency of a ligand binding to a protein is dictated by the binding free energy, which is an intricate interplay between entropy and enthalpy. In addition to providing the atomic resolution structural information, NMR can help to identify protein–ligand interactions that potentially contribute to the enthalpic component of the free energy. NMR can also illuminate dynamic aspects of the interaction, which correspond to the entropic term of the free energy. The ability of NMR to access both terms in the free energy equation stems from the suite of experiments developed to shed light on various aspects that contribute to both entropy and enthalpy, deepening our understanding of the biological function of macromolecules and assisting to target them in physiological conditions. Here we provide a brief account of the contribution of NMR to SBDD, highlighting hallmark examples and discussing the challenges that demand further method development. In the era of integrated biology, the unique ability of NMR to directly ascertain structural and dynamical aspects of macromolecule and monitor changes in these properties upon engaging a ligand can be combined with computational and other structural and biophysical methods to provide a more complete picture of the energetics of drug engagement with the target. Such efforts can be used to engineer better drugs.





Similar content being viewed by others
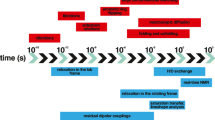


Change history
20 September 2020
Unfortunately, in the original publication, Fig. 5 was published incorrectly. The correct version is given below:
References
Agafonov RV, Wilson C, Otten R, Buosi V, Kern D (2014) Energetic dissection of Gleevec's selectivity toward human tyrosine kinases. Nat Struct Mol Biol 21:848–853
Ahlbach CL et al (2015) Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Future Med Chem 7:2121–2130
Akke M (2012) Conformational dynamics and thermodynamics of protein-ligand binding studied by NMR relaxation. Biochem Soc Trans 40:419–423
Akke M, Brueschweiler R, Palmer AG (1993) NMR order parameters and free energy: an analytical approach and its application to cooperative calcium(2+) binding by calbindin D9k. J Am Chem Soc 115:9832–9833
Alberty RA, Hammes GG (1958) Application of the theory of diffusion-controlled reactions to enzyme kinetics. J Phys Chem 62:154–159
Albrand JP, Birdsall B, Feeney J, Roberts GCK, Burgen ASV (1979) The use of transferred nuclear Overhauser effects in the study of the conformations of small molecules bound to proteins. Int J Biol Macromol 1:37–41
Aldeghi M, Gapsys V, de Groot BL (2018) Accurate estimation of ligand binding affinity changes upon protein mutation. ACS Cent Sci 4:1708–1718
Ambadipudi S, Reddy JG, Biernat J, Mandelkow E, Zweckstetter M (2019) Residue-specific identification of phase separation hot spots of Alzheimer's-related protein tau. Chem Sci 10:6503–6507
Anthis NJ, Clore GM (2015) Visualizing transient dark states by NMR spectroscopy. Q Rev Biophys 48:35–116
Appavoo SD, Huh S, Diaz DB, Yudin AK (2019) Conformational control of macrocycles by remote structural modification. Chem Rev 119:9724–9752
Balaram P, Bothner-By AA, Dadok J (1972) Negative nuclear Overhuaser effects as probes of macromolecular structure. J Am Chem Soc 94:4015–4017
Balazs AYS et al (2019) Free ligand 1D NMR conformational signatures to enhance structure based drug design of a Mcl-1 inhibitor (AZD5991) and other synthetic macrocycles. J Med Chem 62:9418–9437
Ban D, Iconaru LI, Ramanathan A, Zuo J, Kriwacki RW (2017) A small molecule causes a population shift in the conformational landscape of an intrinsically disordered protein. J Am Chem Soc 139:13692–13700
Berlow RB, Dyson HJ, Wright PE (2017) Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 543:447
Bom A, Hope F, Rutherford S, Thomson K (2009) Preclinical pharmacology of sugammadex. J Crit Care 24:29–35
Bonomi M, Pellarin R, Vendruscolo M (2018) Simultaneous determination of protein structure and dynamics using cryo-electron microscopy. Biophys J 114:1604–1613
Bowman GR, Geissler PL (2012) Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc Natl Acad Sci 109:11681
Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S (2015) Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc Natl Acad Sci 112:2734
Bradshaw JM et al (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 11:525–531
Carneiro MG, Ab E, Theisgen S, Siegal G (2017) NMR in structure-based drug design. Essays Biochem 61:485–493
Caro JA et al (2017) Entropy in molecular recognition by proteins. Proc Natl Acad Sci USA 114:6563–6568
Carroll MJ et al (2012) Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat Chem Biol 8:246–252
Chang CE, Chen W, Gilson MK (2007) Ligand configurational entropy and protein binding. Proc Natl Acad Sci USA 104:1534–1539
Chodera JD, Mobley DL (2013) Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu Rev Biophys 42:121–142
Chung BKW, White CJ, Scully CCG, Yudin AK (2016) The reactivity and conformational control of cyclic tetrapeptides derived from aziridine-containing amino acids. Chem Sci 7:6662–6668
Cicero DO, Barbato G, Bazzo R (1995) NMR analysis of molecular flexibility in solution: a new method for the study of complex distributions of rapidly exchanging conformations. Application to a 13-residue peptide with an 8-residue loop. J Am Chem Soc 117:1027–1033
Copeland RA, Pompliano DL, Meek TD (2006) Drug–target residence time and its implications for lead optimization. Nat Rev Drug Discov 5:730–739
Dalvit C et al (2000) Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J Biomol NMR 18:65–68
Dalvit C, Ardini E, Fogliatto GP, Mongelli N, Veronesi M (2004) Reliable high-throughput functional screening with 3-FABS. Drug Discov Today 9:595–602
Das K, Arnold E (2013a) HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol 3:111–118
Das K, Arnold E (2013b) HIV-1 reverse transcriptase and antiviral drug resistance Part 2. Curr Opin Virol 3:119
Dautzenberg FM, Neysari S (2005) Irreversible binding kinetics of neuropeptide Y ligands to Y2 but not to Y1 and Y5 receptors. Pharmacology 75:21–29
Diehl C et al (2010) Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of galectin-3. J Am Chem Soc 132:14577–14589
Dill KA, Bromberg S (2011) Molecular driving forces : statistical thermodynamics in biology, chemistry, physics, and nanoscience. Garland Science, New York
Disse B, Speck GA, Rominger KL, Witek TJ Jr, Hammer R (1999) Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci 64:457–464
Dunitz JD (1994) The entropic cost of bound water in crystals and biomolecules. Science 264:670
Elg M, Gustafsson D, Deinum J (1997) The importance of enzyme inhibition kinetics for the effect of thrombin inhibitors in a rat model of arterial thrombosis. Thromb Haemost 78:1286–1292
Fang Z et al (2018) Inhibition of K-RAS4B by a unique mechanism of action: stabilizing membrane-dependent occlusion of the effector-binding site. Cell Chem Biol 25:1327–1336.e4
Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM (2011) Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature 480:268–272
Fawzi NL, Ying J, Torchia DA, Clore GM (2012) Probing exchange kinetics and atomic resolution dynamics in high-molecular-weight complexes using dark-state exchange saturation transfer NMR spectroscopy. Nat Protoc 7:1523–1533
Fedorov DG, Kitaura K (2009) The fragment molecular orbital method : practical applications to large molecular systems. CRC Press, Boca Raton
Fedorov DG, Nagata T, Kitaura K (2012) Exploring chemistry with the fragment molecular orbital method. Phys Chem Chem Phys 14:7562–7577
Fernandez A, Fraser C, Scott LR (2012) Purposely engineered drug-target mismatches for entropy-based drug optimization. Trends Biotechnol 30:1–7
Ferrari AM, Degliesposti G, Sgobba M, Rastelli G (2007) Validation of an automated procedure for the prediction of relative free energies of binding on a set of aldose reductase inhibitors. Bioorg Med Chem 15:7865–7877
Ferreira de Freitas R, Schapira M (2017) A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 8:1970–1981
Fersht AR et al (1984) Analysis of enzyme structure and activity by protein engineering. Angew Chem Int Ed Engl 23:467–473
Fersht AR et al (1985) Hydrogen bonding and biological specificity analysed by protein engineering. Nature 314:235–238
Fox JM, Zhao M, Fink MJ, Kang K, Whitesides GM (2018) The molecular origin of enthalpy/entropy compensation in biomolecular recognition. Annu Rev Biophys 47:223–250
Frederick KK, Marlow MS, Valentine KG, Wand AJ (2007) Conformational entropy in molecular recognition by proteins. Nature 448:325–329
Freire E (2008) Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today 13:869–874
Freire E (2009) A thermodynamic approach to the affinity optimization of drug candidates. Chem Biol Drug Des 74:468–472
Furuya T, Kamlet AS, Ritter T (2011) Catalysis for fluorination and trifluoromethylation. Nature 473:470–477
Gabrielsson J et al (2009) Early integration of pharmacokinetic and dynamic reasoning is essential for optimal development of lead compounds: strategic considerations. Drug Discov Today 14:358–372
Geist L et al (2017) Direct NMR probing of hydration shells of protein ligand interfaces and its application to drug design. J Med Chem 60:8708–8715
Gil S, Espinosa JF, Parella T (2010) IPAP–HSQMBC: measurement of long-range heteronuclear coupling constants from spin-state selective multiplets. J Magn Reson 207:312–321
Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA (2015) Applications of fluorine in medicinal chemistry. J Med Chem 58:8315–8359
Gilson MK et al (2016) BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res 44:D1045–D1053
Goldbach L et al (2019) Folding then binding vs folding through binding in macrocyclic peptide inhibitors of human pancreatic alpha-amylase. ACS Chem Biol 14:1751–1759
Gorgulla C et al (2020) An open-source drug discovery platform enables ultra-large virtual screens. Nature 580:663–668
Holloway MK et al (1995) A priori prediction of activity for HIV-1 protease inhibitors employing energy minimization in the active site. J Med Chem 38:305–317
Horne WS, Olsen CA, Beierle JM, Montero A, Ghadiri MR (2009) Probing the bioactive conformation of an archetypal natural product HDAC inhibitor with conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew Chem Int Ed 48:4718–4724
Igumenova TI, Frederick KK, Wand AJ (2006) Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem Rev 106:1672–1699
Jain AN et al (2019) Complex macrocycle exploration: parallel, heuristic, and constraint-based conformer generation using ForceGen. J Comput Aided Mol Des 33:531–558
Janin J (1995) Protein-protein recognition. Prog Biophys Mol Biol 64:145–166
Jencks WP (1975) Binding energy, specificity, and enzymic catalysis: the circe effect. Adv Enzymol Relat Areas Mol Biol 43:219–410
Jorge C, Marques BS, Valentine KG, Wand AJ (2019) Characterizing protein hydration dynamics using solution NMR spectroscopy. In: Wand AJ (ed) Methods in enzymology, vol 615. Academic Press, Cambridge, pp 77–101
Joshi P, Vendruscolo M (2015) Druggability of intrinsically disordered proteins. Adv Exp Med Biol 870:383–400
Kasinath V, Sharp KA, Wand AJ (2013) Microscopic insights into the NMR relaxation-based protein conformational entropy meter. J Am Chem Soc 135:15092–15100
Kauzmann W (1959) Some factors in the interpretation of protein denaturation. Adv Protein Chem 14:1–63
Kawasaki Y, Freire E (2011) Finding a better path to drug selectivity. Drug Discov Today 16:985–990
Kay LE, Prestegard JH (1987a) Methyl group dynamics from relaxation of double quantum filtered NMR signals. Application to deoxycholate. J Am Chem Soc 109:3829–3835
Kay LE, Prestegard JH (1987b) Methyl group dynamics from relaxation of double quantum filtered NMR signals. Application to deoxycholate. J Am Chem Soc 109:3829–3835
Keighley W (2011) The need for high throughput kinetics early in the drug discovery process. Drug Discov World Summer 12:39–45
Kitaura K, Ikeo E, Asada T, Nakano T, Uebayasi M (1999) Fragment molecular orbital method: an approximate computational method for large molecules. Chem Phys Lett 313:701–706
Kleckner IR, Foster MP (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim Biophys Acta 1814:942–968
Klotz IM (1959) Protein behavior. Science 129:608
Kostylev MA et al (2018) Liquid and hydrogel phases of PrP(C) linked to conformation shifts and triggered by Alzheimer's amyloid-beta oligomers. Mol Cell 72:426–443.e12
Kövér KE, Forgó P (2004) J-modulated ADEQUATE (JM-ADEQUATE) experiment for accurate measurement of carbon–carbon coupling constants. J Magn Reson 166:47–52
Kovermann M, Rogne P, Wolf-Watz M (2016) Protein dynamics and function from solution state NMR spectroscopy. Q Rev Biophys 49:e6
Kranz M et al (2006) Solution, solid phase and computational structures of apicidin and its backbone-reduced analogs. J Pept Sci 12:383–388
Krishnamurthy VV (1996) Excitation-sculptured indirect-detection experiment (EXSIDE) for long-range CH coupling-constant measurement. J Magn Reson Ser A 121:33–41
Lee C-J et al (2016) Drug design from the cryptic inhibitor envelope. Nat Commun 7:10638
Lee AL, Kinnear SA, Wand AJ (2000) Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nat Struct Biol 7:72–77
Liu Y et al (2017) Unequivocal determination of complex molecular structures using anisotropic NMR measurements. Science 356:eaam5349
Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK (2007) BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res 35:D198–D201
Luchinat E et al (2020) Drug screening in human cells by nmr spectroscopy allows the early assessment of drug potency. Angew Chem Int Ed Engl 59:6535–6539
Luckner SR, Liu N, Am Ende CW, Tonge PJ, Kisker C (2010) A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem 285:14330–14337
Luque I, Freire E (2002) Structural parameterization of the binding enthalpy of small ligands. Proteins: Struct Funct Bioinform 49:181–190
Malde AK, Hill TA, Iyer A, Fairlie DP (2019) Crystal structures of protein-bound cyclic peptides. Chem Rev 119:9861–9914
Manglik A et al (2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161:1101–1111
Markovitch O, Agmon N (2007) Structure and energetics of the hydronium hydration shells. J Phys Chem A 111:2253–2256
Marlow MS, Dogan J, Frederick KK, Valentine KG, Wand AJ (2010) The role of conformational entropy in molecular recognition by calmodulin. Nat Chem Biol 6:352–358
Mauldin RV, Carroll MJ, Lee AL (2009) Dynamic dysfunction in dihydrofolate reductase results from antifolate drug binding: modulation of dynamics within a structural state. Structure 17:386–394
Merino F, Raunser S (2017) Electron cryo-microscopy as a tool for structure-based drug development. Angew Chem Int Ed Engl 56:2846–2860
Milo R, Phillips RB, Orme N, Garland S (2016) Cell biology by the numbers. Garland Science, Taylor & Francis Group, New York, Abingdon
Mizukoshi Y et al (2016a) Improvement of ligand affinity and thermodynamic properties by NMR-based evaluation of local dynamics and surface complementarity in the receptor-bound state. Angew Chem Int Ed Engl 55:14606–14609
Mizukoshi Y et al (2016b) Improvement of ligand affinity and thermodynamic properties by NMR-based evaluation of local dynamics and surface complementarity in the receptor-bound state. Angew Chem Int Ed 55:14606–14609
Morrison C (2018) Constrained peptides' time to shine? Nat Rev Drug Discov 17:531–533
Namanja AT et al (2010) Toward flexibility-activity relationships by NMR spectroscopy: dynamics of Pin1 ligands. J Am Chem Soc 132:5607–5609
Ndukwe IE et al (2019) 13C NMR-based approaches for solving challenging stereochemical problems. Org Lett 21:4072–4076
Nguyen QNN, Schwochert J, Tantillo DJ, Lokey RS (2018) Using (1)H and (13)C NMR chemical shifts to determine cyclic peptide conformations: a combined molecular dynamics and quantum mechanics approach. Phys Chem Chem Phys 20:14003–14012
Nikiforovich GV, Kövér KE, Zhang W-J, Marshall GR (2000) Cyclopentapeptides as flexible conformational templates. J Am Chem Soc 122:3262–3273
Nitsche C, Otting G (2017) Pseudocontact shifts in biomolecular NMR using paramagnetic metal tags. Prog Nucl Magn Reson Spectrosc 98–99:20–49
Nitsche C, Otting G (2018) NMR studies of ligand binding. Curr Opin Struct Biol 48:16–22
Norton SR, Leung WE, Chandrashekaran RI, MacRaild AC (2016) Applications of 19F-NMR in fragment-based drug Discovery. Molecules 21:860
Nucci NV, Pometun MS, Wand AJ (2011) Site-resolved measurement of water-protein interactions by solution NMR. Nat Struct Mol Biol 18:245–249
Ono S et al (2019) Conformation and permeability: cyclic hexapeptide diastereomers. J Chem Inf Model 59:2952–2963
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503:548
Otero-Ramirez ME, Passioura T, Suga H (2018) Structural features and binding modes of thioether-cyclized peptide ligands. Biomedicines 6:116
Otting G, Liepinsh E (1995) Protein hydration viewed by high-resolution NMR spectroscopy: implications for magnetic resonance image contrast. Acc Chem Res 28:171–177
Otting G, Wuethrich K (1989) Studies of protein hydration in aqueous solution by direct NMR observation of individual protein-bound water molecules. J Am Chem Soc 111:1871–1875
Oxenoid K, Chou JJ (2016) A functional NMR for membrane proteins: dynamics, ligand binding, and allosteric modulation. Protein Sci 25:959–973
Öztürk H, Özgür A, Ozkirimli E (2018) DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 34:i821–i829
Page MI (1991) The energetics of intramolecular reactions and enzyme catalysis. Philos Trans R Soc Lond B 332:149–156
Palmer AG 3rd (2001) NMR probes of molecular dynamics: overview and comparison with other techniques. Annu Rev Biophys Biomol Struct 30:129–155
Palmer AG 3rd (2004) NMR characterization of the dynamics of biomacromolecules. Chem Rev 104:3623–3640
Pan AC, Borhani DW, Dror RO, Shaw DE (2013) Molecular determinants of drug-receptor binding kinetics. Drug Discov Today 18:667–673
Perola E, Charifson PS (2004) Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J Med Chem 47:2499–2510
Petros AM et al (2010) Discovery of a potent and selective Bcl-2 inhibitor using SAR by NMR. Bioorg Med Chem Lett 20:6587–6591
Phillips C et al (2011) Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc 133:9696–9699
Pitsawong W et al (2018) Dynamics of human protein kinase Aurora A linked to drug selectivity. eLife 7:e36656
Prosser RS, Kim TH (2015) Nuts and bolts of CF3 and CH3 NMR toward the understanding of conformational exchange of GPCRs. In: Filizola M (ed) G protein-coupled receptors in drug discovery: methods and protocols. Springer, New York, pp 39–51
Punjani A, Fleet DJ (2020) 3D variability analysis: directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images. bioRxiv. https://doi.org/10.1101/2020.04.08.032466
Reibarkh M, Malia TJ, Wagner G (2006a) NMR distinction of single- and multiple-mode binding of small-molecule protein ligands. J Am Chem Soc 128:2160–2161
Reibarkh M, Malia TJ, Hopkins BT, Wagner G (2006b) Identification of individual protein-ligand NOEs in the limit of intermediate exchange. J Biomol NMR 36:1–11
Reibarkh M, Williamson RT, Martin GE, Bermel W (2013) Broadband inversion of 1JCC responses in 1, n-ADEQUATE spectra. J Magn Reson 236:126–133
Reif B et al (1996) ADEQUATE, a new set of experiments to determine the constitution of small molecules at natural abundance. J Magn Reson 118:282–285
Renaud JP et al (2016) Biophysics in drug discovery: impact, challenges and opportunities. Nat Rev Drug Discov 15:679–698
Renaud JP et al (2018) Cryo-EM in drug discovery: achievements, limitations and prospects. Nat Rev Drug Discov 17:471–492
Reynolds CH, Holloway MK (2011) Thermodynamics of ligand binding and efficiency. ACS Med Chem Lett 2:433–437
Robertson IM, Spyracopoulos L, Sykes BD (2009) The evaluation of isotope editing and filtering for protein—ligand interaction elucidation by NMR. In: Puglisi JD (ed) Biophysics and the challenges of emerging threats. Springer, Dordrecht, pp 101–119
Saleh T, Rossi P, Kalodimos CG (2017) Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat Struct Mol Biol 24:893–901
Saur M et al (2020) Fragment-based drug discovery using cryo-EM. Drug Discov Today 25:485–490
Saurí J, Parella T, Espinosa JF (2013) CLIP-HSQMBC: easy measurement of small proton–carbon coupling constants in organic molecules. Org Biomol Chem 11:4473–4478
Saurí J, Parella T, Williamson RT, Martin GE (2017) Improving the performance of J-modulated ADEQUATE experiments through homonuclear decoupling and non-uniform sampling. Magn Reson Chem 55:191–197
Scapin G, Potter CS, Carragher B (2018) Cryo-EM for small molecules discovery, design, understanding, and application. Cell Chem Biol 25:1318–1325
Schreiber SL, Crabtree GR (1992) The mechanism of action of cyclosporin A and FK506. Immunol Today 13:136–142
Sekhar A, Kay LE (2019) An NMR view of protein dynamics in health and disease. Annu Rev Biophys 48:297–319
Sharp KA, Nicholls A, Friedman R, Honig B (1991) Extracting hydrophobic free energies from experimental data: relationship to protein folding and theoretical models [published erratum appears in Biochemistry 1993 May 25;32(20):5490]. Biochemistry 30:9686–9697
Shimada I, Ueda T, Kofuku Y, Eddy MT, Wüthrich K (2019) GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat Rev Drug Discov 18:59–82
Snyder PW et al (2011) Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc Natl Acad Sci USA 108:17889–17894
Souers AJ et al (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19:202–208
Stillinger FH (1973) Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J Solut Chem 2:141–158
Stoffel S, Zhang QW, Li DH, Smith BD, Peng JW (2020) NMR relaxation dispersion reveals macrocycle breathing dynamics in a cyclodextrin-based rotaxane. J Am Chem Soc 142:7413–7424
Sun H, Kay LE, Tugarinov V (2011) An optimized relaxation-based coherence transfer NMR experiment for the measurement of side-chain order in methyl-protonated, highly deuterated proteins. J Phys Chem B 115:14878–14884
Takeuchi K, Wagner G (2006) NMR studies of protein interactions. Curr Opin Struct Biol 16:109–117
Takeuchi K, Tokunaga Y, Imai M, Takahashi H, Shimada I (2014) Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci Rep 4:1–12
Thafar M, Raies AB, Albaradei S, Essack M, Bajic VB (2019) Comparison study of computational prediction tools for drug-target binding affinities. Front Chem 7:782
Tjandra N, Bax A (1997) Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278:1111–1114
Tokunaga Y, Takeuchi K, Takahashi H, Shimada I (2014) Allosteric enhancement of MAP kinase p38α's activity and substrate selectivity by docking interactions. Nat Struct Mol Biol 21:704–711
Tokunaga Y, Takeuchi K, Shimada I (2017) Forbidden coherence transfer of 19F nuclei to quantitatively measure the dynamics of a CF3-containing ligand in receptor-bound states. Molecules 22:1492
Tokunaga Y, Viennet T, Arthanari H, Takeuchi K (2020) Spotlight on the ballet of proteins: the structural dynamic properties of proteins illuminated by solution NMR. Int J Mol Sci 21:1829
Tompa P, Fuxreiter M (2008) Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci 33:2–8
Tonge PJ (2018) Drug-target kinetics in drug discovery. ACS Chem Neurosci 9:29–39
Trigo-Mourino P, Griesinger C, Lee D (2017) Label-free NMR-based dissociation kinetics determination. J Biomol NMR 69:229–235
Tugarinov V, Sprangers R, Kay LE (2007) Probing side-chain dynamics in the proteasome by relaxation violated coherence transfer NMR spectroscopy. J Am Chem Soc 129:1743–1750
Tzeng SR, Kalodimos CG (2009) Dynamic activation of an allosteric regulatory protein. Nature 462:368–372
Vacic V et al (2012) Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput Biol 8:e1002709
Vahidi S et al (2020) An allosteric switch regulates Mycobacterium tuberculosis ClpP1P2 protease function as established by cryo-EM and methyl-TROSY NMR. Proc Natl Acad Sci 117:5895
Valeur E et al (2017) New modalities for challenging targets in drug discovery. Angew Chem Int Ed Engl 56:10294–10323
Vallurupalli P, Bouvignies G, Kay LE (2012) Studying "invisible" excited protein states in slow exchange with a major state conformation. J Am Chem Soc 134:8148–8161
Vallurupalli P, Sekhar A, Yuwen T, Kay LE (2017) Probing conformational dynamics in biomolecules via chemical exchange saturation transfer: a primer. J Biomol NMR 67:243–271
van der Spoel D, van Maaren PJ, Larsson P, Timneanu N (2006) Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J Phys Chem B 110:4393–4398
van Noord JA, Bantje TA, Eland ME, Korducki L, Cornelissen PJ, The Dutch Tiotropium Study Group (2000) A randomised controlled comparison of tiotropium nd ipratropium in the treatment of chronic obstructive pulmonary disease. Thorax 55:289–294
Vinogradov AA, Yin Y, Suga H (2019) Macrocyclic peptides as drug candidates: recent progress and remaining Challenges. J Am Chem Soc 141:4167–4181
Vulpetti A, Dalvit C (2012) Fluorine local environment: from screening to drug design. Drug Discov Today 17:890–897
Wahyudi H, Tantisantisom W, McAlpine SR (2014) Sanguinamide B analogs: identification of active macrocyclic structures. Tetrahedron Lett 55:2389–2393
Wand AJ (2001) Dynamic activation of protein function: a view emerging from NMR spectroscopy. Nat Struct Biol 8:926–931
Wand AJ (2012) The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr Opin Struct Biol 23:75–81
Wand AJ, Sharp KA (2018) Measuring entropy in molecular recognition by proteins. Annu Rev Biophys 47:41–61
Wang J et al (2014) Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem Rev 114:2432–2506
Wells JA, McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450:1001–1009
Williams RJ (1981) Studies of larger motions within proteins by nuclear magnetic resonance spectroscopy. Biochem Soc Symp 46:57–72
Wilson JE et al (2016) Discovery of novel indoline cholesterol ester transfer protein inhibitors (CETP) through a structure-guided approach. ACS Med Chem Lett 7:261–265
Witek J et al (2016) Kinetic models of cyclosporin A in polar and apolar environments reveal multiple congruent conformational states. J Chem Inf Model 56:1547–1562
Witek J et al (2017) Interconversion rates between conformational states as rationale for the membrane permeability of cyclosporines. ChemPhysChem 18:3309–3314
Witek J et al (2019) Rationalization of the membrane permeability differences in a series of analogue cyclic decapeptides. J Chem Inf Model 59:294–308
Wlodawer A, Vondrasek J (1998) Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct 27:249–284
Ye L, Van Eps N, Zimmer M, Ernst OP, Scott Prosser R (2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533:265
Yip KM, Fischer N, Paknia E, Chari A, Stark H (2020) Breaking the next Cryo-EM resolution barrier—atomic resolution determination of proteins! bioRxiv. https://doi.org/10.1101/2020.05.21.106740
Ziarek JJ, Baptista D, Wagner G (2018) Recent developments in solution nuclear magnetic resonance (NMR)-based molecular biology. J Mol Med (Berl) 96:1–8
Acknowledgements
We thank Dr. Shriya S. Srinivasan for the discussions and critical comments on this manuscript. This work is also supported by JSPS-ISF Joint Research Program and JSPS KAKENHI Grant-in-Aid for Challenging Research (Exploratory) 20H03378 and 20H04722 to KT. HA acknowledges funding from NIH (GM136859) and the Claudia Adams Barr Program for Innovative Cancer Research.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dubey, A., Takeuchi, K., Reibarkh, M. et al. The role of NMR in leveraging dynamics and entropy in drug design. J Biomol NMR 74, 479–498 (2020). https://doi.org/10.1007/s10858-020-00335-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-020-00335-9