
Abstract
Collection and storage of crop wild relative (CWR) germplasm is crucial for preserving species genetic diversity and crop improvement. Nevertheless, much of the genetic variation of CWRs is absent in ex situ collections and detailed passport data are often lacking. Here, we focussed on Musa balbisiana, one of the two main progenitor species of many banana cultivars. We investigated the genetic structure of M. balbisiana across its distribution range using microsatellite markers. Accessions stored at the International Musa Germplasm Transit Centre (ITC) ex situ collection were compared with plant material collected from multiple countries and home gardens from Vietnam. Genetic structure analyses revealed that accessions could be divided into three main clusters. Vietnamese and Chinese populations were assigned to a first and second cluster respectively. A third cluster consisted of ITC and home garden accessions. Samples from Papua New Guinea were allocated to the cluster with Chinese populations but were assigned to a separate fourth cluster if the number of allowed clusters was set higher. Only one ITC accession grouped with native M. balbisiana populations and one group of ITC accessions was nearly genetically identical to home garden samples. This questioned their wild status, including accessions used as reference for wild M. balbisiana. Moreover, most ITC accessions and home garden samples were genetically distinct from wild populations. Our results highlight that additional germplasm should be collected from the native distribution range, especially from Northeast India, Myanmar, China, and the Philippines and stored for ex situ conservation at the ITC. The lack of passport data for many M. balbisiana accessions also complicates the interpretation of genetic information in relation to cultivation and historical dispersal routes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crop wild relatives (CWRs) are wild species that are closely related to their associated crop and are a source of genetic diversity for crop improvement. They potentially contain new key alleles against environmental stressors or are desirable for the modification of quantitative and qualitative crop traits such as yield, taste, and shape (Hajjar and Hodgkin 2007; Dempewolf et al. 2017). To safeguard the role of CWRs for future plant breeding, it is very important to collect, identify, protect and use this genetic variation present in CWRs in both in situ and ex situ conservation (Heywood et al. 2007; Maxted and Kell 2009; Castañeda-Álvarez et al. 2016).
In the last decades, several efforts have been undertaken for conservation of CWRs ex situ as there is a growing awareness of their importance for crop improvement and food security (Hajjar and Hodgkin 2007; McCouch 2013; Dempewolf et al. 2017). However, CWRs are still poorly represented in gene banks (Castañeda-Álvarez et al. 2016; Khoury et al. 2021; Mertens et al. 2021b). For example, over 4.1 million accessions are listed in the Genesys global database for crop diversity conserved in gene banks (Genesys 2021) and only 14% of them are considered wild in origin. Based on the European Search Catalogue for Plant Genetic Resources (EURISCO), Ford-Lloyd et al. (2011) demonstrated that only 6% of European CWR species are conserved in ex situ collections. Moreover, many of these species in collections are only represented by a single specimen that is further clonally propagated. This extremely narrow collection basis has several drawbacks: (i) only a subset of the genetic variation is captured from the population where it was sampled from; (ii) there is a risk of genetic drift during regeneration, especially when the original sampling size is low; (iii) when conserved in vitro, somaclonal variation may arise, further differentiating the germplasm accessions from the original wild populations (Krishna et al. 2016); and (iv) ex situ collections are not prone to the same biological (species interactions) and abiotic (climate) processes compared to wild populations, therefore withholding their adaptation to gradual changes in environmental conditions (Meilleur and Hodgkin 2004; Heywood 2016). Moreover, not all species can be conserved ex situ due to either specific ecosystem interactions or limitations in seed storage such as seeds with recalcitrant or intermediate storage classification (Rasmussen et al. 2015).
Besides the need to expand the total number of CWRs in gene banks, the metadata associated with stored CWRs is of vital importance for further use. Although guidelines have been developed for the collection and management of CWR data (e.g. descriptors for uploading passport data to EURISCO or Genesys), such information is often lacking. Moreover, there is currently no standardised format or global portal to access data that are specifically gathered on CWRs (Engels and Thormann 2020).
Bananas (Musaceae) are one of the worlds’ most important staple foods. With over 150 million tonnes being produced annually, they contribute to the income and diets of hundreds of millions of people (FAO 2018, 2019). There are 83 wild banana species (Musa L.) according to the World Checklist for Selected Plant families (WCSP 2021), and over 1,000 varieties have been described (Daniells et al. 2001; Ploetz et al. 2007; Perrier et al. 2011; Ruas et al. 2017). Worldwide, a total of 31 field and in vitro collections conserve 6,772 banana accessions (Ruas et al. 2017), of which 1617 accessions are stored at the International Musa Germplasm Transit Centre (ITC), with 1100 accessions being duplicated and conserved cryogenically (Van den houwe et al. 2020). To date, the ITC has the largest collection of Musa germplasm in the world with the long term security of the banana gene pool as main goal, while also globally providing pest- and disease-free germplasm (Van den houwe et al. 2020). Within the ITC, most acquisitions (84%) are cultivated bananas. Moreover, the 16% wild accessions at the ITC comprise 34 Musaceae species, yet these are often represented by only one clonally reproduced genotype. Next to in vitro collections, many institutes hold banana seed collections, such as the Plant Resources Center in Vietnam and the Millennium Seed Bank in Great Britain (Kallow et al. 2022). Banana seed storage can be a cost-efficient complementary method for the long-term conservation and distribution of banana genetic resources (Li and Pritchard 2009), but many issues remain unresolved regarding their collection, seed banking, and germination (Brown et al. 2017; Kallow et al. 2020b, a; Panis et al. 2020).
Germplasm from wild or cultivated banana material can be requested from gene banks with information coming from the Musa Germplasm Information System (MGIS) (Ruas et al. 2017). Accessions should typically come with passport metadata that includes information on taxonomy, ecology, geography, and ethnobotanical uses. Such data are also helpful to develop breeding programmes and strategies for collecting additional germplasm. Passport data are required to determine what part of the gene pool is insufficiently conserved and to pinpoint favourable geographical regions for additional germplasm collection (Meyer 2015; Weise et al. 2020). However, this information is often missing, unavailable online, or unknown (Dempewolf et al. 2017). For example, georeferenced sampling locations of germplasm accessions are important to carry out a gap analysis to identify which region should be explored or prioritized for future collecting.
Northern Indo-Burma was recently revealed as the region of origin of the banana family (Musaceae) (Janssens et al. 2016) and this region was also marked with a high climatic suitability for many banana species (Mertens et al. 2021b). However, species specific assessments of genetic variation on a large geographic scale have rarely been assessed in this family.
In this study, we focussed on Musa balbisiana Colla, the single progenitor of the BBB cultivar group as well as the donor of the “B genome” to hybrid cultivars with M. acuminata Colla (“A genome”) belonging to the AB, ABB, AAB, AABB, and ABBB groups (Simmonds and Shepherd 1955). Musa balbisiana is a wild diploid species native to (sub)-tropical rainforests ranging from Northeast India to South China and northern Vietnam. Although the species is also present in Taiwan, the Ryukyu islands (Japan), Indonesia, Malaysia, the Philippines, and Papua New Guinea (PNG), these occurrences have been attributed to human-mediated introductions (De Langhe et al. 2015). While M. balbisiana is not parthenocarpic and has seedy fruits, it is regularly cultivated and many parts of the plants are used for food, fodder, fibre, wrapping material or medicine (Kennedy 2009). After being introduced into regions with suitable climatic conditions, M. balbisiana establishes vigorous populations, which are typically called “feral”. Although the Philippines are often considered as part of the native distribution area of M. balbisiana because of its widespread presence across multiple Philippine provinces (Sotto and Rabara 2000), De Langhe et al. (2015) suggested that M. balbisiana accessions were introduced, based on the small variation in local AAB cultivar subgroups that developed there since the start of the cultivation of edible M. acuminata Colla (AA group). In addition, the absence of M. balbisiana in other parts of Marine Southeast Asia and its scattered distribution in PNG may indicate non-natural introductions in the Philippines, but it is no conclusive evidence (De Langhe et al. 2015).
Up to now, few studies have focussed on the genetic diversity present among wild M. balbisiana populations within the native distribution range (Ge et al. 2005; Uma et al. 2006; Wang et al. 2007). Often, genetic research is done on M. balbisiana material obtained from ex situ collections (Ude et al. 2002; Youssef et al. 2011; Bawin et al. 2019), grown outside the native distribution range (Ahmad et al. 2014), or with cultivated material and hybrids (Doloiras-Laraño et al. 2018). Moreover, the results of those studies are difficult to compare, as they use different methods and genetic markers (e.g. AFLP, SSRs, RAPD). More recently, two studies used a set of 18 polymorphic SSR markers to interlink genetic variation in wild M. balbisiana accessions with that in ex situ collections. Bawin et al. (2019) compared the genetic diversity of seeds from wild M. balbisiana populations from Yunnan (China) with those from ex situ seed collections, whereas Kallow et al. (2021) compared seed collections to source populations of M. balbisiana. In the present study, we investigated the genetic structure of M. balbisiana in its complete distribution range (native + feral) in Southeast Asia and Melanesia. With this approach we aim to (i) identify areas in Southeast Asia that require additional collecting; (ii) investigate the extent of the genetic variation in M. balbisiana accessions that are currently stored and maintained in the ITC gene bank; and (iii) examine the importance of passport data for linking genetic data with the putative interpretation of human interference and dispersal routes.
Methods
Taxon sampling
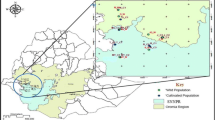
Musa balbisiana samples from different countries were acquired from three different sources (Supplementary Information – Table 1). Firstly, SSR data of Musa balbisiana were retrieved from two previous studies (Bawin et al. 2019; Mertens et al. 2021a). Secondly, 28 different germplasm accessions of M. balbisiana from the ITC (one sample per accession) originating from the Philippines, Indonesia, India, PNG, China, and Thailand were included (Fig. 1). Thirdly, to trace the origin of plants grown in home gardens in Vietnam, leaf material from six M. balbisiana accessions grown in home gardens in South-Central Vietnam and one from a home garden in North Vietnam were selected. Single or multiple plants in gardens or next to houses from villagers were considered as home garden samples.

Locations of M. balbisiana accessions used in this study. Triangles indicate georeferenced accesions from the ITC collection, while squares marked georeferenced samples collected from leaves or seed collections. Areas encircled with a dashed line indicate regions where non-georeferenced ITC collections were presumably collected. Symbol colours specify the country of origin. The dark shaded area represents northern Indo-Burma, the presumed region of origin of the banana family. The red shaded background depicts elevation. (Color figure online)
The dataset included a total of 372 samples from four material types: 225 samples from 17 wild populations of Musa balbisiana, 21 samples from six home gardens, 98 samples retrieved from eight seed collections, and 28 accessions of M. balbisiana from the in vitro collection of the ITC.
The accuracy of the passport data regarding the original sampling location of the 28 ITC accessions varied substantially. Five accessions had accurate passport data with sampling coordinates, whereas four others had a detailed description of their sampling region (e.g. name of the province within the country of origin) without coordinates. Eleven samples only had information on their country of origin, and eight samples had no data on their sampling origin (Supplementary Information – Table 1).
Genotyping and sequencing
DNA extraction
Leaf material from field missions and ITC germplasm was dried using silica gel and DNA was subsequently isolated using a modified cetyltrimethylammonium bromide (CTAB) extraction protocol (Doyle and Doyle 1987). For seed collections, embryos were first excised using a sterile scalpel and were then added to a 10 µl CTAB solution for DNA isolation using the same protocol.
SSR genotyping
We used the same set of 18 microsatellite markers arranged in four multiplexes as described in Bawin et al. (2019) to compare the ITC samples with those used in Bawin et al. (2019) and Mertens et al. (2021a). Markers were coupled to universal primer sequences (Schuelke 2000) and DNA fragments were amplified with PCR using the Type-it Microsatellite PCR Kit (Qiagen, Venlo, The Netherlands). We added 1 µl of diluted PCR sample to a 12 µl HiDi formamide solution mixed with 0.4 µl of MapMarker 500 labelled with DY-632 (Eurogentec, Seraing, Belgium), and 1.5 µl of this product was then genotyped on an ABI 3730 sequencer (Applied Biosystems, Foster City, California, USA) at the Université Libre de Bruxelles (ULB, Belgium). PCR product of reruns and of more recently sampled material (20 µl per sample) was sent to Macrogen (Macrogen Europe, Amsterdam, The Netherlands) for genotyping on an ABI 3730xl system. To deal with potential differences in fragment sizes between the two sequencers, a minimum of ten samples per run were analysed in duplicate. Raw data were scored in Geneious Prime 2021.1.1 using the 3rd order least square sizing method (Kearse et al. 2012). All data obtained from the study of Bawin et al. (2019) were rescored using the same sizing method. Samples showing ambiguous genotyping patterns were genotyped twice to avoid erroneous scoring. Samples with more than 10% missing data were excluded from the analyses.
Sanger sequencing
We additionally screened each ITC accession and three random samples of each population with one nuclear marker (ITS) and three chloroplast markers (rps16, trnL-F, atpB-rbcL) using the PCR protocols described by White et al. (1990), Oxelman et al. (1997), Taberlet et al. (1991), and Chiang et al. (1998) respectively. PCR samples were sent to Macrogen for sequencing (Macrogen Europe, Amsterdam, The Netherlands). Forward and reverse sequences were checked for quality and assembled in Geneious Prime (Kearse et al. 2012). All M. balbisiana sequences were subsequently aligned using the MAFFT Alignment tool (Katoh and Standley 2013) implemented in Geneious with the E-INS-I algorithm, a scoring matrix of 100PAM / K = 2, and a gap open penalty of 1.3. To identify potential hybrids in the ITC collection, Sanger sequences were blasted against the NCBI nucleotide database (NCBI Resource Coordinators 2018). Sequences were additionally uploaded to NCBI GenBank (Accession numbers OK648712-OK649230).
Genetic analyses
As this study aimed to identify areas in the putative native distribution area of M. balbisiana that need additional sampling for ex situ conservation, accessions obtained from outside Asia and PNG (as indicated by accession names or donor institute) or those marked as hybrids in the Musa germplasm information system (MGIS) were excluded from the analyses of genetic structure (Ruas et al. 2017).
Allelic diversity
For each accession, the percentage of polymorphic loci (%P), the average number of different alleles (Na), the average number of unique (private) alleles (Np), and the number of locally common alleles (frequency ≥ 5% in a population) found in less than 25% of all assessed accessions (Lcomm) were calculated using the “allele frequency data parameters” menu in the GenAlEx 6.51 Excel plugin (Peakall and Smouse 2012).
Genetic structure
We calculated a genetic distance matrix based on the codominant genotypic distances and ran a Principal Coordinate Analysis (PCoA) in GenAlEx using the “Distance-Based” menu options. For this analysis, all individuals from each population were used and the data were presented both at the population level (one point per population) and at the individual level (each individual is plotted). As ITC accessions cannot be seen as one population due to their dispersed origin, we refrained from performing an analysis of molecular variance (AMOVA). In another analysis, a similar plot was made including six accessions of unknown origin to see whether their putative origin could be traced back (ITC0080, ITC0211, ITC0212, ITC0246, ITC0247, ITC0271).
Next to PCoA, we used Bayesian clustering in the STRUCTURE software (Pritchard et al. 2000) to infer genetic structure in our sampling. To find the optimal number of clusters (K), we ran an admixture model to allow samples to be assigned to one or multiple clusters. Unequal sampling of populations may lead to inaccurate estimation of K (Wang 2017). To compensate for the presumably low number of accessions per population present in the ITC collection, we reduced at random the number of samples of remaining populations to five. In order to obtain accurate results with biased sampling using STRUCTURE, we used a model with uncorrelated allele frequencies, a separate alpha (a prior of individual ancestry) for each population, and an initial alpha of 1/K (Wang 2017). To infer this K value, we first carried out 10 preliminary runs using a dependent model and correlated allele-frequencies with an initial alpha of 1. For each run, 100,000 Markov Chain Monte Carlo iterations were sampled after a burn-in of 25,000 iterations. To determine the optimal number of clusters, we considered both the ΔK/K and the log posterior probability of the replicates over each K (Evanno et al. 2005) with the online web server of StructureSelector (Li and Liu 2018). This K was then used to optimize the assignment of individuals by running a new admixture model with independent allele frequencies and an initial alpha value of 1/K. Bar plots with the assignment probabilities obtained using STRUCTURE were subsequently made with the CLUMPAK software integrated in StructureSelector (Kopelman et al. 2015; Li and Liu 2018).
Results
DNA barcoding
Screening of both nuclear (ITS) and chloroplast (rps16, trnL-F, atpB-rbcL) markers revealed very little sequence variation between accessions. BLAST resulted in sequence similarities with M. balbisiana ranging from 99.5–100% for all markers. Nonetheless, based on the chloroplast markers, five accessions (ITC0094, ITC0545, ITC1788, ITC1789, and ITC1823) were found to be potential hybrids with M. acuminata (> 99.1% sequence similarity).
Allelic diversity
For the subsequent analyses, the dataset was reduced to accessions presumed to be diploids of M. balbisiana of which the country of origin was known, thus excluding accessions that showed high similarity of chloroplast sequences with M. acuminata. For accession ITC0248 “Singapuri”, the country of origin was inferred to be Singapore. From all assessed microsatellite loci, BB_CT-33, BB_GAA-4, BB_CT-6, and BB_CT-8 were the most informative. While five loci were uninformative (monomorphic) for the samples included in this study (Mbg02, Mbg04, BB_AAC-3, BB_CT-7, and Mbg01), they were retained for all analyses in order to be able to compare results with the studies of Bawin et al. (2019) and Mertens et al. (2021a). The proportion of polymorphic loci (%P) and average number of different alleles (Na) ranged from 0 to 66.67 and 1.000 to 2.167 respectively (Table 1). The proportion of polymorphic loci was lowest in the seed collection of Amami (Japan) followed by Vietnamese populations VNM-N8 and VNM-S1, and the seed collection of PNG, whereas it was highest in the Central Vietnam population VNM-C2. No polymorphic loci were found in the seed collection of Amami and the ITC accessions from PNG. The highest number of private alleles was found in populations of Central Vietnam (VNM-C1, VNM-C2) and a home garden in South Vietnam (VNM-S3). Apart from VNM-S3, M. balbisiana from home gardens in Vietnam and five accessions of the ITC, including the ones sampled from Vietnam (ITC1681, ITC1687), had no locally common alleles. Excluding VNM-S3, no home garden or ITC accession had private alleles.
Genetic structure
Principal coordinate analysis
In the Principal Coordinate Analysis for all populations and accessions with a labelled country of origin, the first two axes explained 51% of the observed variance (37% and 14% respectively). Five main patterns were observed in the PCoA graph with the populations from Vietnam, China, PNG, and Japan plotted together with the distinct ITC and home garden accessions (Fig. 2a). First, populations from China and Vietnam (green: native distribution group) were clearly distinct from home garden samples from northern and southern Vietnam, as well as most of the ITC accessions studied. Second, ITC accessions and the seed collection of PNG (purple: PNG group) were clearly distinct from other accessions. Third, ITC1587 from Indonesia closely grouped together with an accession from Thailand (ITC1120), the Philippines (ITC1787), two ITC accessions from Vietnam (ITC1687, ITC1681), and most of the M. balbisiana sampled from home gardens in Vietnam (VNM-S5-8 and VNM-N9) (yellow group). Fourth, another group of ITC accessions from various countries of origin and one home garden sample from Vietnam (VNM-S3) were genetically similar (pink group). Fifth, only one accessions from ITC (ITC1527) from Xishuangbanna (China) grouped among accessions from the native distribution area. The same patterns were found in the PCoA graph where individual samples from populations were plotted (Fig. 2b). The two first axes explained 20% and 14% of the observed variance respectively. Some accessions from Chinese populations (mainly from CHN-S from Guangdong and CHN-W1 from Yunnan) were genetically relatively close to a set of ITC accessions and VNM-S3 (pink circle in Fig. 2a).

Principal Coordinate Analysis (PCoA) of M. balbisiana samples of known origin based on codominant genotypic distance of the reduced SSR dataset. Symbol colours indicate regions of origin and symbol shape defines sample type. Accessions connected with a line indicate that they were genetically identical for the assessed SSRs. a Each population/accession is represented by one data point. Four groups are indicated and corresponding accessions or population names are represented in the same colour. b Each sample/accession is represented by one data point
The PCoA including the six accessions of unknown origin showed that, apart from one accession (ITC0271), all these accessions grouped together and seemed genetically relatively distinct from other accessions when assessed at the population level (Fig. 3a). However, when screening them at the sample level (Fig. 3b), three accessions of unknown origin (ITC0212, ITC0246 “Cameroun”, ITC0247 “Honduras”) are genetically close to some samples of West China, while ITC0080 and ITC0211 are genetically more distant from other samples.

Principal Coordinate Analysis (PCoA) of M. balbisiana accessions including accessions from unknown geographic origin. Symbol colours indicate known regions of origin and symbol shape defines sample type. Accessions connected with a line indicate that they were genetically identical for the assessed SSRs. a Each population/accession is represented by one data point. b Each sample/accession is represented by one data point
STRUCTURE analyses
In the preliminary run using STRUCTURE with correlated allele frequencies and an initial alpha of 1, the ΔK/K was highest for K = 3 (13.42) and second highest for K = 6 (4.39). The logarithm of the posterior probability of the replicates over each K suggested an optimal K of 19, but the slope of the curve sharply decreased and the standard deviation started to increase for a K larger than 6 (Supplementary Information – Fig. 1). For this reason, K = 6 was used to set an initial alpha of 0.167 (1/6) in the subsequent clustering runs with uncorrelated allele frequencies to obtain a better cluster assignment for each individual. Because an ‘optimal K’ can be quite ambiguous and because ΔK/K clearly showed K = 3 as the preferred number of genetic groups, we here report STRUCTURE plots ranging from K = 3 to K = 6 (Fig. 4a–d).

STRUCTURE bar plots based on 18 SSR markers for a subsampled dataset generated using CLUMPAK with the number of genetic groups ranging from K = 3 to K = 6. Each horizontal line represents one individual and the assignment probabilities were based on all 10 iterations rather than on the run with the highest log likelihood
When K was set to three (Fig. 4a), three well-defined groups were delineated. All Vietnamese populations clustered together in a first group together with the ITC accession originating from Southwest China (Blue), showing little admixture. All other accessions sampled in China grouped together in a second cluster (orange), together with the seed accessions of Amami (Japan), the ITC accessions from PNG, and the seed collection of PNG. A third cluster (green) included all remaining ITC accessions, the home garden accessions from South Vietnam and VNM-N9 from northern Vietnam. Some individuals of VNM-C2 in the first cluster and of CHN-S in the second cluster displayed admixture with the second and third cluster, respectively. One ITC sample (ITC1790) from the Philippines had a hybrid genotype of the second and the third group. When K was set to 4 (Fig. 4b), the accessions from PNG were assigned to a separate fourth cluster (purple) that also contained some individuals from VNM-N4, VNM-N5, and VNM-N6. Accessions from VNM-N10 and the ITC accession from SW China also showed affinity with this genetic group, but showed high levels of admixture. This is all in accordance with the PCoA (Fig. 2a). When K was further increased to 5 or 6 (Fig. 4c and d respectively), we observed some additional sub-structure in Vietnamese populations, mainly in Central and southern Vietnam. Although individuals were more poorly assigned to clusters at higher K-values (data not shown), in general, the main clusters as observed at K = 4 remained clear, with additional sub-structure in some Vietnamese populations.
Discussion
Genetic structure of M. balbisiana accessions in Southeast Asia
In order to place our observed genetic structure within the current knowledge on the history of M. balbisiana movement, it is important to take into account the origin of the species and current knowledge on the introduction of this species across insular Southeast Asia. Our results provide evidence for the genetic structuring of the accessions in four groups: (i) wild Chinese populations, (ii) wild Vietnamese populations, (iii) most ITC and home garden accessions from Vietnam, and (iv) accessions from PNG.
Chinese and Vietnamese populations
A previous study of Mertens et al. (2021a) demonstrated that Chinese and northern Vietnamese populations contained high genetic diversity which is important to conserve while additional sampling in Central Vietnam was recommended based on the high genetic diversity and number of unique alleles in the two sampled populations. Vietnamese populations could be distinguished from Chinese populations, with even further substructuring of Vietnamese populations at higher K-values. In the current study, the same pattern was observed when accessions from other regions were included in the dataset. The seed collection of Amami (Japan) and ITC1527 from Southwest China clustered with Chinese and Vietnamese populations respectively. A Chinese introduction of M. balbisiana in the 16th century in Japan has already been suggested, bringing great economic benefits to the Ryukyu islands (Kennedy 2009; De Langhe et al. 2015). The lack of genetic variation in the 15 samples from Amami, which is part of the Ryukyu islands, supports the assumption of a narrow genetic basis in this region. Furthermore, ITC1527 from Xishuangbanna in southern Yunnan grouped with populations of Vietnam rather than with other Chinese populations. Xishuangbanna is geographically close to northern Vietnam and the composition of the tropical seasonal rainforests of Xishuangbanna and the forests in north-western Vietnam are floristically similar, possibly connecting M. balbisiana populations in China and northern Vietnam (Lü et al. 2010).
ITC accessions and home gardens of Vietnam
Most other ITC and Vietnamese home garden accessions clustered together in the STRUCTURE analyses. Based on the PCoA, they could be further divided into two genetic groups. A first group (Fig. 2a, pink circle) mainly consisted of accessions from the Philippines and one accession from Flores, Indonesia. The accession ITC0248 “Singapuri” was genetically identical to ITC1588 “Lal Velchi” from India and both accessions clustered in this first group (Fig. 3). In the second group, (Fig. 2a, yellow circle), almost all ITC accessions from multiple countries of origin were genetically identical for the studied markers. This group also included six samples from Vietnamese home gardens, suggesting these ITC accessions were likely sampled from cultivated (potentially Vietnamese) rather than wild material and later on distributed to different countries.
Based on our results, we cannot confirm whether Philippine M. balbisiana accessions can be considered cultivated, as proposed by de Langhe et al. (2015), as the genetic distinctness of these accessions from wild populations of China and Vietnam could represent mainland-island differentiation within the natural distribution of the species (Franks 2010). Recently, high phenotypic diversity was found in 97 M. balbisiana accessions from the ex situ collection of the National Plant Genetic Resources Laboratory in Los Baños (Philippines) and the conservation gaps of this collection were highlighted (Sotto and Rabara 2000; Rabara et al. 2020). High genetic diversity was also reported in M. balbisiana in the Philippines, but only cultivars from multiple Philippine collections and genomic groups were considered in their study (Doloiras-Laraño et al. 2018). Linguistic evidence exists that M. balbisiana was likely translocated southwards from South China following a trail to New Guinea over the Philippines. This theory is further supported based on triploids with a component from M. acuminata subsp. banksii, which is not found in more northern parts of the Philippines (Perrier et al. 2009, 2011). Thousands of years of use and semi-cultivation of M. balbisiana might explain why it is widespread in the Philippines and may be the cause of relatively high genetic diversity (De Langhe et al. 2015). Our results provide evidence for a close relationship of Philippine ITC accessions with some individuals from Chinese populations (Fig. 2b) and especially from South China and West China, though individuals were assigned to different clusters. Additional research with more exhaustive sampling is needed to further investigate the relationship between (southern) Chinese and Philippine M. balbisiana accessions.
Though passport data of “Lal Velchi” (ITC1588) were missing, its origin was assessed as a wild accession from India (pers comm. Van den houwe). However, its genetic differentiation from other populations sampled from the native distribution area of mainland Asia and its high genetic similarity to ITC0248 “Singapuri” suggests that this accession might also have been translocated and domesticated in other regions. Suckers of this genotype may have been distributed between villages, also in more southern states and Sri Lanka (Uma 2006). Additional screening of genetic diversity of wild M. balbisiana populations from India is critical, especially because India is considered as a secondary centre of hybridisation of native M. balbisiana with diploid M. acuminata cultivars after their import from Maritime Southeast Asia (Simmonds and Shepherd 1955; Uma 2006; Perrier et al. 2009). Moreover, recent phytolith evidence suggested Sri Lanka might also have been a centre for early banana dispersal and that exchange of banana cultivars between India and Sri Lanka might already have taken place through maritime network connections in the middle of the fifth millennium before present (Premathilake and Hunt 2018).
The second genetic group (Fig. 2, yellow circle) contained five ITC accessions as well as the home garden samples from Vietnam. Strikingly, 7 out of 11 accessions were genetically identical based on the assessed markers. this includes most home garden samples as well as both Vietnamese ITC accessions, ITC1587 “Pisang Klutuk Wulung” from Indonesia, ITC1787 from the Philippines, and ITC1120 “Tani” from Thailand. Because the accession from Thailand is not georeferenced and little passport data are available, we assume that it was not sampled from a native population in Thailand but rather from cultivated material imported from elsewhere. To our knowledge, no real wild populations of M. balbisiana have been reported from northern Thailand. De Langhe et al. (2000) and Simmonds (1956) suggested a human introduction and cultivation of M. balbisiana in evergreen forests of northern Thailand (Nan province), even if populations seem to appear in natural conditions and suitable climate. Similarly, “Pisang Klutuk Wulung” (ITC1587), and M. balbisiana in general, was likely introduced to Java and naturalised there (Simmonds 1956; De Langhe et al. 2015). Based on the patterns of genetic structure, M. balbisiana plants from home gardens in Vietnam were most likely introduced from germplasm accessions or clones from e.g. the genetically similar accessions “Pisang Klutuk Wulung” or “Tani”. Other studies using different markers also showed the genetic relatedness of “Tani” and “Pisang Klutuk Wulung” accessions among other accessions. For accession VNM-S7, according to locals, M. balbisiana was imported from the North after the Vietnam war because it is rather drought resistant and used for feed, typically for cows, though no genetic link with a native population could be found (pers. comm.). This suggests that M. balbisiana used in home gardens might have come from cultivated material rather than from wild populations (Duroy et al. 2016; Jeensae et al. 2021). In contrast to other Philippine accessions, the clustering of ITC1787 with this group and the genetic uniformity with the other accessions within this group suggests that clones were likely shared between different locations, e.g. as in vitro material or as suckers. A similar study in the Democratic Republic of the Congo compared the gene pool of wild populations of Robusta coffee (Coffea canephora Pierre ex A.Froehner) with cultivated accessions from the Institut National des Etudes et Recherches Agronomique (INERA) Yangambi and from local backyards (Vanden Abeele et al. 2021). Coffea canephora plants grown in home gardens were likely directly or indirectly received from INERA breeding programmes and both the accessions from INERA and from backyards were genetically distinct from the local wild gene pool. Hence, it seems that, at least for some crops, planting material from elite cultivars that were bred elsewhere is preferred in backyards above semi-cultivated accessions related to the local gene pool.
The eight ITC accessions for which the country of origin was not known showed additional genetic diversity of M. balbisiana compared to the accessions with a known country of origin, possibly representing populations in a different part of the species’ distribution area. These findings again stress the need for passport data in order to more efficiently collect additional germplasm. Three of those accessions showed genetic similarity to accessions from West China, while one (ITC0271, “Eti Kehel”) showed genetic similarity with some samples from China and two ITC accessions from the Philippines. As M. balbisiana is locally known as Eti-Kehel in Sri Lanka and as ITC0271 was donated to ITC by the Royal Botanic Gardens of Sri Lanka, Sri Lankan origin of this accession can be assumed (Liyanage et al. 1998; Duroy et al. 2015). ITC0271 was genetically most similar to a group containing three Philippine accessions and the accessions ITC1588 “Lal Velchi” from India and ITC0248 “Singapuri”, providing some evidence for the exchange of genetic material between India, the Philippines, and Singapore. Due to the limited number of accessions from the Philippines, India, or other countries of origin apart from China and Vietnam, the country of origin for the other ITC accessions could not be inferred.
Papua New Guinea
In 1956, Simmonds described the appearance of M. balbisiana in New Guinea as widespread and locally abundant in natural habitats, suggesting it could be truly native (Simmonds 1956). Since then, however, it was proposed that PNG does not belong to the native distribution area (Argent 1976; De Langhe et al. 2015). We showed that samples from PNG, both from the seed collection and ITC, were genetically distinct from the other clusters. The very low number of polymorphic loci (0% for both ITC accessions and 11% for the seed collection) further support the hypothesis that M. balbisiana was introduced to PNG thousands of years ago (before ca 3100 years), represented by only one or very few genotypes or “BB” cultivars (Argent 1976). The relatively large genetic differentiation between M. balbisiana accessions from PNG and the Philippines that was observed in this study does not support a dispersal of wild M. balbisiana from south China to PNG over the Philippines. Hence, a wild origin of M. balbisiana in PNG via this dispersal route seems to be less likely.
The value of ITC in distributing Musa balbisiana diversity
Among the 6,772 Musaceae accessions from multiple germplasm collections around the world listed on the Taxonomy Browser of the MGIS, M. balbisiana is represented by 167 accessions (as of May 2021). Of these, 145 are labelled as “wild species or subspecies” (Ruas et al. 2017). Very little passport data are available and only 28 accessions were available for distribution from the ITC and were therefore included in this study. Based on our SSR data analysis we can conclude that most of the M. balbisiana accessions held at ITC were genetically distant to the accessions that were recently collected in the native distribution range of the species (Janssens et al. 2016). Only one accession (ITC1527) sampled from Xishuangbanna in Southwest China grouped with samples of Vietnam and neighbouring Chinese provinces. All other accessions obtained from different countries in Asia and PNG were systematically assigned to separate clusters. This indicates on the one hand that the ITC conserves and distributes unique and valuable genetic diversity of M. balbisiana that is likely not present in the seed collections sampled from Vietnamese populations that are held at the Millennium Seed Bank (MSB) or from seed collections from China. These include Vietnamese seed collections of populations of which leaves were used in this study. On the other hand, very limited germplasm from other countries of the native distribution area of M. balbisiana is available at the ITC or at other germplasm centres, though these populations have relatively high genetic variation within- and among-populations compared to feral populations and cultivated material (Ge et al. 2005; Bawin et al. 2019; Mertens et al. 2021a).
In addition to the one ITC accession from China, nine other ITC accessions originated from a country in which M. balbisiana is assumed to be native. However, the wild origin of these nine ITC accessions is uncertain. Two of those accessions were georeferenced to the Nghe An province of Vietnam (ITC1681 and ITC1687), the same province in which the populations VNM-C1 and VNM-C2 were sampled. Based on the provided coordinates, these ITC accessions were sampled from more densely populated areas in the East of the province in contrast to the populations which were collected in the West, potentially from home gardens instead of a wild population. Two other accessions (ITC1120 and ITC1588) were sampled from Thailand and India, respectively. Both countries are partly covered by the (sub)tropical forests of northern Indo-Burma, which harbour many wild Musa populations (Janssens et al. 2016; Mertens et al. 2021b). While these accessions have been used multiple times in previous studies as representatives of the wild diploid “BB” genomic group, their wild origin remains uncertain due to the lack of passport data (Ruas et al. 2017; Christelová et al. 2017; Zuo et al. 2018; Nakato et al. 2019; Igwe et al. 2021). The five remaining ITC accessionswere of Philippine originand showed relatively high genetic variation. Ongoing genetic screening of wild M. balbisiana at the University of the Philippines Los Baños also suggests high diversity in the Philippines (Gueco, pers. comm.). This high genetic diversity in Philippine M. balbisiana may suggest that the Philippines are part of the species’ native distribution area. Nevertheless, it remains unclear whether this high diversity reflects naturally accumulated genetic variation in wild M. balbisiana populations or increased variation in feral populations. Such increased variation in feral populations can be explained by multiple introductions of M. balbisiana into the Philippines or introgression of genetic material from other Musa species (e.g. M. acuminata) into the Philippine M. balbisiana gene pool (Hufford et al. 2019). Chloroplast sequences with high sequence similarity to M. acuminata of three Philippine ITC M. balbisiana accessions (ITC1788, ITC1789, ITC1823) provide some support that introgression could have taken place.
The lack of sufficient germplasm from the putative regions of origin of this species results in a poor representation of the total genetic diversity of the species at ITC, especially with only one sample from India and Thailand and none from Laos and Myanmar, all countries of which at least some parts are climatically suitable for the species (Janssens et al. 2016; Mertens et al. 2021b; POWO 2022). Current germplasm accessions should be maintained and, where available, passport data should be updated. Because of the limited knowledge on the genetic diversity of M. balbisiana populations in countries such as Myanmar, Thailand, India, Laos, but also the Philippines and Indonesia, additional collecting and genetic screening of populations and existing collections (such as the MSB) with plant material from these countries is required to maximize the genetic representation of the species with minimal resources (e.g. with seed collections).
Optimisation of banana germplasm sampling for conservation and breeding purposes
Based on our findings, there is an urgent need for new and additional sampling of Musa balbisiana germplasm, especially from countries where the species is native. For M. balbisiana specifically, despite its higher resistance to biotic and abiotic stresses compared to M. acuminata (Nelson et al. 2006; Ocan et al. 2008; Vanhove et al. 2012; Mattos-Moreira et al. 2018; Tripathi et al. 2019b), the largest drawback in using this species for breeding is the presence of endogenous banana streak virus (eBSV) in the genome of genotypes containing at least one “B” genome as progenitor. This results in spontaneous infections of the hybrids following abiotic stress, making it a major constraint for use in breeding programmes. However, marker-assisted breeding, the discovery of M. balbisiana plants that contain non-infective eBSV sequences, and recent advances in gene-editing technology are promising to support a more efficient use of M. balbisiana genetic resources (Duroy et al. 2016; Umber et al. 2016; Tripathi et al. 2019a).
The mating system of wild banana species is an important indicator to optimise seed sampling strategies. For (partially) cross-pollinated species such as Musa balbisiana, increasing the number of sampled populations is more effective in maximizing genetic capture than sampling more mother plants per population (Kallow et al. 2021) though within-species variation in mating system should be taken into account. To further maximize genetic capture, large populations distant from anthropogenic activities should be prioritised (Andersson and de Vicente 2010; Almeida-Rocha et al. 2020; Kallow et al. 2021).
Equally important is making sure passport data of germplasm collections are of high quality for optimal use for farmers or breeders, but also that the material is available for research. Optimally, passport data follow a standard of descriptors such as the standard of the multi-crop passport descriptors from FAO and Bioversity International (Alercia et al. 2015). Such data typically include information on the collection and the storage of the germplasm accession itself but should also include a detailed description of the geographical origin and environmental conditions of the sampled location. The status of the accession (wild, semi-wild, cultivated or cultivar) should additionally be recorded as well as phenotypic, morphological, and agronomic traits. When this information is consistently provided for accessions in germplasm collections, their use for research (e.g. for determining conservation gaps) or breeding purposes can more easily be evaluated (Weise et al. 2020).
For bananas, additional information might further promote the collection and use of specific germplasm accessions related to pest management and desirable traits. It is known that the economically most devastating fungal pathogen of banana, the soil borne fungus Fusarium oxysporum f.sp. cubense (Foc), often is symptomless but present in the field which could lead to the spread of Fusarium when locally distributed. Screening wild genotypes of M. balbisiana for resistance against Foc might help in prioritising areas for additional germplasm collection. For example, though five Indian BB-type accessions were found highly resistant or immune to Foc race 1 (Thangavelu et al. 2021), germplasm of Indian M. balbisiana is largely missing at the ITC. Investigating or sampling the root microbiome might give information on whether the fungus is present. Recent studies highlighted a significant change in endophytic microbial and fungal community composition during disease development compared to non-symptomatic plants (Kaushal et al. 2020a, b). Passport data related to the root microbiome could therefore also be important for selecting genetic material for breeding programmes.
Conclusions
Until now, little of the wild M. balbisiana genetic diversity from the native distribution area is captured at the ITC and thus available for distribution and research. By investigating both wild populations as well as germplasm accessions from multiple countries of origin held at the ITC, we found that passport data are often missing and incomplete. The country of origin was unknown for eight out of 28 accessions and only the country was supposedly known for an additional 11 accessions, making it more difficult to evaluate an accession as wild material on the one hand and limiting its use for breeding on the other hand. While considerable genetic variation is found in accessions from the Philippines and wild populations from China and Vietnam, it is clear that M. balbisiana and the genus Musa in general is a very complex group due to multiple and repeated events of migration and intensive cultivation of the species during the last millennia. Assessing the genetic structure revealed that samples could be systematically subdivided into three to six genetic groups, with a clear separation of most ITC accessions and samples from home gardens, samples from PNG, and populations sampled in China and Vietnam. Because most of the distributed material of M. balbisiana from the ITC is genetically similar, we here suggest that more germplasm should be collected from wild populations in China and Vietnam, but especially also in northeastern India, Myanmar, and the Philippines and that existing collections in the world should be genetically screened. High throughput sequencing techniques are necessary to further explore to what extent Philippine accessions can be considered as wild or whether they are more likely introduced from South China for cultivation.
Data availability
Sanger sequences generated in this project were submitted to GenBank (Accession numbers OK648712–OK649230).
Code availability
Not applicable.
References
Ahmad F, Megia R, Poerba Y (2014) Genetic diversity of Musa balbisiana Colla in Indonesia based on AFLP marker. HAYATI J Biosci 21:39–47. https://doi.org/10.4308/hjb.21.1.39
Alercia A, Diulgheroff S, Mackay M (2015) FAO/Bioversity multi-crop passport descriptors V.2.1. Rome, Italy
Almeida-Rocha JM, Soares LASS, Andrade ER et al (2020) The impact of anthropogenic disturbances on the genetic diversity of terrestrial species: A global meta-analysis. Mol Ecol 29:4812–4822. https://doi.org/10.1111/mec.15688
Andersson M, de Vicente M (2010) Banana and plantain (Musa spp.). In: Gene flow between crops and their wild relatives. JHU Press: Baltimore, pp 25–47
Argent GCG (1976) Wild bananas of Papua New Guinea [Ensete, Musa, new taxa]. Notes Roy Bot Gard Edinburgh 35:77–114
Bawin Y, Panis B, Vanden Abeele S et al (2019) Genetic diversity and core subset selection in ex situ seed collections of the banana crop wild relative Musa balbisiana. Plant Genet Resour 17:536–544. https://doi.org/10.1017/S1479262119000376
Brown A, Tumuhimbise R, Amah D et al (2017) Bananas and plantains (Musa spp.). In: Campos H, Caligari PDS (eds) Genetic improvement of tropical crops. Springer, Cham, pp 219–240
Castañeda-Álvarez NP, Khoury CK, Achicanoy HA et al (2016) Global conservation priorities for crop wild relatives. Nat Plants 2:1–6. https://doi.org/10.1038/NPLANTS.2016.22
Chiang TY, Schaal BA, Peng CI (1998) Universal primers for amplification and sequencing a noncoding spacer between the atpB and rbcL genes of chloroplast DNA. Bot Bull Acad Sin 39:245–250
Christelová P, De Langhe E, Hřibová E et al (2017) Molecular and cytological characterization of the global Musa germplasm collection provides insights into the treasure of banana diversity. Biodivers Conserv 26:801–824. https://doi.org/10.1007/s10531-016-1273-9
Daniells J, Jenny C, Karamura D, Tomekpe K (2001) Musalogue: a catalogue of Musa germplasm. In: Arnaud E, Sharrock S (eds) Diversity in the genus Musa. International Network for the Improvement of Banana and Plantain, Montpellier, France
De Langhe E, Wattanachaiyingcharoen D, Volkaert H, Piyapitchard S (2000) Biodiversity of wild Musaceae in northern Thailand. In: Molina AB, Roa VN (eds) Advancing banana and plantain R and D in Asia and the Pacific. International Plant Genetic Resources Institute, Rome
De Langhe E, Perrier X, Donohue M, Denham T (2015) The original banana split: multi-disciplinary implications of the generation of African and Pacific plantains in Island Southeast Asia. Ethnobot Res Appl 14:299–312. https://doi.org/10.17348/era.14.0.299-312
Dempewolf H, Baute G, Anderson J et al (2017) Past and future use of wild relatives in crop breeding. Crop Sci 57:1070–1082. https://doi.org/10.2135/cropsci2016.10.0885
Doloiras-Laraño AD, Garcia RN, Sandoval CMC et al (2018) DNA fingerprinting and genetic diversity analysis of Philippine Saba and other cultivars of Musa balbisiana Colla using simple sequence repeat markers. Philipp J Crop Sci 43:1–11
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Duroy PO, Perrier X, Laboureau N et al (2016) How endogenous plant pararetroviruses shed light on Musa evolution. Ann Bot 117:625–641. https://doi.org/10.1093/aob/mcw011
Duroy P-O, Laboureau N, Seguin J, et al (2015) Endogenous banana streak virus sequences (eBSV) are likely transcriptionally silenced in the resistant seedy diploid Musa balbisiana Pisang Klutuk Wulung (PKW). 15èmes Rencontres Virol. Végétale
Engels JMM, Thormann I (2020) Main challenges and actions needed to improve conservation and sustainable use of our crop wild relatives. Plants 9:968. https://doi.org/10.3390/plants9080968
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
FAO (2018) Banana Market Review: Preliminary results for 2018. In: Food Agric. Organ. United Nations. http://www.fao.org/fileadmin/templates/est/COMM_MARKETS_MONITORING/Bananas/Documents/Banana_Market_Review_Prelim_Results_2018.pdf
FAO (2019) FAOSTAT Database. http://www.fao.org/faostat/en/#home. Accessed 15 Feb 2021
Ford-Lloyd BV, Schmidt M, Armstrong SJ et al (2011) Crop wild relatives—undervalued, underutilized and under threat? Bioscience 61:559–565. https://doi.org/10.1525/bio.2011.61.7.10
Franks SJ (2010) Genetics, evolution, and conservation of island plants. J Plant Biol 53:1–9. https://doi.org/10.1007/s12374-009-9086-y
Ge XJ, Liu MH, Wang WK et al (2005) Population structure of wild bananas, Musa balbisiana, in China determined by SSR fingerprinting and cpDNA PCR-RFLP. Mol Ecol 14:933–944. https://doi.org/10.1111/j.1365-294X.2005.02467.x
Genesys (2021) Genesys is an online platform where you can find information about Plant Genetic Resources for Food and Agriculture (PGRFA) conserved in genebanks worldwide. https://www.genesys-pgr.org/. Accessed 18 May 2021
Hajjar R, Hodgkin T (2007) The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica 156:1–13. https://doi.org/10.1007/s10681-007-9363-0
Heywood VH (2016) In situ conservation of plant species – an unattainable goal? Isr J Plant Sci 63:211–231. https://doi.org/10.1080/07929978.2015.1035605
Heywood V, Casas A, Ford-Lloyd B et al (2007) Conservation and sustainable use of crop wild relatives. Agric Ecosyst Environ 121:245–255. https://doi.org/10.1016/j.agee.2006.12.014
Hufford MB, Berny Mier Y, Teran JC, Gepts P (2019) Crop biodiversity: an unfinished magnum opus of nature. Annu Rev Plant Biol 70:727–751. https://doi.org/10.1146/annurev-arplant-042817-040240
Igwe DO, Ihearahu OC, Osano AA et al (2021) Genetic diversity and population assessment of Musa L. (Musaceae) employing CDDP markers. Plant Mol Biol Report. https://doi.org/10.1007/s11105-021-01290-x
Janssens SB, Vandelook F, De Langhe E et al (2016) Evolutionary dynamics and biogeography of Musaceae reveal a correlation between the diversification of the banana family and the geological and climatic history of Southeast Asia. New Phytol 210:1453–1465. https://doi.org/10.1111/nph.13856
Jeensae R, Kongsiri N, Fluch S et al (2021) Cultivar specific gene pool may play an important role in Musa acuminata Colla evolution. Genet Resour Crop Evol 68:1589–1601. https://doi.org/10.1007/s10722-020-01088-y
Kallow S, Davies R, Panis B et al (2020a) Regulation of seed germination by diurnally alternating temperatures in disturbance-adapted banana crop wild relatives (Musa acuminata). Seed Sci Res 30:238–248. https://doi.org/10.1017/S0960258520000471
Kallow S, Longin K, Sleziak NF et al (2020b) Challenges for ex situ conservation of wild bananas: seeds collected in Papua New Guinea have variable levels of desiccation tolerance. Plants 9:1–21. https://doi.org/10.3390/plants9091243
Kallow S, Panis B, Vu DT et al (2021) Maximizing genetic representation in seed collections from populations of self and cross-pollinated banana wild relatives. BMC Plant Biol 21:415. https://doi.org/10.1186/s12870-021-03142-y
Kallow S, Mertens A, Janssens SB et al (2022) Banana seed genetic resources for food security: status, constraints, and future priorities. Food Energy Secur. https://doi.org/10.1002/fes3.345
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kaushal M, Mahuku G, Swennen R (2020a) Metagenomic insights of the root colonizing microbiome associated with symptomatic and non-symptomatic bananas in Fusarium wilt infected fields. Plants 9:263. https://doi.org/10.3390/plants9020263
Kaushal M, Swennen R, Mahuku G (2020) Unlocking the microbiome communities of banana (Musa spp.) under disease stressed (Fusarium wilt) and non-stressed conditions. Microorganisms 8:443. https://doi.org/10.3390/microorganisms8030443
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kennedy J (2009) Bananas and people in the homeland of genus Musa: not just pretty fruit. Ethnobot Res Appl 7:179. https://doi.org/10.17348/era.7.0.179-197
Khoury CK, Carver D, Greene SL et al (2021) Crop wild relatives of the United States require urgent conservation action. Proc Natl Acad Sci U S A 117:33351–33357. https://doi.org/10.1073/PNAS.2007029117
Kopelman NM, Mayzel J, Jakobsson M et al (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Krishna H, Alizadeh M, Singh D et al (2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6:54. https://doi.org/10.1007/s13205-016-0389-7
Li YL, Liu JX (2018) StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177. https://doi.org/10.1111/1755-0998.12719
Li D-Z, Pritchard HW (2009) The science and economics of ex situ plant conservation. Trends Plant Sci 14:614–621. https://doi.org/10.1016/j.tplants.2009.09.005
Liyanage ASU, Manawaprema MMC, Mendis MH (1998) Differentiation of A & B genome of banana and plantain (Musa spp.) by esterase enzyme. J Natl Sci Counc Sri Lanka 26:125–131. https://doi.org/10.4038/jnsfsr.v26i2.3560
Lü XT, Yin JX, Tang JW (2010) Structure, tree species diversity and composition of tropical seasonal rainforests in Xishuangbanna, South-West China. J Trop For Sci 22:260–270
Mattos-Moreira LA, Ferreira CF, Amorim EP et al (2018) Differentially expressed proteins associated with drought tolerance in bananas (Musa spp.). Acta Physiol Plant 40:60. https://doi.org/10.1007/s11738-018-2638-3
Maxted N, Kell SP (2009) Establishment of a global network for the in situ conservation of crop wild relatives: status and needs. Rome: Commission on Genetic Resources for Food and Agriculture, FAO. http://www.fao.org/docrep/013/i1500e/i1500e18d.pdf
McCouch S (2013) Feeding the future. Nature 499:13–17
Meilleur BA, Hodgkin T (2004) In situ conservation of crop wild relatives: status and trends. Biodivers Conserv 13:663–684. https://doi.org/10.1023/B:BIOC.0000011719.03230.17
Mertens A, Bawin Y, Vanden Abeele S et al (2021a) Genetic diversity and structure of Musa balbisiana populations in Vietnam and its implications for the conservation of banana crop wild relatives. PLoS ONE 16:e0253255. https://doi.org/10.1371/journal.pone.0253255
Mertens A, Swennen R, Rønsted N et al (2021) Conservation status assessment of banana crop wild relatives using species distribution modelling. Divers Distrib. https://doi.org/10.1111/ddi.13233
Meyer RS (2015) Encouraging metadata curation in the diversity seek initiative. Nat Plants 1:1–2. https://doi.org/10.1038/nplants.2015.99
Nakato GV, Christelová P, Were E et al (2019) Sources of resistance in Musa to Xanthomonas campestris pv. musacearum, the causal agent of banana Xanthomonas wilt. Plant Pathol 68:49–59. https://doi.org/10.1111/ppa.12945
NCBI Resource Coordinators (2018) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 46:D8–D13. https://doi.org/10.1093/nar/gkx1095
Nelson SC, Ploetz RC, Kepler AK (2006) Musa species (banana and plantain), ver. 2.2. In: Elevitch CR (ed) Species profiles for Pacific Island agroforestry. permanent agriculture resources (PAR), Hōlualoa, Hawai‘i, pp 1–33
Ocan D, Mukasa HH, Rubaihayo PR et al (2008) Effects of banana weevil damage on plant growth and yield of East African Musa genotypes. J Appl Biosci 9:407–415
Oxelman B, Lidén M, Berglund D (1997) Chloroplast rps16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Plant Syst Evol 206:393–410. https://doi.org/10.1007/BF00987959
Panis B, Kallow S, Janssens SB (2020) Seed germination, preservation and population genetics of wild Musa germplasm. In: Kema G, Drenth A (eds) Achieving sustainable cultivation of bananas Volume 2: germplasm and genetic improvement. Burleigh Dodds, Cambridge
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Perrier X, Bakry F, Carreel F et al (2009) Combining biological approaches to shed light on the evolution of edible bananas. Ethnobot Res Appl 7:199–216. https://doi.org/10.17348/era.7.0.199-216
Perrier X, De Langhe E, Donohue M et al (2011) Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc Natl Acad Sci 108:11311–11318. https://doi.org/10.1073/pnas.1102001108
Ploetz RC, Kepler AK, Daniells J, Nelson SC (2007) Banana and plantain - an overview with emphasis on Pacific island cultivars, ver. 1. In: Elevitch CR (ed) Species Profiles for Pacific Island Agroforestry. Permanent Agriculture Resources (PAR), Hōlualoa, Hawai‘i
POWO (2022) Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. http://www.plantsoftheworldonline.org/. Accessed 18 Jan 2022
Premathilake R, Hunt CO (2018) Earliest Musa banana from the late Quaternary sequence at Fahien Rock Shelter in Sri Lanka. J Quat Sci 33:624–638. https://doi.org/10.1002/jqs.3041
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rabara RC, Sotto RC, Salas EAL (2020) Species distribution modeling and phenotypic diversity reveals collection gap in the Musa balbisiana germplasm conservation in Philippines. Asian J Agric 4:60–71. https://doi.org/10.13057/asianjagric/g040203
Rasmussen HN, Dixon KW, Jersáková J, Těšitelová T (2015) Germination and seedling establishment in orchids: a complex of requirements. Ann Bot 116:391–402. https://doi.org/10.1093/aob/mcv087
Ruas M, Guignon V, Sempere G et al (2017) MGIS: managing banana (Musa spp.) genetic resources information and high-throughput genotyping data. Database 2017:1–12. https://doi.org/10.1093/database/bax046
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Simmonds NW (1956) Botanical results of the banana collecting expedition, 1954–5. Kew Bull 11:463–489. https://doi.org/10.2307/4109131
Simmonds NW, Shepherd K (1955) The taxonomy and origins of the cultivated bananas. Bot J Linn Soc 55:302–312. https://doi.org/10.1111/j.1095-8339.1955.tb00015.x
Sotto RC, Rabara RC (2000) Morphological diversity of Musa balbisiana Colla in the Philippines. InfoMusa 9:28–30
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Thangavelu R, Saraswathi MS, Uma S et al (2021) Identification of sources resistant to a virulent Fusarium wilt strain (VCG 0124) infecting Cavendish bananas. Sci Rep 11:1–14. https://doi.org/10.1038/s41598-021-82666-7
Tripathi JN, Ntui VO, Ron M et al (2019) CRISPR/Cas9 editing of endogenous banana streak virus in the B genome of Musa spp. overcomes a major challenge in banana breeding. Commun Biol 2:46. https://doi.org/10.1038/s42003-019-0288-7
Tripathi L, Tripathi JN, Shah T et al (2019) Molecular basis of disease resistance in banana progenitor Musa balbisiana against Xanthomonas campestris pv. musacearum. Sci Rep 9:7007. https://doi.org/10.1038/s41598-019-43421-1
Ude G, Pillay M, Nwakanma D, Tenkouano A (2002) Genetic Diversity in Musa acuminata Colla and Musa balbisiana Colla and some of their natural hybrids using AFLP Markers. Theor Appl Genet 104:1246–1252. https://doi.org/10.1007/s00122-002-0914-4
Uma S, Siva SA, Saraswathi MS et al (2006) Variation and intraspecific relationships in Indian wild Musa balbisiana (BB) population as evidenced by random amplified polymorphic DNA. Genet Resour Crop Evol 53:349–355. https://doi.org/10.1007/s10722-004-0576-y
Uma S (2006) Farmers’ knowledge of wild Musa in India. Food Agric Organ United Nations 46
Umber M, Pichaut J-P, Farinas B et al (2016) Marker-assisted breeding of Musa balbisiana genitors devoid of infectious endogenous Banana streak virus sequences. Mol Breed 36:74. https://doi.org/10.1007/s11032-016-0493-8
van den Houwe I, Chase R, Sardos J et al (2020) Safeguarding and using global banana diversity: a holistic approach. CABI Agric Biosci 1:1–22. https://doi.org/10.1186/s43170-020-00015-6
Vanden Abeele S, Steven JB, Assimonyio AJ et al (2021) Genetic diversity of wild and cultivated Coffea canephora in northeastern DR Congo and 1 the implications for conservation. Am J Bot. https://doi.org/10.1101/2021.08.09.455630
Vanhove AC, Vermaelen W, Panis B et al (2012) Screening the banana biodiversity for drought tolerance: can an in vitro growth model and proteomics be used as a tool to discover tolerant varieties and understand homeostasis. Front Plant Sci 3:1–10. https://doi.org/10.3389/fpls.2012.00176
Wang J (2017) The computer program STRUCTURE for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17:981–990. https://doi.org/10.1111/1755-0998.12650
Wang X-L, Chiang T-Y, Roux N et al (2007) Genetic diversity of wild banana (Musa balbisiana Colla) in China as revealed by AFLP markers. Genet Resour Crop Evol 54:1125–1132. https://doi.org/10.1007/s10722-006-9004-9
WCSP (2021) World checklist of selected plant families. In: R. Bot. Gard. Kew. http://wcsp.science.kew.org/. Accessed 5 May 2021
Weise S, Lohwasser U, Oppermann M (2020) Document or lose it—on the importance of information management for genetic resources conservation in genebanks. Plants 9:1–13. https://doi.org/10.3390/plants9081050
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols. Elsevier, pp 315–322
Youssef M, James AC, Rivera-Madrid R et al (2011) Musa genetic diversity revealed by SRAP and AFLP. Mol Biotechnol 47:189–199. https://doi.org/10.1007/s12033-010-9328-8
Zuo C, Deng G, Li B et al (2018) Germplasm screening of Musa spp. for resistance to Fusarium oxysporum f. sp. cubense tropical race 4 (Foc TR4). Eur J Plant Pathol 151:723–734. https://doi.org/10.1007/s10658-017-1406-3
Acknowledgements
The authors would like to thank Wim Baert, Lynn Delgat and Sander de Backer (Meise Botanic Garden) as well as Pieter Asselman (UGhent) for their aid in molecular lab work. Moreover, we thank David Eyland and Ines Van den houwe for their knowledge and insight in ex situ conservation methods of Musa germplasm.
Funding
The Research Foundation Flanders (FWO) funded this research through the Flanders Bilateral Research Cooperation with Vietnam project with reference “G0D9318N”. Funding was received from the Vietnamese National Foundation for Science and Technology development under grant number “FWO.106-NN.2017.02”, and from the Bill and Melinda Gates foundation via the BBTV mitigation project “OPP1130226”. Additional financial support was given by the CGIAR Fund, and in particular the CGIAR Research Program Roots, Tubers and Bananas (RTB-CRP).
Author information
Authors and Affiliations
Contributions
AM, RS, DTV, FV, and SBJ conceived the study. AM, YB, SK, DTV, TDV, HTM, and SBJ were responsible for data curation. AM, YB, SVA analysed the data. AM, YB, SK, RS, FV, SBJ validated the results. AM wrote the original draft and visualised the data. AM, YB, SVA, SK, RS, DTV, TDV, HTM, FV, and SBJ reviewed and edited the manuscript. RS, DTV, FV, and SBJ acquired funding and were responsible for project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Consent for publication
All authors participated in, read and approved the final version of the article before publication.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mertens, A., Bawin, Y., Vanden Abeele, S. et al. Phylogeography and conservation gaps of Musa balbisiana Colla genetic diversity revealed by microsatellite markers. Genet Resour Crop Evol 69, 2515–2534 (2022). https://doi.org/10.1007/s10722-022-01389-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-022-01389-4