
Abstract
Selection for multiple traits is a highly challenging task for breeders due to potential unfavorable associations between characters. Fusarium head blight FHB, being one of the most relevant diseases affecting durum wheat frequently shows in this respect an unfavorable correlation with morpho-agronomical traits like plant height (PH) and heading date (HD). In this study, we used a cross-validation scheme to assess the prediction ability of the genomic predictions (GP) for FHB severity relying on genomic best linear unbiased prediction models in a diverse panel of 178 durum wheat lines evaluated across five environments. Additionally, we compared three types of approaches to include HD and PH as covariates into the analysis: (1) correcting FHB severity values before training GP models, (2) tuning the GP model parameters that included multi-trait alternatives, and (3) adjusting the genomic-based predictions by restriction indexes. Models that weighted genomic estimated breeding values (GEBV) by restriction indexes as well as models that predicted FHBms values corrected by regression-based methods were efficient alternatives in diminishing the HD trade-off, nonetheless they were also associated with large reductions in prediction ability for FHB severity. After a simulated round of genomic selection, considering HD as fixed effect in the GP model were the most suitable alternative to select a higher proportion of genotypes moderately resistant with lower-than-average HD and PH estimations. Hence, an appropriate GP model given unfavorable association between characters should combine high predictabilities and adequate reduction of undesired trade-offs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fusarium head blight FHB, also called scab, is one of the most calamitous diseases affecting cereals such as wheat, barley, maize and oat (Walter et al. 2010; Beres et al. 2018). FHB is mainly caused by Fusarium graminearum and Fusarium culmorum with the former being considered as the fourth most economic and scientifically important plant fungal pathogen worldwide (Dean et al. 2012). The disease’ effects comprise losses in grain yield and quality as well as contamination by mycotoxins like deoxynivalenol (DON) and its acetylated derivatives, which in turn are a serious threat for human and animal health and malting purposes (Darwish et al. 2014; Dweba et al. 2017; Fung and Clark 2004). In comparison to hexaploid bread wheat (Triticum aestivum L.), the tetraploid durum wheat (Triticum turgidum L. ssp. durum) is particularly susceptible to the Fusarium species complex which makes the development of resistant varieties a major breeding goal. Durum wheat is a cereal crop with an annual global production of 39 million of tons corresponding to 5% of the total wheat production (Kadkol and Sissons 2016). Besides bread and bulgur, other essential foodstuffs like couscous and pasta, consumed by hundreds millions people worldwide, are prepared based on semolina that is the granular milled product of durum grains (Fiedler et al. 2017; Tuberosa and Pozniak 2014). A certain level of control over FHB can be achieved with a series of cultural, biological and chemical control measures plus cultivar resistance (Pirgozliev et al. 2003). Moreover, and within a Genotype \(\times\) Environment \(\times\) Management (\(\hbox {G} \times \hbox {E} \times \hbox {M}\)) interaction framework, the development and adoption of cultivars tolerant to FHB is and will remain to be the cornerstone of any strategy aiming to manage this disease (Beres et al. 2018).
FHB resistance (FHBr), is a trait whose quantitative inheritance is controlled by many loci each with small effect which leads to a slow genetic gain per unit of time. More than 500 QTL have been described for FHB resistance on all 21 chromosomes of hexaploid wheat (Buerstmayr et al. 2009; Jia et al. 2018; Liu et al. 2009; Venske et al. 2019), which is not the case in tetraploid wheats where only a small number of loci are reported (Prat et al. 2014). For the latter wheat species FHBr loci have been mapped on all chromosomes except 1A, 1B and 5A, though all of them possess only small or moderate effects compared to the major resistance loci in hexaploid wheat e.g. Fhb1 located on chromosome 3BS, Fhb2 located on 6BS and Qfhs.ifa-5A all derived from the Chinese resistance cultivar “Sumai 3” (Prat et al. 2014; Zhao et al. 2018). In addition, within the durum gene pool there is a narrow genetic variation for FHBr in both the mostly susceptible elite cultivars (Buerstmayr et al. 2003; Miedaner and Longin 2013) and landraces, while among the latter only few have shown some elevated level of resistance (Huhn et al. 2012; Talas et al. 2011). Other wild wheat tetraploids relatives (T. turgidum ssp. dicoccoides, T. turgidum ssp. dicoccum, and T. turgidum ssp. carthlicum) have shown moderate to high resistance levels (Oliver et al. 2007, 2008) although the incorporation of loci from such sources into elite germplasm faces some obstacles like linkage drag, unfavorable epistatic interactions or pleiotropic effects (Kumar et al. 2018; Prat et al. 2014; Zhu et al. 2016). Nevertheless, a couple of studies in this direction have been conducted and reported successful pyramiding of resistance genes from hexaploid wheat together with native durum loci (Prat et al. 2014; Zhao et al. 2018).
Marker assisted selection (MAS) has proved to be a suitable and effective instrument for breeders when targeting a trait with small number of large effect genes like the stripe (yellow) rust resistance gene Yr15 in durum wheat (Yaniv et al. 2015). The actual implementation of MAS for quantitative inherited traits with an underlying complex genetic architecture is though less feasible, while genomic prediction (GP) has overcome such limitations since it uses genome-wide markers to estimate the breeding values of unobserved traits in a panel of selection candidates targeting also the multitude of minor QTL (Meuwissen et al. 2001). Precisely, GP models involving FHB resistance in hexaploid wheat (Arruda et al. 2015, 2016b; Mirdita et al. 2015; Rutkoski et al. 2012; Schulthess et al. 2017; Dong et al. 2018) and in durum wheat (Miedaner et al. 2017; Steiner et al. 2018) have shown higher prediction accuracy and larger selection gain than traditional MAS approaches. With few exceptions (Schulthess et al. 2017; Steiner et al. 2018), suggestions and proofs of the advantages of including correlated traits in GP models have been barely explored for FHBr in wheat, even though the well-known associations with others traits like heading date or plant height are obvious. In this regard, earlier flowering genotypes are generally more susceptible to FHB and most of the published evidence support the existence of such negative and significant effect in both bread wheat (Gervais et al. 2003; He et al. 2016; McCartney et al. 2016; Paillard et al. 2004; Schmolke et al. 2005; Yi et al. 2018) and durum wheat (Buerstmayr et al. 2012; Miedaner et al. 2017; Miedaner and Longin 2013; Prat et al. 2017). Loci affecting both traits were found to overlap specifically on chromosomes 4A, 5B, 6A, 6D and 7B in various of the mentioned studies, although the correlation FHBr-heading date might be also highly dependent on environmental factors like temperature, rainfall and humidity (Miedaner and Longin 2013). Another trait studied in the host response to FHB is plant height where previous findings suggest a clear negative and significant trade-off between shortness and resistance to FHB in bread and durum wheat (Buerstmayr et al. 2012; Miedaner and Longin 2013; Prat et al. 2017; Talas et al. 2011). One of the most relevant height-reducing mutant alleles, Rht-B1b, has been intensively introgressed into elite durum wheat germplasm since the 1960s’ and important loci for FHBr have been mapped at the Rht-B1 position on chromosome 4B, (Buerstmayr et al. 2012; Miedaner et al. 2017; Prat et al. 2017; Steiner et al. 2018) suggesting a pleiotropic effect of the latter on susceptibility to FHB (Srinivasachary et al. 2009).
In view of the findings described by Miedaner et al. (2017) regarding to the potential and improvements needed to implement GP methods for FHB, the scope of such endeavor has thus to be widened to deal with the multiple involved traits. Hence, the main objectives of this study were (1) to evaluate the implementation of different approaches in order to include the records of two covariates namely heading date and plant height into GP models and (2) to assess the performance of the developed GP models in terms of prediction accuracy and selection gain for all involved traits.
Materials and methods
Plant material
The international diversity panel in matter contained 170 winter and 14 spring types, including modern and old cultivars as well as current breeding lines. Field trials were conducted at the experimental stations Heidfeldhof (Hoh) and Oberer Lindenhof (Oli) of the University of Hohenheim, Stuttgart, Germany both in the cropping season 2013 and 2014 as well as at the experimental site of the Department of Agrobiotechnology Tulln in Austria (Tul) during 2014. The combination of location and year provided five environments (Hoh13, Hoh14, Oli13, Oli14, Tul14), in which experiments were laid out as \(\upalpha\)-lattice designs with three replications in the locations Hoh, Oli and two replications in Tul.
The Fusarium inoculation procedure and its details can be found in the studies of Miedaner et al. (1996) and Miedaner et al. (2017). In short, the whole experiment was inoculated by \(2 \times 10^{5}\) conidiospores \(\hbox {ml}^{-1}\) with a machine-driven small-plot field sprayer (Hoh, Oli) or a motor-driven backpack sprayer (Tul) several times during flowering. The inoculation dates were spread across the whole flowering period of the experiment in a way that each genotype was inoculated at least once at full flowering. In Tul, the crop canopy was kept moist by mist irrigation during 20 hr after inoculations. Fusarium head blight (FHB) severity was scored several times by the visual evaluation of all spikelets in a plot rating from 1 to 9, where 1 stands for no visible symptoms and 2 to 9 respectively means: < 5%, 5–15%, 15–25%, 25–45%, 45–65%, 65–85%, 85–95% and \(>\,\)95%. Mean FHB severity (FHBms) will be the term referred in this study to the arithmetic mean of six individual plot ratings measured from the beginning of the symptoms development (11–15 days after inoculation) and repeated in a 3-days interval until the beginning of the yellow ripening stage. Heading date (HD) was noted as the day in the year when 75% of the ears of the plot emerged to 75% and plant height (PH) as the cm from the ground to the middle of the ear at EC70 stage.
Phenotypic data analysis
A two-step approach, as described by Möhring and Piepho (2009), was implemented in order to obtain the Best Linear Unbiased Estimates (BLUE) of each genotype for each of the traits. Firstly, each environment was analyzed by a linear mixed model of the form:
where \({y}_{ikm}\) is the phenotypic observation of a trait of interest, \({\mu }\) is the overall mean, \({g}_{i}\) is the effect of the ith genotype, \({r}_{k}\) is the effect of the kth replicate, \({b}_{\textit{km}}\) is the effect of the mth block nested within the kth replicate, and \({e}_{\textit{ikm}}\) is the residual effect. All the effects except \({g}_{{i}}\) were considered as random. In a second step, BLUEs were calculated across the multi-environment trails using the linear mixed model:
where \({y}_{\textit{ij}}\) is the BLUEs from the first step, \({E}_{{j}}\) is the effect of the jth environment and \({e}_{\textit{ij}}\) is the residual term accounting for Genotype \(\times\) Environment interaction and the residual effect. The effect of genotypes was considered here as fixed as in the first model.
Variance components were estimated by restricted maximum likelihood (REML) to estimate the repeatability and entry-mean heritability, (\(\hbox {H}^{2}\)), within each environment and across them as described by Piepho and Möhring (2007) (Eq. 19) using instead the model described in section 2.2 in Miedaner et al. (2017). The models were implemented in the package sommer for R (Covarrubias-Pazaran 2016). Accessions’ id and the estimated BLUEs for each environment can be found as supplementary material.
Correction of the FHBms estimators
Three methods were taken to correct BLUE of FBHms in each of the environments and across them. The first approach, ADJ.A, considered the residuals from the linear regression of FHBms on the covariates HD and PH, used both separate and simultaneously; in a general form following the equation:
where y is a vector of BLUEs for FHBms (dependent variable), \({1 \beta }_{0}\) is the intercept, while \({x}_{\textit{HD}}\) and \({x}_{\textit{PH}}\) are the BLUEs of the covariates HD and PH estimated within each environment by model (1), \(\beta _{\textit{HD}}\) and \(\beta _{\textit{PH}}\) are the regression coefficients and \(\epsilon\) are the residuals. Finally, the mean of the FHBms was added to the vector of residuals to be use as response variable in the subsequent analysis.
In the second method, denoted as ADJ.B, we investigated the approach suggested by Emrich et al. (2008), Miedaner et al. (2006), and included plant height and heading date as fixed covariates when calculating BLUEs resulting in an extension of model (1) to:
where \(c1_{ikm}\) and \(c2_{ikm}\) modeled the plot basis observations of HD and PH, same as in above method \(\beta _{\textit{HD}}\) and \(\beta _{\textit{PH}}\) are the regression coefficients, while analogues to the adjustments made by model (3) either both traits were considered simultaneously or alone. The corrected FHBms values obtained from this method were defined as the effect \({g}_{i}\) from model (4) plus the overall mean of the FHBms.
The third correction method ADJ.C was adopted from Rapp et al. (2018), and consisted in the combination of the target trait and the covariate(s) in a multi-variate mixed model, following this equation:
where each of effects had the same nomenclature as used in models (1) and (2), except for the superscript T which denotes that each term is an array of \(n \times t\) dimension being t the number of traits involved. The initial assumption is that all the effects are considered as random and each follows a multi-variate normal MVN distribution, like the one for the genetic effects \({g}_{\textit{FHBms}}, {g}_{\textit{HD}}, g_{\textit{P}{\mathrm{H}}}\):
where diagonal \(\sigma ^{2}\) terms stands for the respective genetic variances; the genetic correlation of FHBms with HD is: \({r}_{1}\), with PH: \({r}_{2}\) and between covariates: \({r}_{3}\). Then, the residuals from the model (5) were taken, similarly as in Thorwarth et al. (2018), as the corrected values for FHBms according to:
Genotypic data
After a refinement step to exclude markers either with a minor allele frequency (MAF) of < 5% or with a percentage of missing data higher than 20%, as stated in Sieber et al. (2017); a final set of 23,542 DArT (Diversity Arrays Technology Yarralunla, Australia) markers and 7070 single nucleotide polymorphism (SNPs) markers were yielded. Missing data was imputed as the average score of each marker by the ”mean” method included in rrBLUP R package (Endelman 2011). Marker data is available for downloading as supplementary material.
Genomic predictions
Genomic best-linear unbiased predictions were obtained from the following model:
where y contains the calculated FHBms estimates with or without any adjustment; \(\beta\) is the vector of the fixed effects containing the overall mean; g is the vector of genomic estimated breeding values (GEVB) \([g\sim N(0,G\sigma _{g}^{2})]\) with \(\hbox {g}^{2}\) accounting for the genotypic variance estimated by REML and G stands for the genomic relationship matrix obtained according to the description of VanRaden (2008): \(G=ZZ'/2\sum p_{j}(1-p_{j})\) where Z is a \(n \times m\) matrix of m markers and n individuals with elements \(z_{ij}=x_{ij}-2p_{j}+1\) and \({x}_{{\mathrm{ij}}}\) being the value of a given allele for the ith genotype at the jth locus, and \({p}_{j}\) the allele frequency of the jth marker. X and Z are the design matrices for fixed and random effects respectively and, e is a vector containing the residuals \([e\sim N(0,\sigma ^2_e)]\).
The prediction ability (PA) was calculated by a five-fold cross validation (CV) scheme as the correlation between the GEBVs and the corresponding BLUEs. Briefly, the panel was randomly divided in five equally sized subsets, then four out of those were used as the training set and the left one as the validation set. The procedure was repeated 40 times totaling 200 cross validation runs. Mean comparisons between the PAs under any model were estimated with Tukey’s HSD (honest significant difference) test (Tukey 1949) with an alpha level of 5%.
Tuning GBLUP model parameters
The following GBLUP model reformulations included the approach taken by Song et al. (2017), where the covariates effects were directly integrated and considered as fixed in the GBLUP model (8). This approach will be referred later on as G + COV method. In addition, multi-trait GBLUP models (MT.GP) were executed following this general equation:
where \({y}_{t}\) is the array for t traits, namely FHBms, HD and/or PH, containing the BLUE values; \({g}_{t}\) is the array of genetic effects GEBVs \([g\sim MVN(0,\sum _g \bigotimes G)]\) with as the complete unstructured variance-covariance matrix \(\begin{pmatrix} \sigma ^2_{g1} &{} cov_{g1,2} &{} cov_{g1,3} \\ cov_{g1,2} &{} \sigma ^2_{g2} &{} cov_{g2,3} \\ cov_{g1,3} &{} cov_{g2,3} &{}\sigma ^2_{g3} \\ \end{pmatrix}\) alongside with its design matrix \({M}_{t}\). The former terms \(\sigma ^2_{g1}\), \(\sigma ^2_{g2}\) and \(\sigma ^2_{g3}\) are the genetic variances of FHBms, HD and PH; \({cov}_{\textit{g1,2}}\), \({cov}_{\textit{g1,3}}\) are the covariance of FHBms with HD and PH respectively, and \({cov}_{\textit{g2,3}}\) is the covariance between the covariates. The residual array follows a distribution: \([e\sim MVN(0,\sum _e \bigotimes\, I_n)]\) where \({I}_{{n}}\) stands for an \(n \times n\) identity matrix and for the variance-covariance matrix similar to the previous one but accounting for the residuals between traits.
Lastly we evaluated the so called trait assisted genomic prediction (TA.GP) (Fernandes et al. 2018; Michel et al. 2017) in which for each CV run, unlike as in the MT.GP model framework described above, the covariate estimators of the validation set were included into the training set and represented pre-existing information about the genotype performance.
Index selection
Genomic selection indexes were afterwards constructed out of the GEBVs obtained in the MT.GP models. Briefly, we used a desired gains index developed by Pešek and Baker (1969) but restricting gains for the covariates and thus allowing only response to selection towards the target trait.
where \({g}_{\textit{1}}\), \({g}_{{2}}\) and \({g}_{{3}}\) are the GEBVs obtained from multi-trait model (9), b is the array containing the indexes for each trait obtained as \(b=\sum _g^{-1}\times a\), with a being the vector with the respective weights keeping always \({a}_{\textit{FHBms}}\) as 1 and \({a}_{\textit{HD}}\), \({a}_{\textit{PH}}\) equal to zero. The genomic variance-covariance matrix \(\varSigma _{\mathrm{g}}\) was derived from model (9). Finally, an alternative \(\varSigma _{\mathrm{g}}\) matrix was derived by calculating the variances and covariances between the GEBVs obtained from the single trait models ST.GP from model (8). Table 1 condense all the GP approaches previously mentioned in the preceding sections.
Expected selection gain
The differences between the mean of BLUE estimators of the best 5, 10, 15 and 20% performing lines and the overall mean for a given validation set were calculated in each CV run to estimate the expected selection gain (\({G}_{\textit{e}}\)) The mentioned differences were standardized, in order to make comparisons between traits, according to:
where \(\mu _{Trait}^{sel}\) is the trait mean of the selected genotypes ranked based on the FHBms GEBVs and \(\mu _{Trait}^{all}\) stands for the overall mean of the respective trait. When any correction approach was applied to perform the predictions, subscript Trait referred always to the non-adjusted estimations of FHBms. The standardized \({G}_{\textit{e}}\) was also obtained choosing lines based on the ranking on the phenotypic estimation of FHBms (phenotypic selection).
Results
Phenotypic analysis
Plant height records of genotypes in 2014 season were in average greater than in 2013, in which Hoh was the location with the shortest plant stand while Tul had the tallest one and in the across environment analysis plant height was, on average approximately 81.02 cm (Table 2). For the sake of the subsequent analyses we decided to exclude six of the extremely tallest genotypes categorized as outliers based on their across environments’ BLUEs. These “outgroup” lines included several genetic resources with exotic genetic background from the US, Canada, Bulgaria and Ukraine: DGE-1, Agathe, AC Navigator, Cirpan 13, No3026 and Nursith. On average, the heading date was 155 days after January with a significant later heading behavior in 2013. High entry-mean heritabilities are reported here for both covariates analyzed and, moreover, their genetic variances were always higher than both genotype \(\times\) environment and residual variances. In addition, a significant and direct association between both covariates were detected in the environments Hoh13 and Tul14 (Table 2).
The artificial inoculation of Fusarium was successful in all environments, and a broad FHBms variation could be found within the individual environments as well as in the across trail environments analysis ranging from resistant to highly susceptible genotypes. Tul14 and Oli14 were the most and least affected environments, respectively, and the later was the environment with the largest FHBms variation and heritability (Table 2). The updated top resistant lines included the Italian variety ”Belfuggito” released in the 1960’s, the Russian cultivar ”Amazonka”, the cultivar ”Soldur” and several breeding lines from breeding program of the University of Hohenheim. FHBms had a moderate to high heritability, even though its genotypic variance component which was just slightly higher than both the residual and the \(\hbox {G} \times \hbox {E}\) components (Table 2), thus confirming the good quality of the scoring for Fusarium in this multi-environment study.
Both covariates plant height (PH) and heading date (HD), exhibited significantly negative correlations with FHBms of \(\hbox {r}=-\,0.25\) and \(\hbox {r}=-\,0.63\) (\({p}<0.001\)), respectively. Despite of dropping six outlier genotypes for plant height PH, its correlation with FHBms was still highly significant also for the single environment Hoh13. FHBms was in all environments but Tul14 as well as in the multi environment (MET) analysis highly associated with HD as seen by coefficients higher than \(\mathrm{r}=-\,0.60\).
Correction effects
The variability of FHBms BLUEs decreased when they were corrected by either HD or HD plus PH in terms of their coefficient of variation under any of the methods and the same effect was detected for their heritabilities. A comparison between their distributions revealed how their frequencies increased around the mean value being the more pronounced case the \(\hbox {FHBms}^{{\mathrm{ADJ.C}}}\) BLUEs (Supplemental Figure 1).
Amongst the correction methods considering only HD as covariate, ADJ.A and ADJ.C performed nearly similar, both adjusting completely for the unfavorable FHBms-HD correlation while ADJ.B reduced it until non-significance only for the environment Oli13. The former methods though led the coefficient of the FHBms-PH trade-off to significance (\(p <0.05\)). Almost the same first mentioned pattern was found when FHBms’ BLUEs were adjusted exclusively for PH under the former methods however, ADJ.B significantly increased the correlation towards PH except for the environments Hoh14 and Oli14 (Table 3).
The adjustment of the FHBms’ BLUEs considering simultaneously both covariates was evaluated only when worth it i.e. the environment Hoh13 and the MET case. ADJ.A was the only capable to effectively decrease the magnitude of correlation coefficients towards both covariates and turn them non-significant. In the other hand, ADJ.B and ADJ.C merely reduced the \(\sim\) HD coefficients’ magnitudes and regarding PH both methods raised the coefficients in the environment Hoh13 and the former method did so in the MET case (Table 3).
Genomic predictions
Prediction abilities
Prediction abilities of FHBms, across environments ranged from \(\hbox {PA}=0.61\) in Oli14 to \(\hbox {PA}=0.69\) in Hoh14 (Supplemental Table 1). However, when predictions accounted for the \(\hbox {G} \times \hbox {E}\) interaction term, the prediction ability reached a significant higher value of PA = 0.75. The above values corresponded to the FHB mean severity estimators predicted without any correction or parameter tuning in the GBLUP model. It must be mentioned that both prediction abilities and expected selection gain \(\hbox {G}_{\mathrm{e}}\) rates for all the models were calculated taking as reference the non-adjusted estimators of FHBms. This consideration turned out to be a remarkable point since, specifically for the correction methods, it allowed us to compare PAs from any type of approach.
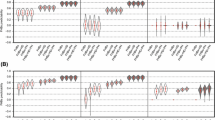
Significant lower mean prediction abilities were obtained when the corrected FHBms estimators were used for genomic prediction as expected since these estimators deviated from the non-adjusted FHBms. In fact, this occurred irrespective of the applied adjustment method, except when PH was the covariate included (Fig. 1 left panel). Analogously, if the \(\sim\)PH models are ignored since their predictabilities are comparable to the reference model, prediction abilities under the ADJ.B method were less diminished and they, in turn, outperformed their equivalents from both ADJ.A and ADJ.C methods. In the other hand, what stands out in Fig. 1 (right panel) is the significant improvement in the prediction abilities of the trait-assisted TA.GP approach, reaching PA = 0.80, when HD was included as covariate. There was no predictability upsurge for any of the multi-trait MT.GP models and even for such version involving both covariates a slight diminution in mean predictability was found. Our so-called G + HD and G + HD + PH alternatives had poorer predictabilities than the multi-trait GP models though greater than their respective ADJ.A and ADJ.C correction models versions.

Stem plots for the mean values of prediction ability PA (above the zero line) and correlation with the covariates (below the zero line) obtained from 40 cycles of five-fold cross validation of genomic prediction for Fusarium head blight mean severity (FHBms) and the covariates heading date (HD) and plant height (PH). Prediction abilities for three FHBms correction methods (left panel) and three GBLUP model parameter tuning (right panel) including multi-trait versions, are illustrated as the correlation of phenotypically estimated BLUEs of both the target trait and covariates with the GEBVs for the target trait. Mean PAs and correlation scores for the non-adjusted values are highlighted in orange bars and the rest of the models/methods are gray colored and decreasingly sorted by the within method average PA. Mean PAs were compared with Tukey’s HSD test with \(\upalpha =0.05\) and displayed in compact letters display fashion. Correlations with *, **, *** superscripts stand for significance level at \(\upalpha =0.05\), 0.01 and 0.001 respectively. Every model is denoted by its abbreviation (see Table 2) followed by “\(\sim\)” symbol and the covariate(s) incorporated

Stem plots for the mean values of prediction ability PA (above the zero line) and correlation with the covariates (below the zero line) obtained from 40 cycles of five-fold cross validation of genomic prediction (GP) for Fusarium head blight mean severity (FHBms) and the covariates heading date (HD) and plant height (PH). In each panel with gray bars are shown the scores for different selection indexes applied over the single and multi-trait GP models’ outputs. In the right panel, the scores for the GP model of the non-adjusted FHBms are presented with orange bars. In the left panel, the multi-trait genomic GP models including the covariates both together and separately are presented with orange bars. Mean PAs were compared with Tukey’s HSD test with \(\upalpha =0.05\) and displayed in compact letters display fashion. Correlation coefficients with *, **, *** superscripts stand for significance level at \(\upalpha =0.05\), 0.01 and 0.001 respectively. Every model is denoted by its abbreviation (see Table 2) followed by “\(\sim\)” symbol and the covariate(s) incorporated

Models performance in terms of expected selection gain (\(\hbox {G}_e\)). Models that predicted FHBms corrected values (left panel), GBLUP model tuning (central panel) and application of restricted selection indexes (right panel) when predicting FHBms –dotted lines–. \(\hbox {G}_e\) values are displayed for FHBms and heading date HD across several selection intensities. In each of the facets are included also the Ge scores for the non-adjusted FHBms predictions –solid lines- and for the selection based exclusively on the phenotypic BLUE values PS –two dashed lines. Every model is denoted by its abbreviation (see Table 2) followed the covariate(s) incorporated

Layout of 178 durum wheat genotypes according to their FHBms and HD BLUE values. After a single round of simulated genomic-based selection aiming the 20% most resistant lines, the probability for a genotype to be selected based on 40 cross-validation cycles is highlighted in a blue-red scale. The mean values of the selected genotypes in each panel are cross-pointed and the overall mean values are marked in the non-adjusted panels. Every panel is labeled with the model abbreviation (see Table 1)
In addition, relative low \(\hbox {PA}=0.60\) resulted when GEBVs obtained from either MT.GP\(\sim\)HD or MT.GP\(\sim\)HD+PH were weighed by restricted selection indexes (Fig. 2 right panel), however the lowermost predictabilities amongst all the methods were obtained when the restricted indexes were derived from ST.GP’ GEBVs involving both HD and HD plus PH versions (Fig. 2 left panel). Lastly, only for the TA.GP approach a slightly predictability increase was detected when the three traits at issue were fit taking the NO.ADJ GP model as reference and the tri-variate versions of both ADJ.C and IDX.ST models were the only ones for which a significant lower prediction ability was observed in a within version’s model comparison.
Correlation with covariates
Genome-based estimators for non-adjustment FHBms were negligibly associated (\(\hbox {r}=-0.14, p > 0.05\)) with PH though still highly correlated (\(\hbox {r}=-0.47, p < 0.001\)) with HD. Genomic predictions under the correction method ADJ.B presented still significant (\({p}< 0.01\)) correlations towards HD, nevertheless the genomic predictions using ADJ.A and ADJ.C methods -omitting their \(\sim\)PH versions- were able to decline the correlation with HD to marginal non-significant levels. Regarding the trade-offs with PH, FHBms’ predictions became significant (\({p}< 0.05\)) when both PH and HD + PH were corrected for in method ADJ.B (Fig. 1 left panel).
All versions of MT.GP and TA.GP models’ predictions were highly associated (\({p}< 0.001\)) with HD, and for the latter model correlation coefficients up to \(\hbox {r}=-0.85\) and \(\hbox {r}=-0.38\) were detected for HD and PH respectively (Fig. 1 right panel). The only MT approach for which a non-significant trade-off between FHBms and PH was found was MT.GP\(\sim\)HD. On the other hand, the method G + COV presented no relevant correlation coefficients between their predictions and both covariates, excepting when PH was the incorporated trait. Non-significant correlations with both PH and HD were observed when predictions obtained either from ST or MT models were weighted by restricted indexes (Fig. 2).
Expected selection gain
A single round of genomic-based selection was performed targeting the 5, 10, 15 and 20% of the most resistant genotypes, and their standardized expected selection gain values are presented in Fig. 3 ticked by approach type and both traits FHBms and HD. Selection based on the most accurate model (TA.GP) showed a larger indirect response towards HD than the conventional GP model for uncorrected FHBms values and such response was even comparable to the one coming by means of phenotypic selection when more than 5% of the lines were selected (Fig. 3 central panel). In contrast, if the GEBVs from ST.GP models were scaled by restricted indexes, their gain differentials in heading time was almost reduced to zero (Fig. 3 right panel). ADJ.A and ADJ.C methods followed IDX.ST model being the most effective alternatives to counteract the gains in heading date (Fig. 3 left panel). G + COV and IDX.MT methods performed similar decreasing by about a half standard deviation the unfavorable expected selection gain towards a later average heading date. Expected selection gains for FHBms amongst approaches kept the same prediction abilities’ ranking with the TA.GP model notably increasing the disease resistance gains. Moreover, confirmed after the simulation of the genomic selection of the best 20% genotypes proportion, the latter was the only method which significantly decreased the FHBms from the GP model with no modifications (Fig. 4). Contrarily, the rest of methods increased in average the susceptibility with exception of the multi-trait MT.GP model for which no meaningful change was detected. Additionally, from Fig. 4 it can be advised that all methods, excluding MT.GP and TA.GP, had the capability to choose more genotypes having both susceptibility and heading date estimators under the overall mean than the reference non-adjusted model.
Discussion
Simultaneous selection for multiple traits is an exigent task in plant breeding, hence some considerations have to made in order to exploit favorable or to adjust unfavorable trade-offs between traits. The present research focused on the prediction of FHB severity in the framework of its correlation with plant height and heading date, using a diverse durum wheat panel as a case study. The investigated alternatives for addressing these relationships ranged from corrections applied prior and post genomic prediction either through adjustments of FHBms estimations or usage of restriction indexes, respectively, as well as the tuning of GP model parameters including multi-trait GP alternatives.
The highly quantitative nature of FHB resistance regarding this case study panel was unveiled by Miedaner et al., (2017) via genome-wide association mapping, reporting nine loci explaining between 1% to 14% of the genetic variance. Combining minor FHBr loci detected in durum varieties with major QTL from bread wheat has been showed certain efficiency in decrease the levels of susceptibility e.g. Prat et al. (2017) reported that concurrently introgressing the major locus Fhb1 with either loci on chromosome 2BL or 5AL could even overcome the negative effects on FHBr of the Rht-B1b allele and, Zhao et al. (2018) combined the introgressed QTL Qfhb.ndwp-7A from the Chinese line PI 277012 with both minor loci on chromosomes 2A and 7A to significantly increase FHB resistance. Such allele combination and deployment in durum populations has been typically achieved by phenotypic selection or MAS in the past, although genomic selection might nowadays be prefigured as a recommended strategy to capture the genetic variation generated by many small-effect QTL and might assist breeders by shortening the breeding cycle or improve the selection gain in early stages where typically more resistant lines could in this way be selected. Moreover, among the studies demonstrating the higher efficiency of GS over traditional FHBr breeding strategies, Mirdita et al. (2015) evidenced a GS predictability three times superior than MAS in the absence of major QTL using a large set of 2325 European winter wheat genotypes evaluated in 11 environments and, Arruda et al. (2016a, b) and Steiner et al. (2018) also showed the same accuracy boost that was even increased when significant FHBr loci were included as fixed effects in the GP models. Predictability of MAS in the present dataset reached 0.65, reported by Miedaner et al. (2017), being even higher than some of our investigated approaches (Figs. 1, 2). However, the latter MAS’ feature could be an overestimation due to the relatedness between lines which in turn is a drawback that GS could overcome as revealed for instance by independent versus cross validation sampling comparisons, that makes GP models’ performance more stable in cases like European wheat breeding programs with slow allele frequency dynamics Jiang et al. (2017).
Correction of covariates tradeoffs as an alternative in GP
Roughly eight QTL for plant height explaining minor proportions of phenotypic variation and not being localized nearby any FHBr locus were revealed for the panel in study. Apart from those loci, the Rht-B1 (Rht1) locus explained more than half of the phenotypic variation in plant stature. It has been verified, for other durum germplasms as well, that Rht1 can increase or even duplicate measurements of FHB severity (Buerstmayr et al. 2012; Prat et al. 2017; Talas et al. 2011). We partially mitigated this trade-off by leaving out six out of nine of the tallest lines carrying the Rht-B1a wild type allele which met the outlier exclusion criteria, by doing so the range and variance of FHB severity ratings did not dramatically change and the correlation coefficient was reduced from \(-\,0.37\) to \(-\,0.25\) (p\(< 0.001\)). The latter exclusion could be taken as a step in order to focus on genomic predictions for elite durum wheat rather than genetic resources. Unlike plant height, for the tandem FHB severity-days to heading/flowering, both positive (Clark et al. 2016; Prat et al. 2017; Steiner et al. 2004; Zhao et al. 2018) and negative (Buerstmayr et al. 2012; Gervais et al. 2003; He et al. 2016; McCartney et al. 2016; Paillard et al. 2004; Schmolke et al. 2005; Somers et al. 2003; Yi et al. 2018) type of correlations have been reported in bread and durum wheat. Co-localization of these traits’ loci have been found in durum on chromosomes 4AL and 6AS according to Prat et al., 2017 and on chromosome 7B to Buerstmayr et al. (2012). In all environments analyzed in this study as well as in the across environments analysis strong negative FHBms-HD trade-offs were detected with the exception of Tul14 (Table 2), a circumstance that may be attributable to the moist-keeping conditions during flowering in this location and both its lower HD repeatability and range (e.g. just 9.5 days compared with 20.5 days in Hoh13). In addition, HD loci detected for this panel were not in LD respect to FHB resistance QTL yet when FHBms BLUES were adjusted for this covariate (Miedaner et al. 2017), therefore the great influence of specific differences in weather factors or ripening and possible pleiotropy events might be plausible explanations for this trade-off.
Some alternatives have been investigated to assess true genotypic effects of FHB resistance and dissect it from a passive mechanism of resistance such as plant height or development stage i.e. flowering time. For instance Klahr et al. (2007) used a covariance analysis and detected the only stable QTL QFhb.hs-5B non-affected by plant height and heading date in a RIL population of European bread wheat. Likewise, Miedaner et al. (2006) adjusted the FHB ratings to the effect of heading date and evaluated the effect of QTL introgressed from resistant donors in European elite spring wheat lines. Other covariance considerations highlight additional advantages over traditional mapping methods when trying to identify QTL, i.e. He et al. (2016) and Lu and Lillemo (2014) included HD and PH as covariates into the QTL mapping algorithms, while Yi et al. (2018) suggested a conditional QTL mapping on either HD or PH. In the context of genome-wide association mapping and genomic prediction studies for FHB resistance in hexa- and tetraploid wheat, several studies (Arruda et al. 2015, 2016a, b; Miedaner et al. 2017) have used the strategy first proposed by Emrich et al. (2008) and included the trait HD as a quantitative covariate in the mixed linear model from where the adjusted BLUE/BLUP values were obtained. The former covariance analysis was also examined in this study under the method ADJ.B (4) for which the unexpected higher FHBms-PH correlations obtained after correction for this covariate in addition to the unsatisfactory association levels of the \(\hbox {FHBms}^{\mathrm{ADJ.B}}\) predictions discourage considering this method for either low or moderately correlated traits. Furthermore, as proposed by Song et al. (2017), our so-called G + COV approach evaluated the inclusion of the covariates as fixed effects in the GP model stated in [8] and it turned out to be the most efficient method to accomplish both higher predictabilities and negligible trade-offs (\({p}> 0.05\)) with the covariates regardless whether HD alone or together with PH were involved.
Regression-based correction methods are frequently employed in a plant breeding perspective to assess negative trade-offs between major agronomic traits e.g. disease resistance and both maturity and plant height (Bormann et al. 2004; Bradshaw et al. 2004) or grain yield and protein content (Monaghan et al. 2001). In fact, one of the most extended applications of regression-based adjustments addressing the latter traits in wheat are the so-called grain protein deviations GPD, which are defined as the residuals of regressing protein content on grain yield (Rapp et al. 2018; Thorwarth et al. 2018). The ADJ.A and ADJ.C methods took advantage of this procedure as did the correction of FHBms based on regression coefficients derived from either single or multi-variate mixed models on a plot basis, respectively. The fact that the predictions for the FHBms values corrected by the latter methods were among the poorest in terms of predictability can be explained since these adjustments deviated the most from the original FHBms; although, this attribute led, in turn, to a highly effective reduction of the respective trade-offs both at BLUEs and GEBVs levels.
Single-trait predictions comparison with multi-trait models
Multi-trait genomic prediction MT.GP models tested in this study provided no predictability’ advantage over the reference non-adjusted model. These observation is supported by other empirical studies, where the usage of multi-trait GP models did not necessarily result in an increase in prediction abilities (Fernandes et al. 2018; Schulthess et al. 2017; Guo et al. 2014), but is in disagreement with reports that showed important performance’ improvements when either i) higher heritabilities for the indicator traits and/or ii) significant correlations between traits were evidenced (Calus and Veerkamp 2011; Guo et al. 2014; Jia and Jannink 2012). In a simulation study Calus and Veerkamp (2011) showed that only the inclusion of traits with genetic correlations stronger than 0.5 and higher heritabilities can lead to greater predictabilities and Schulthess et al. (2017) suggested that the benefits of MT.GP models could be limited since achieving last mentioned requirements might be somewhat unrealistic, as was evinced for our case where no pronounced differences in heritabilities were found. Plant height having moderately high phenotypic associations with FHBms in this study, did not seem to play a major role in multi-trait GP models as expected if compared to, for instance, the evidence showed in Lado et al. (2018) who have demonstrated that the inclusion of a third mildly correlated trait into MT.GP models was not suitable to increase predictabilities in comparison to the inclusion of a single highly correlated indicator trait.
The multi-trait GP approach type that improved the prediction ability was the one for which phenotypic records of the correlated covariates were already available in the validation population when predicting the target trait i.e. the trait-assisted selection model TA.GP Fernandes et al. (2018) also known as phenotypic imputation Jia and Jannink (2012). This predictability’ upsurge was evinced for both the TA.GP model fitting HD alone and HD+PH simultaneously, which was significant in the former case. A similar effect has been noticed in several studies with different crops (Fernandes et al. 2018; Jia and Jannink 2012; Rutkoski et al. 2012, 2016; Schulthess et al. 2017; Wang et al. 2017), although the degree of association between the target trait’ predictions and the covariates has not been extensively assessed e.g. in Fernandes et al. (2018) since the trade-offs between yield biomass and either moisture or plant height in sorghum were not of concern. While predictions based on both approaches MT.GP and TA.GP exhibited the higher predictabilities among the alternatives studied here, the former had similar correlation levels with the covariates than the NO.ADJ model but the TA.GP models exhibited coefficients up to 44% higher respect to HD and regarding to PH it was even more than tripled in the TA.GP\(\sim\)PH model. Moreover, it was demonstrated after selection simulation that under TA.GP model none of the moderately resistant and early flowering lines would have been selected. Using this phenotypic imputation for multi-trait GP, Steiner et al. (2018) revealed an important increase in the trade-off FHB severity-PH when both PH and PH+FD (flowering date) were fit in the GP models since in their analyzed panel the only moderately correlated trait with FHB severity was plant height. The actual usage of such previous generated records, that are routinely assessed in earlier generations during variety development for GP models should therefore be examined in the light of the indirect responses which rise for such scenarios.
Weighting predictions by genomic restriction indexes
Final products delivered by breeders in form of new cultivars must satisfy a certain number of requirements. In this context, a durum cultivar is desirable if it displays moderate resistance levels to Fusarium, high yields, short stature and early flowering. Breeders have been traditionally conducted multi-trait selection by methods like tandem selection, independent culling levels and index selection (Dudley 1997), while usage of selection indexes is generally more efficient than the former ones (Wang et al. 2018). Since the net merit index proposed by Smith (1936) and Hazel (1943) many modifications like restriction index have appeared, where the aim is to improve a given trait while keeping the response to others at zero (Kempthorne and Nordskog 1959). Similarly, Pešek and Baker (1969) presented an approach to achieve a certain rate of desired gains for a set of traits. In our case, weighting the GEBVs of FHBms, HD and PH obtained in ST.GP models by employing this desired gain index as restriction indexes had the best performance in annulling any response towards the respective indicator traits HD and PH though with the subsequent largest predictability penalization for FHBms. However, the utter goal in a multi-trait GP framework may not necessarily be the cancellation of the unfavorable correlations towards the indicator traits but rather their reduction till negligible levels of either magnitude or significance, as it was accomplished here using the restriction indexes ponderating the GEBVs derived from MT.GP. In the same regard, the use of this genomic indexes deflated the enlarged FHB severity-PH trade-off detected after either ST or MT genomic-based predictions in a durum wheat panel (Steiner et al. 2018). The authors of the mentioned study did not detect significant differences between the index usage in either single or multiple trait models in terms of predictability although on average they were significantly lower than their unweighted counterparts. In this regard, it must be said that the most plausible models through which both the less drastic predictability penalization and non-significant \(\sim\)HD correlation levels were attained appeared to be G + HD and IDX.MT\(\sim\)HD, although some features would give the former certain advantage: (1) its slightly higher predictability, (2) due to it is less computationally demanding and, (3) since it was more prone to avoid the selection of the earliest flowering-moderately susceptible genotypes after the genomic selection simulation round (Fig. 4).
Conclusion
Highly significant associations of FHB severity and the indicator traits PH and HD estimations were detected, although the trade-off with HD turned out to be the only one relevant and persisted after genomic predictions. Including PH as the only covariate in genomic prediction models resulted in imperceptible changes in the GP performances, except when PH was fit in the trait-assisted GP model for which a significant decline in predictability was detected. Incorporating simultaneously both covariates did not improve the predictability and for the models dealing with multi-trait frameworks such configuration led to worse predictabilities. The approaches that corrected FHBms phenotypic estimations by regression-based methods and weighting the FHBms’ genomic breeding values by restriction indexes derived from single trait GP models were the most effective alternatives controlling the indirect responses towards the covariates and even decreased by up to two standard deviations the most important trade-off. Multi-trait genomic prediction approaches significantly outperformed in predictability the non-adjusted reference model only when HD records for the validation population were already available, which led in turn to the largest rise in the correlation between HD and FHBms. Fitting HD as fixed effect in the GP model (G + COV) and correcting the FHBms’ genomic estimations using restriction indexes derived from multi-trait GP models achieved smaller predictabilities but were on the other hand capable to reduce HD trade-offs, and therefore represent alternative models with the highest relevance in this study.
References
Arruda M, Brown PJ, Lipka AE, Krill AM, Thurber C, Kolb FL (2015) Genomic selection for predicting Fusarium head blight resistance in a wheat breeding program. Plant Genome. https://doi.org/10.3835/plantgenome2015.01.0003
Arruda M, Brown P, Brown-Guedira G, Krill AM, Thurber C, Merrill KR, Foresman BJ, Kolb FL (2016a) Genome-wide association mapping of Fusarium head blight resistance in wheat using genotyping-by-sequencing. Plant Genome. https://doi.org/10.3835/plantgenome2015.04.0028
Arruda M, Lipka AE, Brown PJ, Krill AM, Thurber C, Brown-Guedira G, Dong Y, Foresman BJ, Kolb FL (2016b) Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol Breed 36(7):84. https://doi.org/10.1007/s11032-016-0508-5
Beres BL, Brûlé-Babel AL, Ye Z, Graf RJ, Turkington TK, Harding MW, Kutcher HR, Hooker DC (2018) Exploring genotype $\times $ environment $\times $ management synergies to manage fusarium head blight in wheat. Can J Plant Pathol 40(2):179–188. https://doi.org/10.1080/07060661.2018.1445661
Bormann CA, Rickert AM, Castillo Ruiz RA, Paal J, Lübeck J, Strahwald J, Buhr K, Gebhardt C (2004) Tagging quantitative trait loci for maturity-corrected late blight resistance in tetraploid potato with PCR-based candidate gene markers. Mol Plant Microbe Interact 17(10):1126–1138. https://doi.org/10.1094/MPMI.2004.17.10.1126
Bradshaw JE, Pande B, Bryan GJ, Hackett CA, McLean K, Stewart HE, Waugh R (2004) Interval mapping of quantitative trait loci for resistance to late blight [Phytophthora infestans (Mont.) de Bary], height and maturity in a tetraploid population of potato (Solanum tuberosum subsp. tuberosum). Genetics 168(2):983–95. https://doi.org/10.1534/genetics.104.030056
Buerstmayr H, Steiner B, Hartl L, Griesser M, Angerer N, Lengauer D, Miedaner T, Schneider B, Lemmens M (2003) Molecular mapping of QTLs for Fusarium head blight resistance in spring wheat. II. Resistance to fungal penetration and spread. Theor Appl Genet 107(3):503–508. https://doi.org/10.1007/s00122-003-1272-6
Buerstmayr H, Ban T, Anderson JA (2009) QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: a review. Plant Breed 128(1):1–26
Buerstmayr M, Huber K, Heckmann J, Steiner B, Nelson JC, Buerstmayr H (2012) Mapping of QTL for Fusarium head blight resistance and morphological and developmental traits in three backcross populations derived from Triticum dicoccum $\times $ Triticum durum. Theor Appl Genet 125(8):1751–1765. https://doi.org/10.1007/s00122-012-1951-2
Calus MP, Veerkamp RF (2011) Accuracy of multi-trait genomic selection using different methods. Genet Sel Evol 43(1):26. https://doi.org/10.1186/1297-9686-43-26
Clark AJ, Sarti-Dvorjak D, Brown-Guedira G, Dong Y, Baik BK, Van Sanford DA (2016) Identifying rare FHB-resistant segregants in intransigent backcross and F2 winter wheat populations. Front Microbiol 7:277. https://doi.org/10.3389/fmicb.2016.00277
Covarrubias-Pazaran G (2016) Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE. https://doi.org/10.1371/journal.pone.0156744
Darwish WS, Ikenaka Y, Nakayama SM, Ishizuka M (2014) An overview on mycotoxin contamination of foods in Africa. J Vet Med Sci 76(6):789. https://doi.org/10.1292/JVMS.13-0563
Dean R, Van Kan JA, Preotrious ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickmann M, Kahmann R, Ellis J, Foster GD (2012) The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13(4):414–430. https://doi.org/10.1111/j.1364-3703.2011.00783.x
Dong H, Wang R, Yuan Y, Anderson J, Pumphrey M, Zhang Z, Chen J (2018) Evaluation of the potential for genomic selection to improve spring wheat resistance to Fusarium head blight in the Pacific Northwest. Front Plant Sci 9:911. https://doi.org/10.3389/fpls.2018.00911
Dudley JW (1997) Quantitative genetics and plant breeding. Adv Agron 59:1–23. https://doi.org/10.1016/S0065-2113(08)60051-6
Dweba C, Figlan S, Shimelis H, Motaung T, Sydenham S, Mwadzingeni L, Tsilo T (2017) Fusarium head blight of wheat: pathogenesis and control strategies. Crop Prot 91:114–122. https://doi.org/10.1016/J.CROPRO.2016.10.002
Emrich K, Wilde F, Miedaner T, Piepho HP (2008) REML approach for adjusting the Fusarium head blight rating to a phenological date in inoculated selection experiments of wheat. Theor Appl Genet 117(1):65–73. https://doi.org/10.1007/s00122-008-0753-z
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome J 4(3):250. https://doi.org/10.3835/plantgenome2011.08.0024
Fernandes SB, Dias KOG, Ferreira DF, Brown PJ (2018) Efficiency of multi-trait, indirect, and trait-assisted genomic selection for improvement of biomass sorghum. Theor Appl Genet 131(3):747–755. https://doi.org/10.1007/s00122-017-3033-y
Fiedler JD, Salsman E, Liu Y, de Jiménez M, Hegstad JB, Chen B, Manthey FA, Chao S, Xu S, Elias EM (2017) Genome-wide association and prediction of grain and semolina quality traits in durum wheat breeding populations. Plant Genome. https://doi.org/10.3835/plantgenome2017.05.0038
Fung F, Clark RF (2004) Health effects of mycotoxins: a toxicological overview. J Toxicol Clin Toxicol 42(2):217–234. https://doi.org/10.1081/CLT-120030947
Gervais L, Dedryver F, Morlais JY, Bodusseau V, Negre S, Bilous M, Groos C, Trottet M (2003) Mapping of quantitative trait loci for field resistance to Fusarium head blight in an European winter wheat. Theor Appl Genet 106(6):961–970. https://doi.org/10.1007/s00122-002-1160-5
Guo G, Zhao F, Wang Y, Zhang Y, Du L, Su G (2014) Comparison of single-trait and multiple-trait genomic prediction models. BMC Genet. https://doi.org/10.1186/1471-2156-15-30
Hazel LN (1943) The genetic basis for constructing selection indexes. Genetics 28(6):476–90
He X, Lillemo M, Shi J, Wu J, Bjørnstad Å, Belova T, Dreisigacker S, Duveiller E, Singh P (2016) QTL characterization of Fusarium head blight resistance in CIMMYT bread wheat line Soru#1. PLoS ONE 11(6):e0158052. https://doi.org/10.1371/journal.pone.0158052
Huhn MR, Elias EM, Ghavami F, Kianian SF, Chao S, Zhong S, Alamri MS, Yahyaoui A, Mergoum M (2012) Tetraploid Tunisian wheat germplasm as a new source of Fusarium head blight resistance. Crop Sci 52(1):136. https://doi.org/10.2135/cropsci2011.05.0263
Jia Y, Jannink JL (2012) Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 192(4):1513. https://doi.org/10.1534/genetics.112.144246
Jia H, Zhou J, Xue S, Li G, Yan H, Ran C, Zhang Y, Shi J, Jia L, Wang X, Luo J, Ma Z (2018) A journey to understand wheat Fusarium head blight resistance in the Chinese wheat landrace Wangshuibai. Crop J 6(1):48–59. https://doi.org/10.1016/J.CJ.2017.09.006
Jiang Y, Schulthess AW, Rodemann B, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Ganal MW, Röder MS, Reif JC (2017) Validating the prediction accuracies of marker-assisted and genomic selection of Fusarium head blight resistance in wheat using an independent sample. Theor Appl Genet 130(3):471–482. https://doi.org/10.1007/s00122-016-2827-7
Kadkol GP, Sissons M (2016) Durum wheat: overview. In: Wrigley C, Corke H, Seetharaman K, Faubion J (eds) Encyclopedia of Food Grains, 2nd edn. Oxford, Academic Press, pp 117–124
Kempthorne O, Nordskog AW (1959) Restricted selection indices. Biometrics 15(1):10. https://doi.org/10.2307/2527598
Klahr A, Zimmermann G, Wenzel G, Mohler V (2007) Effects of environment, disease progress, plant height and heading date on the detection of QTLs for resistance to Fusarium head blight in an European winter wheat cross. Euphytica 154(1–2):17–28. https://doi.org/10.1007/s10681-006-9264-7
Kumar A, Sandhu N, Dixit S, Yadav S, Swamy BPM, Shamsudin NAA (2018) Marker-assisted selection strategy to pyramid two or more QTLs for quantitative trait-grain yield under drought. Rice 11(1):35. https://doi.org/10.1186/s12284-018-0227-0
Lado B, Vázquez D, Quincke M, Silva P, Aguilar I, Gutiérrez L (2018) Resource allocation optimization with multi-trait genomic prediction for bread wheat (Triticum aestivum L.) baking quality. Theor Appl Genet 131(12):2719–2731. https://doi.org/10.1007/s00122-018-3186-3
Liu S, Hall MD, Griffey CA, McKendry AL (2009) Meta-analysis of QTL associated with Fusarium head blight resistance in wheat. Crop Sci 49:1955–1968. https://doi.org/10.2135/cropsci2009.03.0115
Lu Q, Lillemo M (2014) Molecular mapping of adult plant resistance to Parastagonospora nodorum leaf blotch in bread wheat lines ‘Shanghai-3/Catbird’ and ‘Naxos’. Theor Appl Genet 127(12):2635–2644. https://doi.org/10.1007/s00122-014-2404-x
McCartney CA, Brûlé-Babel AL, Fedak G, Martin RA, McCallum BD, Gilbert J, Hiebert CW, Pozniak C (2016) Fusarium head blight resistance QTL in the spring wheat cross Kenyon/86ISMN 2137. Front Microbiol 7:1542. https://doi.org/10.3389/fmicb.2016.01542
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–29
Michel S, Ametz C, Gungor H, Akgöl B, Epure D, Grausgruber H, Löschenberger F, Buerstmayr H (2017) Genomic assisted selection for enhancing line breeding: merging genomic and phenotypic selection in winter wheat breeding programs with preliminary yield trials. Theor Appl Genet 130(2):363–376. https://doi.org/10.1007/s00122-016-2818-8
Miedaner T, Longin CFH (2013) Genetic variation for resistance to Fusarium head blight in winter durum material. Crop Pasture Sci 65(1):46–51. https://doi.org/10.1071/CP13170
Miedaner T, Gang G, Geiger HH (1996) Quantitative-genetic basis of aggressiveness of 42 isolates of Fusarium culmorum for winter rye head blight. Plant Dis 118:97–100
Miedaner T, Wilde F, Steiner B, Buerstmayr H, Korzun V, Ebmeyer E (2006) Stacking quantitative trait loci (QTL) for Fusarium head blight resistance from non-adapted sources in an European elite spring wheat background and assessing their effects on deoxynivalenol (DON) content and disease severity. Theor Appl Genet 112(3):562–569. https://doi.org/10.1007/s00122-005-0163-4
Miedaner T, Sieber AN, Desaint H, Buerstmayr H, Longin CFH, Würschum T (2017) The potential of genomic-assisted breeding to improve Fusarium head blight resistance in winter durum wheat. Plant Breed 136(5):610–619. https://doi.org/10.1111/pbr.12515
Mirdita V, He S, Zhao Y, Korzun V, Bothe R, Ebmeyer E, Reif JC, Jiang Y (2015) Potential and limits of whole genome prediction of resistance to Fusarium head blight and Septoria tritici blotch in a vast Central European elite winter wheat population. Theor Appl Genet 128(12):2471–2481. https://doi.org/10.1007/s00122-015-2602-1
Möhring J, Piepho HP (2009) Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci 49(6):1977. https://doi.org/10.2135/cropsci2009.02.0083
Monaghan JM, Snape JW, Chojecki AJS, Kettlewell PS (2001) The use of grain protein deviation for identifying wheat cultivars with high grain protein concentration and yield. Euphytica 122(2):309–317. https://doi.org/10.1023/A:1012961703208
Oliver RE, Stack RW, Miller JD, Cai X (2007) Reaction of wild emmer wheat accessions to Fusarium head blight. Crop Sci 47(2):893. https://doi.org/10.2135/cropsci2006.08.0531
Oliver RE, Cai X, Friesen TL, Halley S, Stack RW, Xu SS (2008) Evaluation of Fusarium head blight resistance in tetraploid wheat ( L.). Crop Sci 48(1):213. https://doi.org/10.2135/cropsci2007.03.0129
Paillard S, Schnurbusch T, Tiwari R, Messmer M, Winzeler M, Keller B, Schachermayr G (2004) QTL analysis of resistance to Fusarium head blight in Swiss winter wheat (Triticum aestivum L.). Theor Appl Genet 109(2):323–332. https://doi.org/10.1007/s00122-004-1628-6
Pešek J, Baker RJ (1969) Desired improvement in relation to selection indices. Can J Plant Sci 49(6):803–804. https://doi.org/10.4141/cjps69-137
Piepho HP, Möhring J (2007) Computing heritability and selection response from unbalanced plant breeding trials. Genetics. https://doi.org/10.1534/genetics.107.074229
Pirgozliev SR, Edwards SG, Hare MC, Jenkinson P (2003) Strategies for the control of Fusarium head blight in cereals. Eur J Plant Pathol 109(7):731–742. https://doi.org/10.1023/A:1026034509247
Prat N, Buerstmayr M, Steiner B, Robert O, Buerstmayr H (2014) Current knowledge on resistance to Fusarium head blight in tetraploid wheat. Mol Breed 34(4):1689–1699. https://doi.org/10.1007/s11032-014-0184-2
Prat N, Guilbert C, Prah U, Wachter E, Steiner B, Langin T, Robert O, Buerstmayr H (2017) QTL mapping of Fusarium head blight resistance in three related durum wheat populations. Theor Appl Genet 130(1):13–27. https://doi.org/10.1007/s00122-016-2785-0
Rapp M, Lein V, Lacoudre F, Lafferty J, Müller E, Vida G, Bozhanova V, Ibraliu A, Thorwarth P, Piepho HP, Leiser WL, Würschum T, Longin CFH (2018) Simultaneous improvement of grain yield and protein content in durum wheat by different phenotypic indices and genomic selection. Theor Appl Genet 131(6):1315–1329. https://doi.org/10.1007/s00122-018-3080-z
Rutkoski J, Benson J, Jia Y, Brown-Guedira G, Jannink JL, Sorrells M (2012) Evaluation of genomic prediction methods for Fusarium head blight resistance in wheat. Plant Genome J 5(2):51. https://doi.org/10.3835/plantgenome2012.02.0001
Rutkoski J, Poland J, Mondal S, Autrique E, Pérez LG, Crossa J, Reynolds M, Singh R (2016) Canopy temperature and vegetation indices from high-throughput phenotyping improve accuracy of pedigree and genomic selection for grain yield in wheat. G3 (Bethesda, Md) 6(9):2799–808. https://doi.org/10.1534/g3.116.032888
Schmolke M, Zimmermann G, Buerstmayr H, Schweizer G, Miedaner T, Korzun V, Ebmeyer E, Hartl L (2005) Molecular mapping of Fusarium head blight resistance in the winter wheat population Dream/Lynx. Theor Appl Genet 111(4):747–756. https://doi.org/10.1007/s00122-005-2060-2
Schulthess AW, Zhao Y, Longin CFH, Reif JC (2017) Advantages and limitations of multiple-trait genomic prediction for Fusarium head blight severity in hybrid wheat (Triticum aestivum L.). Theor Appl Genet. https://doi.org/10.1007/s00122-017-3029-7
Sieber AN, Longin CFH, Würschum T (2017) Molecular characterization of winter durum wheat (Triticum durum) based on a genotyping-by-sequencing approach. Plant Genet Resour 15(01):36–44. https://doi.org/10.1017/S1479262115000349
Smith HF (1936) A discriminant function for plant selection. Ann Eugen 7(3):240–250. https://doi.org/10.1111/j.1469-1809.1936.tb02143.x
Somers DJ, Fedak G, Savard M (2003) Molecular mapping of novel genes controlling Fusarium head blight resistance and deoxynivalenol accumulation in spring wheat. Genome 46(4):555–564. https://doi.org/10.1139/g03-033
Song J, Carver BF, Powers C, Yan L, Klápště J, El-Kassaby YA, Chen C (2017) Practical application of genomic selection in a doubled-haploid winter wheat breeding program. Mol Breed 37(10):117. https://doi.org/10.1007/s11032-017-0715-8
Srinivasachary Gosman N, Steed A, Hollins TW, Bayles R, Jennings P, Nicholson P (2009) Semi-dwarfing Rht-B1 and Rht-D1 loci of wheat differ significantly in their influence on resistance to Fusarium head blight. Theor Appl Genet 118(4):695–702. https://doi.org/10.1007/s00122-008-0930-0
Steiner B, Lemmens M, Griesser M, Scholz U, Schondelmaier J, Buerstmayr H (2004) Molecular mapping of resistance to Fusarium head blight in the spring wheat cultivar Frontana. Theor Appl Genet 109(1):215–224. https://doi.org/10.1007/s00122-004-1620-1
Steiner B, Michel S, Maccaferri M, Lemmens M, Tuberosa R, Buerstmayr H (2018) Exploring and exploiting the genetic variation of Fusarium head blight resistance for genomic-assisted breeding in the elite durum wheat gene pool. Theoret Appl Genet. https://doi.org/10.1007/s00122-018-3253-9
Talas F, Longin CFH, Miedaner T (2011) Sources of resistance to Fusarium head blight within Syrian durum wheat landraces. Plant Breed 130(3):398–400. https://doi.org/10.1111/j.1439-0523.2011.01867.x
Thorwarth P, Piepho HP, Zhao Y, Ebmeyer E, Schacht J, Schachschneider R, Kazman E, Reif JC, Würschum T, Longin CFH (2018) Higher grain yield and higher grain protein deviation underline the potential of hybrid wheat for a sustainable agriculture. Plant Breed. https://doi.org/10.1111/pbr.12588
Tuberosa R, Pozniak C (2014) Durum wheat genomics comes of age. Mol Breed 34(4):1527–1530. https://doi.org/10.1007/s11032-014-0188-y
Tukey JW (1949) Comparing individual means in the analysis of variance. Biometrics 5(2):99. https://doi.org/10.2307/3001913
VanRaden P (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91(11):4414–4423. https://doi.org/10.3168/JDS.2007-0980
Venske E, dos Santos RS, Farias DdR, Rother V, da Maia LC, Pegoraro C, Costa de Oliveira A (2019) Meta-analysis of the QTLome of Fusarium head blight resistance in bread wheat: refining the current puzzle. Front Plant Sci 10:727. https://doi.org/10.3389/fpls.2019.00727
Walter S, Nicholson P, Doohan FM (2010) Action and reaction of host and pathogen during Fusarium head blight disease. New Phytol 185(1):54–66. https://doi.org/10.1111/j.1469-8137.2009.03041.x
Wang X, Li L, Yang Z, Zheng X, Yu S, Xu C, Hu Z (2017) Predicting rice hybrid performance using univariate and multivariate GBLUP models based on North Carolina mating design II. Heredity 118(3):302–310. https://doi.org/10.1038/hdy.2016.87
Wang X, Xu Y, Hu Z, Xu C (2018) Genomic selection methods for crop improvement: current status and prospects. Crop J 6(4):330–340. https://doi.org/10.1016/J.CJ.2018.03.001
Yaniv E, Raats D, Ronin Y, Korol AB, Grama A, Bariana H, Dubcovsky J, Schulman AH, Fahima T (2015) Evaluation of marker-assisted selection for the stripe rust resistance gene Yr15, introgressed from wild emmer wheat. Mol Breed 35(1):43. https://doi.org/10.1007/s11032-015-0238-0
Yi X, Cheng J, Jiang Z, Hu W, Bie T, Gao D, Li D, Wu R, Li Y, Chen S, Cheng X, Liu J, Zhang Y, Cheng S (2018) Genetic analysis of Fusarium head blight resistance in CIMMYT bread wheat line C615 using traditional and conditional QTL mapping. Front Plant Sci 9:573. https://doi.org/10.3389/fpls.2018.00573
Zhao M, Leng Y, Chao S, Xu SS, Zhong S (2018) Molecular mapping of QTL for Fusarium head blight resistance introgressed into durum wheat. Theor Appl Genet 131(9):1939–1951. https://doi.org/10.1007/s00122-018-3124-4
Zhu X, Zhong S, Cai X (2016) Effects of D-genome chromosomes and their A/B-genome homoeologs on Fusarium head blight resistance in durum wheat. Crop Sci 56(3):1049. https://doi.org/10.2135/cropsci2015.09.0535
Acknowledgements
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU). We thank Matthias Fidesser for the excellent technical support and supervising the field trials. This project has received funding from the Deutsche Forschungsgemeinschaft (Grant ID: LO 1816/2-1) and the European Union’s Horizon 2020 research and innovation program under the Marie Słodowska-Curie Grant Agreement No. 674964.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moreno-Amores, J., Michel, S., Miedaner, T. et al. Genomic predictions for Fusarium head blight resistance in a diverse durum wheat panel: an effective incorporation of plant height and heading date as covariates. Euphytica 216, 22 (2020). https://doi.org/10.1007/s10681-019-2551-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-019-2551-x