
Abstract
Understanding the exchange of individuals between wildlife populations, particularly those with naturally fragmented habitats, is important for the effective management of these species. This is of particular consequence when the species is of conservation concern, and isolated populations may be lost due to pressures from predation or competition, or catastrophic events such as wildfire. Here we demonstrate the use kinship and population structure analysis to show potential recent movement between colonies in metapopulations of yellow-footed rock-wallaby (Petrogale xanthopus Gray 1854) at two sites in the Grey Range of Queensland, and at four sites in the Gawler Ranges of South Australia. These colonies are also compared to a single colony from the Flinders Ranges, a connected landscape of rock-wallaby habitat. Using reduced representation next-generation sequencing, we acquired and filtered a set of ~ 17,000 single-nucleotide polymorphisms to examine population genetic variation, structure and relationships within populations, and also identify putative migrants. Initial STRUCTURE analysis re-confirmed each population should be considered separately. Tests of population genetic variation identify several colonies appearing to be experiencing genetic erosion, also with low calculated effective population sizes (Ne = 4.5–36.6). Pairwise comparisons of individual relatedness (relatedness coeffiecients; r) implied several contemporary movement events between colonies within both the Gawler and Grey Ranges (r > 0.125), which was then affirmed with tests for putative first generation migrants. These results are of particular note in South Australia, where threat abatement (management of key predators and competitors) may facilitate dispersion. Additionally, in Queensland, colonies are separated by anthropogenic barriers: predator exclusion fencing designed to exclude dingoes (Canis familiaris) from grazing land, which may hinder dispersal. This work highlights the usefulness of population genetics to inform management outcomes in wildlife, in this case, highlighting the need for threatened species management at the landscape level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A metapopulation is the group of spatially distinct populations of the same species connected by movement of individuals (Wells and Richmond 1995). For species in such systems, movement behaviours are essential, as they insure against risk of extinction from negative pressures (such as predation, competition and catastrophic events; Holyoak and Lawler 1996), and protect against the negative genetic effects of inbreeding (Olivieri et al. 1995; Perrin and Mazalov 1999). Movement behaviour also gives rise to potential recolonization of “empty” sites or the formation of new populations at suitable sites (Hanski 1998). Understanding such movement is therefore critical for effective species management.
For timid and reclusive species, it can be hard to determine whether individuals are moving between isolated colonies. In these species, the use of genetic techniques to identify the relationships between individuals of a population could be used to infer recent movement events, and be extremely useful to understanding barriers to dispersal (Escoda et al. 2019).
The yellow-footed rock-wallaby (Petrogale xanthopus Gray, 1854) is a threatened macropod found in the semi-arid zone of Australia. There are two subspecies of P. xanthopus: P. x. xanthopus is found in several remote mountain ranges of South Australia (SA; Threatened Species Scientific Committee 2016a), and also a single population in New South Wales (NSW; Lim and Giles 1987), and P. x. celeris is found in the Grey Range in Queensland (QLD; Threatened Species Scientific Committee 2016b). The distribution of both subspecies is assumed to have significantly decreased since European settlement (Copley 1983). Rock-wallabies (Petrogale spp.) have high habitat specificity, only occupying complex rocky habitats (see Gordon et al. 1993; Lim and Giles 1987; Smith and Allen 2021; Telfer et al. 2008). As a result of this habitat specificity, suitable rock-wallaby habitat is naturally fragmented, forming metapopulations (Lethbridge et al. 2019; Lethbridge and Strauss 2015; Murray et al. 2008; Ruykys and Lancaster 2015). Primary evidence of movement between colonies of P. xanthopus is surprisingly scarce given the quantity of literature describing the species, particularly in South Australia, though it is not completely absent. A study using tracking collars and ear-marked rock-wallabies in Queensland found evidence of a single movement event over 36 months (Sharp 2002), and another (also with collars) revealed several long distance transient movements of released captive-bred rock-wallabies (Lapidge 2001). The latter also inferred several dispersal events from wild rock-wallabies that were trapped at the previously empty reintroduction sites.
The limitations of radio tracking and capture-mark-recapture to detect all movement behaviours is evident—both rely on the captured individual moving during the study period. In addition to this, the ability to detect or recapture individuals that have moved is further limited by the distance of the movement. Detecting long distance movements, such as natal dispersal, with such techniques is understandably infrequent for P. xanthopus. The use of genetic data would mitigate both these shortcomings, as it would detect any contemporary movement event between colonies at any distance, as long as two related individuals are sampled. To this end, a recent study assessing the genetic variation of P. x. xanthopus identified 13 putative first generation migrations of between 2 and 60 km over connected habitat in the Flinders ranges using population assignment methods based on the individuals’ genetic structure gained through microsatellite analysis (n = 194, Potter et al. 2020). Potter et al. (2020) built on earlier genetic analysis of dispersal (also using microsatellite analysis), that had concluded that yellow-footed rock-wallabies rarely move between colonies, even within connected habitat (Pope et al. 1996), although the methods employed were reliable, this earlier study was limited by sample size. Advancements in genetic technologies such as next-generation sequencing (NGS), and the development of more robust methods of determining kinship values also provides greater confidence in the results of kinship analysis. This confidence can allow for more reliable inferences to be drawn between kinship values and the probability of a contemporary movement between colonies (Escoda et al. 2017). These early P. xanthopus studies were also performed without or with limited threat abatement practices (effective management of key predators and feral competitors) in place, which may be critical to allow rock-wallaby movement behaviours (Pope et al. 1996, 1998).
While the recent publications on the same species are highly informative, a greater understanding of P. xanthopus inter-colony movement is needed to understand potential metapopulation dynamics for the purpose of informing management of the species. The distribution of P. x. celeris has recently (2016) been heavily subdivided by agricultural exclusion fences (Smith et al. 2020c). While the fences may ultimately be of a benefit to the species’ long term persistence (Smith et al. 2020c), exclusion fencing has the potential to isolate colonies, which may lead to genetic consequences (Smith et al. 2020a, b, c). The conservation of P. x. xanthopus may also be reliant on an understanding of inter-colony relations and connectivity. Predation from red foxes (Vulpes vulpes), competition with feral goats (Capra hircus) and catastrophes can all contribute to extinction risk of the entire metapopulation by restricting dispersal behaviours between colonies. Understanding the interchange of the individuals between sites would give insight into the effect of pest species control on metapopulation dynamics and the utility of broad scale pest management in the effective conservation of P. xanthopus.
Given the cryptic nature of the species, its disjunct distribution, and threats facing both subspecies, here we use P. xanthopus to demonstrate the usefulness of reduced representation next-generation sequencing to study metapopulation movement, population structure and how outcomes from such studies can help inform threatened species management. Our objective is to assess if there has been recent movement of P. xanthopus between geographically distinct colonies, using population structure analysis, pairwise relatedness coefficients and identification of first-generation migrants. We then discuss the value of kinship analysis to infer species movement within potential metapopulations, and how this analysis affects the management of P. xanthopus, both in South Australia and Queensland.
Materials and methods
Study sites
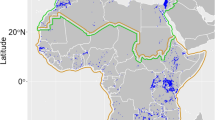
Petrogale xanthopus were trapped and ear biopsies were obtained at four sites in the Gawler Ranges, and one site in the Flinders Ranges (South Australia) by Lethbridge and Andrews (2014) and later Lethbridge (unpublished report), under a S.A. Wildlife Ethics approval: S23997-19, and at two sites in the Grey Range, near Quilpie in central-western Queensland, under a University of Southern Queensland Animal Ethics approval: USQ-17REA011 (Fig. 1). The Gawler Ranges (GWL), the Flinders Ranges (FLD) and the Grey Range (GRY) will hereafter be referred to as ‘populations’. Trapping sites within populations will be referred to as ‘colonies’. Trapping in the Flinders and Gawler Ranges (South Australia) took place from August 2012 to September 2016, and in the Grey Range (Queensland) in 2018 and 2019. The two colonies in Queensland are separated by 8 km, including approximately 5 km of unsuitable habitat. Historically this gap between the two colonies was thought to be traversable by P. xanthopus, but an exclusion fence was erected 2 years prior to sampling to alleviate the pressures of wild dog predation on sheep grazing properties. The colonies in the Gawler Ranges are not disrupted by fencing but vary in distance from Yandinga, a theorised source/refuge population (Fig. 1). The Gawler Ranges populations were also historically disrupted by exclusion fencing (circa 1920s), but spatial and temporal information on this fencing is minimal/non-existent.

Yellow-footed rock-wallaby trapping sites. The main map displays the locations of the Gawler Ranges, Flinders Ranges and Grey Range. Inset A shows the locations of the 4 colonies trapped in the Gawler Ranges (SA). Inset B shows the location of 2 colonies trapped in the Grey Range (QLD). Inset C shows the context of the main map within the broader global region. Waukawoodna Gap (SA; the only trap site within the Flinders Ranges) is marked by a solid square (■) on the main map
Trapping, sampling, extraction and sequencing
Rock-wallabies were trapped using established methods in soft-walled treadle cage. A tissue biopsy was removed from each rock-wallaby’s ear using a 3 mm punch. The sample is taken from the opposite ear to an ear-tag employed for potential future visual identification in the field. Morphometric data was also collected before each rock-wallaby was microchipped for identification on recapture. Tissue samples were placed in Longmire’s buffer solution (Queensland samples—Longmire et al. 1997) or 100% ethanol (South Australian samples) and stored in a cool place until extraction. DNA was extracted from 95 samples (Table 1) using a salt-based ethanol extraction (Ebert and Andrew 2014) and quantified on a Qubit™ Flurometer and quality controlled using a NanoDrop™ 3000. One sample from the Flinders Ranges site was not of high enough quality for accurate sequencing (based on quality control steps) and therefore was removed from the samples sent for sequencing. Samples were then sent to Diversity Arrays Technology (DArT P/L) in Canberra, ACT for sequencing via their DArTseq service (Kilian et al. 2012). The protocol involves technical replicates, which provides an empirical measure of the repeatability of the resulting loci. DArT conducted filtering and reference-free clustering of reads, followed by genotyping using their proprietary analysis pipeline (see https://www.diversityarrays.com). Three samples did not meet DArT’s internal quality control thresholds, and therefore data was not returned for these samples.
Data filtering
The single-nucleotide polymorphism (SNP) data returned by DArT was first explored in R (version 3.6.2) using the dartR v1.1.11 package and following the workflow suggested in the package documentation (Gruber et al. 2019a, b). The reports generated by this initial analysis were used to inform filtering thresholds (Tables S1–S5, Figs. S1, S2). The primary dataset was later subdividedbased on population structure results. This was completed to look for hierarchical structuring (see Results) and for further analysis of kinship (see (v) Coefficients of relationship, relatedness networks). As these subsets reflect the source populations, these datasets were named after the population. The filtered, total dataset will be referred to as the primary dataset (Table 1). Monomorphic loci are automatically removed during this sub-setting. Data were first filtered for the average repeatability of each locus (loci with repeatability > 0.95 were kept), and the call rate by locus (> 0.95) and by individual (> 0.90). The data was then filtered to remove over-split loci (< 0.2 Hamming distance), and for observed heterozygosity greater than 0.6. Finally, loci with minor allele frequencies lower than 0.05 were removed before all metrics were recalculated. Filtering thresholds were chosen based on the recommendations of Gruber et al. (2019b) and O’Leary et al. (2018).
Relatedness estimates range from 0 (totally unrelated individuals) to 1 (clones). Preliminary data exploration identified four samples with high pairwise relatedness to other samples, which is not consistent with outbreeding and random mating. These values may have arisen in a number of ways: monozygotic twinning, duplicate sampling or pipetting error prior to sequencing. Twins are known to occur in Petrogale assimilis (Spencer and Marsh 1997), and the twinning rate in Macropus is low, but non-zero (Inns 1980; Norbury 1986; van Oorschot and Cooper 1989). Additionally, pouch young have DNA samples taken, but are not ear tagged/microchipped and may have been sampled again as adults. Nevertheless, as pipetting error cannot be excluded, we removed samples with high pairwise kinship estimates prior to all analyses. The sample of higher quality was kept and the sample of lower quality was removed prior to filtering and final analysis.
Population structure, statistics, genetic distances, and PCoA
Population structure analysis was performed using STRUCTURE (v2.3.4; Pritchard et al. 2000). Parameter settings varied by dataset. The primary dataset was first analysed from for all clusters (K) from 1 to 10, with 10 independent repeats of 100,000 MCMC (Markov Chain Monte Carlo) iterations after a 10,000 iteration burn-in. Based on these results, Datasets 2–4 (subsets of the primary dataset) were analysed, using more robust methods. Each dataset was analysed from K = 1–10, after 50,000 burn-in and 500,000 MCMC iterations with sampling populations as priori. K values were estimated through an assessment of both Pritchard’s model likelihood method (Pritchard et al. 2000) and Evanno’s ΔK method (Evanno et al. 2005) implemented in Structure Harvester (Earl and vonHoldt 2012), and scrutinised following recommendations detailed in Cullingham et al. (2020) for K = 2 results. The production of population structure bar plots was performed in CLUMPAK (v1.1; Kopelman et al. 2015). Also, Principle Coordinate Analysis (PCoA) was performed using Euclidean distance in R with dartR. Each dataset was analysed and informative dimensions examined. Plots of the two most informative axes of each population were generated with ggplot2 v3.3.2 (Wickham 2006).
Mean observed heterozygosity (HO), expected heterozygosity (HE), population Allelic Richness (rarefied allelic counts, per locus and population; AR), and inbreeding coefficients (FIS) were reported using hierfstat v0.04–22 in R (basic.stats function) with 98% confidence intervals (1000 bootstraps). Population divergence was explored using several approaches; pairwise private and fixed alleles, Euclidean and Nei’s (Nei 1972) genetic distance matrices as well as pairwise FST. Pairwise FST values were also estimated using StAMPP (1000 bootstraps, 95% CI), which follows Weir and Cockerham’s (Weir and Cockerham 1984) methods. An unrooted neighbour joining tree was constructed from a Euclidian distance matrix before effective population sizes (\({N}_{e}\)) were estimated for each colony using NeEstimator (v2.1; Do et al. 2014) using the Linkage Disequilibrium (random mating) methods for a minimum allele frequency at 0.05, 0.02 and 0.01 with jack-knife confidence intervals (Jones et al. 2016).
Coefficients of relationship, relatedness networks
The R package SNPRelate v1.16.0 (Zheng 2013) was first used to calculate Identity-By-State (IBS) fractions for each pair of rock-wallabies in each of the Datasets 1–4. Hierarchical cluster analysis using the average link (UPGMA) method was then performed on each of the IBS matrices to produce a genetic distance trees. Kinship was estimated independently in Datasets 2–4, to ensure that later filtering was not biased by sample size, and that appropriate population-specific allele frequencies were used. The primary dataset contains SNPs that are monomorphic in individual populations and kinship values can be inflated by these fixed alleles. Identity-By-Descent coefficients (calculated by Maximum Likelihood Estimation) were estimated in R using the SNPRelate package (Zheng 2013). Pairwise kinship values were estimated based on these IBD coefficients, also using SNPRelate. Kinship values vary from 0 to 0.5 so, for ease of comprehension, coefficients of relationship (r), which vary from 0 to 1, were calculated by doubling kinship values (Wright 1922). Full siblings and parent–offspring pairs are expected to have r values of approximately 0.5, half siblings 0.25, and first cousins 0.125, and so on. To visualise close r values geographically, the program GEPHI (v0.9.2; Bastian, Heymann and Jacomy, 2009) was used to produce relatedness networks with the plugin GeoLayout. Individuals were treated as nodes and relatedness estimates as edge values, weighted by increasing r. These visualisations were limited to coefficients of relationship greater than 0.0625 (i.e. 1/16th, e.g. first-cousin once removed, half-first-cousin).
First-generation migration
GeneClass2 (Piry et al. 2004) was used to identify presumed first generation (f0) migrants. This was performed using the Rannala and Mountain (1997) methods with Monte-Carlo resampling at both the 0.01 and 0.05 probability threshold and all loci. This approach follows the same methods as Potter et al. (2020). The distance between the trapping (source) colony and the putative origin colony (as well as the site map, Fig. 1) was also then generated in ArcMap (v10.5.1; Environmental Systems Research Institute 2019).
Results
Population structure, statistics, genetic distances, and PCoA
The Gawler sites (Coolgundibie, Organ Pipes, Stone Dam and Yandinga) consistently had the lowest observed and expected heterozygosity (HO and HE, respectively), followed by the Gray Range population and Flinders Ranges sites (Table 2; Fig. S3).. FIS (inbreeding coefficient; calculated as \(F=1-{H}_{O}/{H}_{E}\)) showed several colonies to have heterozygote excess (negative values) and several to be deficient in heterozygotes. FIS values were indicative of heterozygote deficiency in the Yandinga and Organ Pipes colonies of the Gawler Ranges and Waukawoodna Gap (Table 2). All other colonies excepting the Alaric Colony, where FIS was not significantly different from 0, showed evidence of heterozygote excess (Table 2). These results were also reflected in the results of allelic richness, which was highest within the Flinders Ranges colony, followed by the Grey Range and finally Gawler Ranges (Table 2). Pairwise FST was greatest between the Coolgundibie and Ray colonies, and the lowest value was between the geographically nearby Yandinga and Organ Pipes colonies (Table 3). Average FST by population was 0.03 within Grey Range colonies, and 0.04 within Gawler Ranges. Average pairwise FST was greatest between the Grey and Gawler Ranges (\(\overline{{F }_{ST}}\)= 0.63). Flinders Ranges to Grey Range comparisons resulted in slightly higher FST values (\(\overline{{F }_{ST}}\) = 0.47) than Flinders Ranges to Gawler Ranges (\(\overline{{F }_{ST}}\) = 0.35). All FST values with the exception of Organ pipes-Yandinga were significant (p < 0.001; Table 3).
The neighbour-joining tree clearly separates colonies by population (Fig. 2). This was also evident using several other different genetic distance measures (Supplementary Information), as well as through Principal Coordinates Analysis (Fig. 3). The first two eigenvalues in the PCoA of the primary dataset corresponded to 47.1% and 12.0% (Fig. S5) of the variation between samples.

Unrooted neighbour-joining tree of all YFRW populations. Neighbour joining tree generated from Euclidean genetic distance matrix of YFRW. Circles indicate population groups based on mountain ranges, Gawler ranges—left, Flinders Ranges—centre, Grey Range—right

PCoA by Dataset. Principal Coordinate analysis of each Dataset, respectively, coloured by colony. X- and Y-axes show the first and second most informative eigenvalues, correspondingly, and their percentage contributions (in parentheses) to total variance
Effective population size (Ne) estimates ranged from 4.5 for the Stone Dam colony, to 36.6 for the Yandinga colony in the Gawler Ranges. In the Flinders Ranges, the Waukawoodna Gap colony had an Ne of 21.4, and in the Grey Range, the Ray and Alaric colonies had Ne estimates of 5.1 and 21.3, respectively (Table 2).
After STRUCTURE analysis, Evanno’s ΔK method from STRUCTURE HARVESTER indicated that K = 2 was the best supported K value for the primary dataset 1; however, the maximum L(K), pairwise FST values (Table 3), neighbour-joining trees (Fig. 2), genetic distances (Table S7-10), PCoA results (Fig. 3) and other analyses all gave K = 3 greater support. Therefore, delimitations of K = 3 (Fig. 4) were used to subset the primary dataset into populations for further investigation of hierachical population structure and kinship analysis.

STRUCTURE bar plots for the Primary Dataset, Galwer Range Dataset, and Grey Range Dataset. (1) Bar plots showing the major clusters for K = 2 and K = 3 (all 10/10 iterations, i.e. no minor clustering). K = 2 was most supported by Evanno’s ΔK method, though K = 3 was better supported by other methods of examining population structure. (2) Bar plots showing K = 2 and K = 3 for the Gawler Dataset and (4) K = 2 and K = 6 for the Grey Range Datasetfrom structure analysis are also shown. Graphs of ΔK and L(K) over each value of K and bar plots of all clusters (K = 1–10) can be found in the Supplementary material
PCoA analysis of the Gawler ranges showed some potential structure (Fig. 3) though the percentage contributions of each axis to differentiation were relatively low (Supplementary Information). Genetic distance trees showed little structure. Bayesian cluster analysis with STRUCTURE showed the greatest support for K = 2 in the Gawler Ranges, but again K = 3 (Fig. 4), which also had a high ΔK value, appeared to be better supported by other analysis. The colonies from the Grey Range also showed some limited genetic differentiation from PCoA, distance trees and STRUCTURE analysis (K = 2 and K = 6, supported from ΔK and L(K)). These results were also supported by pairwise FST values, which were low, but still significant (p < 0.001; Table 3).
Coefficients of relationship, relatedness networks
Average intra-population relatedness (\(\overline{r }\)) was greatest in the Gawler Ranges (0.031). The Gawler Ranges populations had 419 relationships greater than r = 0 (out of a potential 1035 pairwise relationships, 40.5%), and 161 relationships greater than the r = 0.0625 threshold (15.5%; Fig. 5). This was followed by the Grey Range where average \(\overline{r }\)=0.027, with 67 of the 300 possible relationships greater than r = 0 (22.3%), and 30 greater than the r-threshold (10.0%). Finally, in the Flinders Ranges, 25 out of a possible 136 relationships were greater than 0 (18.4%), with 15 relationships greater than the threshold (11.0%; \(\overline{r }\)=0.027), (see Figs. S10, S15, S17 and S22).

YFRW relatedness networks generated from IBD estimates. Visual representation of relatedness estimates greater than 0.0625 generated though Identity by Descent (IBD) analysis. Coloured circles (nodes) show individuals of each population, relatedness lines (edges) are weighted by the strength of the relationship (not comparable between populations). Grouped node locations correspond to geographic locations (Fig. 1)
First-generation putative migration
Putative migration here is defined as shared sequence similarities (based on genetic distance, allele frequency and Bayesian criterion; Piry et al. 2004) exhibited geographically, thus suggesting a permanent movement event from one site to another, rather than the more general definition of ecological migration, which relates to season and resource. Out of 71 individuals (46 in the Gawler Ranges and 25 in the Grey Range) 17 were identified as putative f0 putative migrants (Table 4). Of the 15 potential migrations within the Gawler Ranges, seven (41.1%) were between Organ Pipes and Yandinga. Two rock-wallabies caught at Stone Dam appeared to be immigrants, one from Yandinga and one from Organ Pipes, and three emigrants of Stone Dam were caught at other colonies (two at Yandinga, one at Organ Pipes). Lastly, there were three occurrences of putative migration to Coolgundibie, from the Yandinga (two) and Organ Pipes (one) colonies. These putative migrations were of ~ 13.32 and ~ 15.70 km, respectively. There were no occurrences of putative migrants from Coolgundibie caught at any other trapped site in the Gawler Ranges. Within the Grey Range population two rock-wallabies were identified as potential f0 putative migrants; both suggest emigration from Alaric to the Ray colony.
Figures from reports used for filtering, distance matrices, scree plots of Eigen values of PCoA, output figures of Structure Harvester, bar plots of all values of K from CLUMPAK, histograms of relationship coefficients, outputs of GeneClass2 and other additional results for all datasets can be found in the Supplementary Information. The data underlying this analysis is also available online (Smith et al. 2020a, b, c).
Discussion and summary
Key findings
This study aimed to examine contemporary movement between disjunct colonies of P. xanthopus with varying levels of connectedness in both South Australia and Queensland. We found though the examination of population structure, kinship analysis and maximum likelihood analysis of putative migration that there has likely been recent contemporary movement. Individuals with kinship values greater than 0.0625 were common between colonies within the Gawler Ranges with several movements greater than 13 km, and despite an approximately 5 km gap of unsuitable habitat, related individuals were present from the two sampled locations in the Grey Range. Further to this, multiple putative f0 migrations were identified within both the Gawler and Grey Ranges. From these results it is clear that P. xanthopus are more mobile within mountain ranges than previously assumed. The results from the Gawler ranges colonies in particular add weight to recently published results also showing several potential long distance migration events of P. xanthopus in the Flinders Ranges (Potter et al. 2020). This previous study primarily focused the genetic variation of colonies, but provided insight into potential movements between colonies that was more thoroughly examined here. Greater mobility of rock-wallabies also fits closer with metapopulation theory, where movement between semi-isolated demes and back into locally extinct or new sites helps maintain the genetic and ecological viability of the broader population (Hanski 1998). This new understanding is likely to affect future management of the species.
Population structure
The initial analysis of population structure across all samples (Primary Dataset) showed that each mountain range should be considered independent of the others when considering genetic monitoring within the species. These results are in agreement with the literature on P. xanthopus genetics and phylogeny which splits the South Australian and Queensland species into subspecies, and shows that separate mountain ranges/populations are genetically distinct (Eldridge 1997; Pope et al. 1996, 1998; Potter et al. 2012, 2020). Populations of P. xanthopus were shown to have an order of magnitude greater pairwise FST values between mountain ranges than within mountain ranges (Table 3), and all other methods employed for determining genetic differentiation between populations and individuals supported this result (Figs. 2, 3, Fig. S4 and Tables S6–S10). Given the ΔK method has the propensity to lead to over identification of K = 2 (Cullingham et al. 2020; Janes et al. 2017), STRUCTURE and other methods indicated that3 was best supported K value). From the initial structure analysis there was also evidence of some population substructuring, leading us to examine fine-scale structure in data subsets based on each of the populations. This showed that there was indeed some genetic structuring between colonies of each population, though not nearly as distinct as between mountain ranges (Figs. 3, 4). Fine scale structure is also supported by the recent analysis of microsatellite loci, which showed some structure within colonies within the Flinders Ranges (Potter et al. 2020).
Genetic diversity
Greater genetic diversity in populations is closely linked to a population’s ability to withstand genetic pressures associated with small population size and to adapt to changing environmental conditions (Frankham 2005). Our results in rock-wallabies showed that the smaller, less geographically connected, populations in the Gawler and Grey Ranges have lower genetic diversity (HO and AR) than those of the Flinders Ranges (Waukawoodna site—Table 2). This outcome is in concordance with assumptions that genetic diversity should be positively correlated with population size (Frankham 1996). Significance testing of FIS also revealed all colonies (with the exception of Alaric in the Grey Range) significantly deviate from zero, the null value, indicating either heterozygote deficiency (Yandinga, Organ Pipes, Waukawoodna) or heterozygote excess (Ray, Coolgundibie and Stone Dam). While the theory behind inbreeding leading to heterozygote deficiency is well established (Buri 1956), the processes leading to heterozygote excess are less clear. Though there are several potential explanations (referenced and discussed in Stevens et al. 2007), this case is likely due to the small population sizes. Binomial sampling error can cause differences in allele frequencies of male and female breeders, leading to heterozygote excess in their progeny (Luikart and Cornuet 1999; Robertson 1965; Waples 2015). This explanation of heterozygote excess in these rock-wallaby colonies is supported by the estimates of effective population size (Ne), which are substantially smaller in the corresponding colonies (Table 2).
Inter-colony relationships and movement
The first step employed here to examine how P. xanthopus move between colonies was assessing pairwise relatedness (r) between all individuals of a population. The analysis revealed relationships greater than the 0.065 (i.e. a common ancestor within 4 generations) threshold between all colonies within each population (excluding the Flinders Ranges), and one relationship of ~ 0.5 (indicating a full-sibling or parent-progeny relationship) between two colonies in the Gawler Ranges.. These relationships also indicate that there may be greater mobility between populations than previously recognised based on early genetic assessments (Pope et al. 1996).
While these results indicate recent movement between the colonies, inter-colony r values that are further apart than full-sibling or parent-offspring (less than ~ 0.5) may be a result of stepwise movement events through intermediate and unsampled colonies. To identify the likely colonies-of-origin, putative f0 migrants were identified in GeneClass2. The program identified 15 potential first generation migrants in the Gawler Ranges, and two in the Grey Range (Table 4). As might have been postulated from the analysis of population structure, movement between Organ Pipes and Yandinga constituted a large percentage of putative migrants. These colonies are separated by only 2.9 km of suitable habitat (Fig. 1). As P. xanthopus are known to move up to 1.5 km to water points (Sharp 2011), this regular exchange of individuals was expected. Less predictable were the putative migrants into and from Stone Dam, and those from the Yandinga/Organ Pipes area, found at Coolgundibie. Interestingly, no f0 migrants were identified as having dispersed in the reverse direction, i.e. Coolgundibie to any other colony in the Gawler Ranges. Despite this exception, within the Gawler Ranges rock-wallabies appear to move readily to areas of connected habitat, even over relatively large distances.
Within the Grey Range population, only two migrants were detected, with both indicating the individual moved from Alaric to Ray. These movements are of particular relevance, both to the understanding of species population dynamics, and to future management of the species in Queensland. The area of the Grey Range that these two colonies reside in is at the very Southern end of P. x. celeris’ distribution. The Alaric colony is located on the Canaway Fault, a line of connected rock-wallaby habitat/cliffs that run the North–South. The Ray colony, however, is approximately 8 km distant on fragmented rocky outcrops known as The Matrix. As previously mentioned, the shortest distance between suitable rock-wallaby habitat between the Canaway Fault and The Matrix is ~ 5 km of open, flat, farmland. Moreover, this dispersal route is now impeded by an exclusion fence (Fig. 1).
Management implications
Early assessments of the Gawler ranges had not found colonies of P. xanthopus at Coolgundibie (Lethbridge 2004a) or Stone Dam (Lethbridge et al. 2012; Lethbridge et al. 2010). In part, samples were collected from these colonies to assess whether theylikely represent recolonisation events from the other established colonies under threat abatement. The high kinship values between samples from these colonies and those of Yandinga and Organ Pipes suggest that movement reported here likely reflect recolonisation as a result of 25 years of goat and fox control (and some kangaroo management) and a major drought in the Gawler Ranges (Lethbridge et al. 2019). Integrated pest management (of feral rabbits Oryctolagus cuniculus, foxes, goats, cats Felis catus and weeds) may have driven this pulse of movement as a reduced number of predators, and also a shortage of resources in core P. xanthopus colonies, induced density-dependant dispersal behaviours. Continued recovery of rock-wallabies under these conditions likely depends on the threat abatement being widespread enough to facilitate further recolonisation events at sites that, without abatement, would be unsuitable.
For P. x. celeris, the rapid expansion of pest-exclusion fencing in the area (Smith et al. 2020c) potentially divides colonies that have previously relied on movement to maintain greater levels of genetic diversity, such as that of the Flinders Ranges (Table 2). In all cases, identified f0 migrants (Table 4) were also those with higher inter-colony kinship values in the relatedness network (Fig. 5). The fence between the two colonies was completed in 2016, so either the rock-wallabies were able to pass across the exclusion fence, or the rock-wallabies relocated colonies prior to the exclusion fences construction. The fences have proved highly effective at preventing the movement of other species (RAPAD and QFPI 2018), and aging the potential migrants based on tail-length data (Lethbridge 2004b) showed that dispersal pre-2016 was possible. It would be reasonable to assume that the migrants moved colonies pre-fence, and that the population in Queensland is now fragmented by exclusion fencing (Smith et al. 2020c). Indications that the rapid reduction in P. xanthopus distribution over the past century has already had negative genetic impacts on population viability which makes this assessment all-the-more troubling (Potter et al. 2020). This is particularly true of colonies, such as the Ray colony, that were already not part of connected and continuous rock-wallaby habitat. To allay the potential negative impacts, the genetic variation of isolated populations would have to be monitored into the future and mitigation strategies such as meta-population management be employed if/when the populations appear to be experiencing genetic erosion. Meta-population management has been successfully employed for the maintenance of genetic variation issues caused by fencing in the past (Boast et al. 2018; Miller et al. 2015; Schroeder 2019).
Limitations
While most FIS values deviated significantly, they may not reflect the true value in the populations. Estimation of both FIS and Ne assumes random mating and random sampling in the population; both assumptions may have been violated here. For P. xanthopus there is evidence of social structure, dominance behaviours and philopatry (Lapidge 2001; Potter et al. 2020; Sharp 2002) and territorial defence (or more specifically, trap bait defence) which may result in non-random sampling. Additionally, clear delimitations between sampling populations is needed to avoid Wahlund effects (De Meeûs, 2018; Wahlund 1928; Waples 2015). As it appears colonies have an exchange of individuals (Fig. 5), colonies likely represent demes of a metapopulation, and this may have altered FIS estimates of colonies.
This study also lacked a proper ‘control’ site, which demonstrably functions as a healthy metapopulation, and replication within the separate populations. Understandably, even with sufficient time and resources, finding and sampling populations and colonies to remove this limitation is difficult in threatened species.
Future research
It is clear from the results above that for the effective conservation and recovery of P. xanthopus, the management of the species needs to be considered at a broader scale. The long term conservation of the species relies on the ability of individuals to immigrate into neighbouring colonies for the maintenance of genetic variation (Potter et al. 2020). Further to this, the recovery of the species relies on an increase in the species distribution as potentially suitable habitats become available as a result of broad scale management of threatening processes (Lethbridge et al. 2012; Lethbridge et al. 2019). Future research should seek to elucidate whether recolonised sites continue to be genetically supplemented from other colonies in the metapopulation. Additionally, as new sites arise, genetic assignment of colonising rock-wallabies should be made a priority to establish source populations. More specifically for P. x. celeris, continuing genetic monitoring should be implemented alongside long term studies of behaviour (and simulations of metapopulation movement and genetics) both to ensure genetic viability of the population is maintained and as a study of the long term effects of anthropogenic barriers on species genetics and behaviour (Smith et al. 2020a, b, c).
Molecular ecology tools can often be overlooked in favour of more traditional methods of assessing species. The above study clearly demonstrates the application of genetic ecology methods, specifically kinship analysis, to help inform management of a threatened species. By applying these more appropriate methods for a cryptic and disjunct species we were able to infer regular movement between colonies where previous studies failed to find movements or significantly underestimated their regularity. Studies of species that have had similar short comings should consider how genetic analysis could overcome these obstacles and better inform the species management.
Data availability
Data accessibility: Metadata (Sampling locations), and genotypes are available at GitHub: https://github.com/D-A-Smith/YFRWgenetics. https://doi.org/10.5281/zenodo.7412787.
Change history
24 February 2023
Missing Open Access funding information has been added in the Funding Note.
References
Bastian M, Heymann S, Jacomy M (2009). Gephi: an open source software for exploring and manipulating networks. In: Proceedings of the Third international AAAI conference on weblogs and social media. (San Jose, California).
Boast L, Chelysheva E, van der Merwe V, Schmidt-Küntzel A, Walker E, Cilliers D et al (2018) Cheetah translocation and reintroduction programs: past, present, and future. In: Marker L, Boast L, Schmidt-Kuentzel A (eds) Cheetahs: biology and conservation. Academic Press, London, pp 275–289
Buri P (1956) Gene frequency in small populations of mutant Drosophila. Evolution 10(4):367–402
Copley P (1983) Studies on the yellow-footed rock-wallaby, Petrogale xanthopus Gray (Marsupialia: Macropodidae) I. Distribution in South Australia. Wildl Res 10(1):47–61
Cullingham C, Miller J, Peery R, Dupuis J, Malenfant R, Gorrell J, Janes J (2020) Confidently identifying the correct K value using the ΔK method: when does K = 2? Mol Ecol 29(5):862–869. https://doi.org/10.1111/mec.15374
De Meeûs T (2018) Revisiting FIS, FST, Wahlund effects, and null alleles. J Hered 109(4):446–456
Do C, Waples R, Peel D, Macbeth G, Tillett B, Ovenden J (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14(1):209–214
Earl D, vonHoldt B (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Ebert D, Andrew R (2014) DNA extraction from tissue preserved in ethanol or Longmires lysis buffer. School of environmental and rural science. University of New England, Australia
Eldridge M (1997) Restriction analysis of mitochondrial DNA from the yellow-footed rock-wallaby, Petrogale xanthopus: implications for management. Wildl Res 24(3):289–294
Environmental Systems Research Institute (2019) ArcMap (Version 10.5.1). USA: ESRI. Retrieved from https://desktop.arcgis.com/en/arcmap/
Escoda L, González-Esteban J, Gómez A, Castresana J (2017) Using relatedness networks to infer contemporary dispersal: application to the endangered mammal Galemys pyrenaicus. Mol Ecol 26(13):3343–3357. https://doi.org/10.1111/mec.14133
Escoda L, Fernández-González Á, Castresana J (2019) Quantitative analysis of connectivity in populations of a semi-aquatic mammal using kinship categories and network assortativity. Mol Ecol Resour 19(2):310–326. https://doi.org/10.1111/1755-0998.12967
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10(6):1500–1508
Frankham R (2005) Stress and adaptation in conservation genetics. J Evol Biol 18(4):750–755
Gordon G, McRae P, Lim L, Reimer D, Porter G (1993) The conservation status of the yellow-footed rock-wallaby in Queensland. Oryx 27(3):159–168
Gruber B, Georges A, Unmack P, Clark L, Berry O (2019a) dartR: importing and analysing SNP and silicodart data generated by genome-wide restriction fragment analysis. Retrieved from https://CRAN.R-project.org/package=dartR
Gruber B, Georges A, Unmack P, Clark L, Berry O (2019b) Introduction to dartR. Retrieved from https://CRAN.R-project.org/package=dartR (Unpublished)
Hanski I (1998) Metapopulation dynamics. Nature 396:41–49
Holyoak M, Lawler S (1996) Persistence of an extinction-prone predator-prey interaction through metapopulation dynamics. Ecology 77(6):1867–1879
Inns R (1980) Occurrence of twins in macropod marsupials. Search 11:118–119
Janes J, Miller J, Dupuis J, Malenfant R, Gorrell J, Cullingham C, Andrew R (2017) The K= 2 conundrum. Mol Ecol 26(14):3594–3602
Jones A, Ovenden J, Wang Y (2016) Improved confidence intervals for the linkage disequilibrium method for estimating effective population size. Heredity 117(4):217–223
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Pompanon F, Bonin A (eds) Data production and analysis in population genomics. Humana Press, USA, pp 67–89
Kopelman N, Mayzel J, Jakobsson M, Rosenberg N, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191
Lapidge S (2001) Reintroduction biology of yellow-footed rock wallabies (Petrogale xanthopus celeris and P. x. xanthopus). (Doctoral Thesis), University of Sydney, Australia
Lethbridge M (2004a) A survey of the yellow-footed rock-wallaby in the Gawler Ranges and recommendations for further monitoring. A report to Flinders University, Australia
Lethbridge M (2004b) Tail length to age P. xanthopus (software). Ecoknowlegde, Australia
Lethbridge M, Andrews L (2014). Yandinga gorge yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) trapping, Gawler Ranges National Park July 2014. EcoKnowledge report to Department of Environment, Water and Natural Resources: Australia
Lethbridge M, Strauss J (2015) A novel dispersal algorithm in individual-based, spatially-explicit population viability analysis: a new role for genetic measures in model testing? Environ Model Softw 68:83–97
Lethbridge M, Harper M, Strauss J (2010). An analysis of the population viability of the yellow-footed rock-wallaby at selected sites in South Australia report to Flinders University & DEH Bounceback Program: Adelaide, Australia
Lethbridge M, Andrews L, Harper M (2012). An analysis of mark-resighting of the yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) in the Gawler Ranges, South Australia EcoKnowledge report to Department of Environment, Water and Natural Resources: Australia
Lethbridge M, Shute E, Wells C, Stead M (2019) Population trends of the yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) and feral goats (Capra hircus) from aerial survey data in the Flinders and Olary Ranges, South Australia. EcoKnowledge report to Department for Environment and Water: Australia
Lim L, Giles J (1987) Studies on the yellow-footed rock-wallaby, Petrogale xanthopus Gray (Marsupialia: Macropodidae). 3. Distribution and management in western New South Wales. Wildl Res 14(2):147–161
Longmire J, Maltbie M, Baker R (1997) Use of “lysis buffer” in DNA isolation and its implication for museum collections. Occasional Pap Museum 163:1–3
Luikart G, Cornuet J (1999) Estimating the effective number of breeders from heterozygote excess in progeny. Genetics 151(3):1211–1216
Miller S, Harper C, Bloomer P, Hofmeyr J, Funston P (2015) Fenced and fragmented: conservation value of managed metapopulations. PLoS ONE 10(12):1–16. https://doi.org/10.1371/journal.pone.0144605
Murray J, Low Choy S, McAlpine C, Possingham H, Goldizen A (2008) The importance of ecological scale for wildlife conservation in naturally fragmented environments: a case study of the brush-tailed rock-wallaby (Petrogale penicillata). Biol Cons 141(1):7–22. https://doi.org/10.1016/j.biocon.2007.07.020
Nei M (1972) Genetic distance between populations. Am Nat 106(949):283–292
Norbury G (1986) Twins in the western grey kangaroo, Macropus fuliginosus (Marsupialia: Macropodidae), in north-western Victoria. Aust Mammal 10(1–2):33
O’Leary S, Puritz J, Willis S, Hollenbeck C, Portnoy D (2018) These aren’t the loci you’e looking for: principles of effective SNP filtering for molecular ecologists. Mol Ecol 27(16):3193–3206. https://doi.org/10.1111/mec.14792
Olivieri I, Michalakis Y, Gouyon P (1995) Metapopulation genetics and the evolution of dispersal. Am Nat 146(2):202–228
Perrin N, Mazalov V (1999) Dispersal and inbreeding avoidance. Am Nat 154(3):282–292
Piry S, Alapetite A, Cornuet J, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95(6):536–539
Pope L, Sharp A, Moritz C (1996) Population structure of the yellow-footed rock-wallaby Petrogale xanthopus (Gray, 1854) inferred from mtDNA sequences and microsatellite loci. Mol Ecol 5(5):629–640
Pope L, Sharp A, Moritz C (1998) The genetic diversity and distinctiveness of the yellow-footed rock-wallaby Petrogale xanthopus (Gray, 1854) in New South Wales. Pac Conserv Biol 4(2):164–169
Potter S, Cooper S, Metcalfe C, Taggart D, Eldridge M (2012) Phylogenetic relationships of rock-wallabies, Petrogale (Marsupialia: Macropodidae) and their biogeographic history within Australia. Mol Phylogenet Evol 62(2):640–652
Potter S, Neaves L, Lethbridge M, Eldridge M (2020) Understanding historical demographic processes to inform contemporary conservation of an arid zone specialist: the yellow-footed rock-wallaby. Genes 11(154):1–25
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rannala B, Mountain J (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci 94(17):9197–9201
RAPAD, QFPI (2018) RAPAD cluster fencing project round 1: march 2016–april 2018 final report: section 3. RAPAD and QFPI report to Queensland Government: Australia
Robertson A (1965) The interpretation of genotypic ratios in domestic animal populations. Anim Sci 7(3):319–324
Ruykys L, Lancaster M (2015) Population structure and genetic diversity of the black-footed rock-wallaby (Petrogale lateralis MacDonnell Ranges race). Aust J Zool 63(2):91–100
Schroeder M (2019) Cheetah (Acinonyx jubatus) mortality and survival in fenced reserves as part of a managed metapopulation across South Africa. (Doctoral Thesis), University of Cape Town, South Africa
Sharp A (2002). The ecology and conservation biology of the yellow-footed rock-wallaby. (Doctoral Thesis), University of Queensland, Australia
Sharp A (2011) Drinking behaviour of yellow-footed rock-wallabies (Petrogale xanthopus celeris) in semiarid Queensland. Aust Mammal 33(2):189–194
Smith D, Allen B (2021) Habitat use by yellow-footed rock-wallabies in predator exclusion fences. J Arid Environ. https://doi.org/10.1016/j.jaridenv.2020.104329
Smith D, King R, Allen B (2020a) Impacts of exclusion fencing on target and non-target fauna: a global review. Biol Rev. https://doi.org/10.1111/brv.12631
Smith D, Lethbridge M, Allen B, Andrew R (2020b) Data from: inferring inter-colony movement within metapopulations of yellow-footed rock-wallabies using estimates of kinship. https://doi.org/10.5281/zenodo.7412787
Smith D, Waddell K, Allen B (2020c) Expansion of vertebrate pest exclusion fencing and its potential benefits for threatened fauna recovery in Australia. Animals. https://doi.org/10.3390/ani10091550
Spencer P, Marsh H (1997) Microsatellite DNA fingerprinting confirms dizygotic twinning and paternity in the allied rock-wallaby, Petrogale assimilis (Marsupialia: Macropodidae). Aust Mammal 19:279–280
Stevens L, Salomon B, Sun G (2007) Microsatellite variability and heterozygote excess in Elymus trachycaulus populations from British Columbia in Canada. Biochem Syst Ecol 35(11):725–736
Telfer W, Griffiths A, Bowman D (2008) The habitat requirements of four sympatric rock-dwelling macropods of the Australian monsoon tropics. Austral Ecol 33(8):1033–1044
Threatened Species Scientific Committee (2016a) Conservation advice for Petrogale xanthopus xanthopus, yellow-footed rock-wallaby (SA and NSW) created by Threatened Species Scientific Committee (Australia)
Threatened Species Scientific Committee (2016b) Conservation advice: Petrogale xanthopus celeris, yellow-footed rock-wallaby (central-western Queensland). Canberra, Australia: Threatened Species Scientific Committee. Retrieved from http://www.environment.gov.au/biodiversity/threatened/species/pubs/87608-conservation-advice-05052016b.pdf
van Oorschot R, Cooper D (1989) Twinning in the genus Macropus, especially M. Eugenii (Marsupialia: Macropodidae). Aust Mammal 12(1–2):83–84
Wahlund S (1928) Composition of populations and correlation appearances viewed in relation to the studies of inheritance. Hereditas 11:65–106
Waples R (2015) Testing for Hardy-Weinberg proportions: have we lost the plot? J Hered 106(1):1–19
Weir B, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
Wells J, Richmond M (1995) Populations, metapopulations, and species populations: what are they and who should care? Wildl Soc Bull 23(3):458–462
Wickham H (2006) An introduction to ggplot: an implementation of the grammar of graphics in R. Statistics
Wright S (1922) Coefficients of inbreeding and relationship. Am Nat 56(645):330–338
Zheng X (2013). Package ‘SNPRelate’ (Version 0.9.15). Retrieved from http://corearray.sourceforge.net/
Acknowledgements
DS was funded by a Research Stipend Scholarship for the completion of his PhD, for which this manuscript forms a part. This research project was funded by the Holsworth Endowment, for which we are incredibly grateful. We would like to thank the reviewers for their constructive and helpful comments. We would also like to thank the rock-wallabies for (mostly) cooperating, and their DNA contribution. Finally, the animals sampled for this project were captured on Aboriginal lands. The authors would like to acknowledge the traditional owners of Australia, Australia’s First Peoples, for their past and continuing care of the land and waters, and their contribution to modern Australia.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. Funding was provided by Holsworth Wildlife Research Endowment.
Author information
Authors and Affiliations
Contributions
DS and MRL collected tissues samples from Queensland and South Australia, respectively. The methods and study design was decided on by DS and RLA as part of a broader project conceptualised by DS and BLA. DS performed all analysis. The main body of the writing was completed by DS. All authors contributed to the editing and review of the manuscript pre-submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, D.A., Lethbridge, M.R., Allen, B.L. et al. Inferring inter-colony movement within metapopulations of yellow-footed rock-wallabies using estimates of kinship. Conserv Genet 24, 265–278 (2023). https://doi.org/10.1007/s10592-022-01498-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01498-8