
Abstract
The evolution of the multi-copy family of ribosomal RNA (rRNA) genes is unique in regard to its genetics and genome evolution. Paradoxically, rRNA genes are highly homogenized within and between individuals, yet they are globally distinct between species. Here, we discuss the implications for models of rRNA gene evolution in light of our recent discoveries that ribosomes bearing rRNA sequence variants can affect gene expression and physiology and that intra-individual rRNA alleles exhibit both context- and tissue-specific expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The ribosome is an RNA-protein assembly responsible for the translation of the genetic information encoded in messenger RNA (mRNA) into proteins. The ribosomal RNA (rRNA) component of the intact ribosome, which constitutes its universally conserved, functional core (Noller 2005), is principally encoded by the ribosomal DNA (rDNA) operon (Parks et al. 2018; Grummt 2003). Most eukaryotic genomes encode hundreds of copies of the rDNA operon arranged in tandem repeats on the acrocentric arms of multiple chromosomes (Eickbush and Eickbush 2007). These operons encode the functional rRNAs found in the fully assembled ribosome as well as non-coding RNAs of potentially diverse physiological functions (Bierhoff et al. 2014).
To meet cellular demands for rapid protein synthesis during cellular growth and proliferation, rDNA operons are highly transcribed by RNA Polymerase I. Pre-rRNA transcripts (47S in higher eukaryotic organisms) are processed into the functional rRNAs (5.8S, 18S, and 28S) present within the ribosome through a highly complex multi-step process referred to as ribosome biogenesis (Peña et al. 2017). In addition to its interactions with hundreds of ribosome biogenesis factors, the rRNA components of the assembled ribosome must precisely interact with more than 80 ribosomal proteins and a multitude of translation factors and other components within the cellular milieu (Simsek et al. 2017). Correspondingly, rRNA is thought to be highly intolerant to sequence variation within a given species, thus requiring the homogenization of allelic variation to maintain a diversity of structure-function relationships. Despite this constraint, both the sequence and structure of rRNAs have been remodeled, sometimes dramatically, over evolutionary time (Petrov et al. 2014; Bernier et al. 2018). The physiological impacts of rRNA sequence variation during the evolution of rRNA genes have thus been proven to be a special case with respect to conventional notions of evolution.
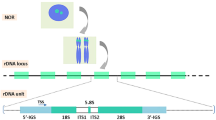
Two accepted scientific observations give rise to an apparent paradox in rRNA evolution: on the one hand, rRNA genes are highly similar within and between individuals of a given species (Eickbush and Eickbush 2007). On the other hand, they are sufficiently divergent to distinguish one species from another using rRNA genes alone, including closely related primates (Arnheim et al. 1980). How then can the hundreds of rRNA genes be maintained as highly homologous across individuals and generations, while significantly diverging to a new consensus sequence during the emergence of a new species? If rRNA genes are homogenized, then the appearance of a new consensus rRNA sequence with the emergence of a new species must have arisen from a de novo mutation in one gene, which then quickly spread to become ubiquitous to all other copies of rRNA across non-homologous chromosomes, leaving little room for selection in the interim (Fig. 1). To be inheritable, these events must occur in meiotic cell lineages.

How do the hundreds of copies of rRNA genes evolve during speciation? Schematic model of a speciation event. Cells (black circles) containing translating ribosomes bearing different rRNA alleles (gray, blue, or yellow) correspond to representative individuals (white or shaded primate outlines) depicted during speciation. In a model of concerted evolution, wherein rRNA variants are only deleterious or genetic drift, the rRNA genes are maintained homologous while the species evolves, but the consensus rRNA gene in the emergent new species differs from the ancestor due to some sudden meiotic recombination event, which spreads the variant to all copies of the rRNA gene in the genome. Here, we propose an update to the model of concerted evolution, wherein rRNA variants can contribute beneficially to cell physiology and certain variants can be selected for as the species evolves
Concerted evolution, a genetics model proposed in the wake of these observations, seeks to provide a framework for rRNA evolution that entails a combination of gene conversion and unequal chromosome crossover (Eickbush and Eickbush 2007), mechanisms of recombination, and DNA repair that induce genome rearrangements among repetitive DNA (Parks et al. 2015; Hastings et al. 2009). An assumption underlying concerted evolution is that the precise nucleotide sequence of rRNA must be maintained for proper functioning of the ribosome (Eickbush and Eickbush 2007; Brown et al. 1972). In its present form, concerted evolution regards rRNA sequence variants as inconsequential genetic drift or deleterious mutations (Eickbush and Eickbush 2007; Brown et al. 1972).
Mounting evidence hints that the evolution of rRNA sequence variation is more complicated than the rapid spread of mutations across rRNA genes and the simple avoidance of deleterious mutations. In 1980, Arnheim et al. reported an evolutionarily conserved intra-individual 28S rRNA sequence polymorphism in four primate species in which the same nucleotide polymorphism was present in the 28S rRNA gene of only a subset of each individual’s rRNA genes (Arnheim et al. 1980). In human samples, this sequence polymorphism was estimated to be present in 30% of the genes (Krystal et al. 1981). The authors noted that concerted evolution could not explain the existence of such a conserved intra-individual polymorphism and speculated that a selection pressure prevented removal of this variant through homogenization. Subsequent discoveries of intra-individual nucleotide polymorphisms in human rRNA genes led Gonzalez et al. to speculate that rRNA variants may regulate gene expression, such that small genotypic differences in rRNA genes may have substantial phenotypic consequences (Gonzalez et al. 1985). As rRNA gene clusters had been evidenced to exhibit tissue- and condition-specific expression (de Capoa et al. 1985a; de Capoa et al. 1985b), the ramifications of such findings appeared profound, albeit difficult to explore.
Consistent with the notion of concerted evolution, we recently performed a focused analysis on the functional rRNA genes using relatively short-read length human whole genome sequencing data from diverse populations worldwide to show that mammalian rRNA genes are, in general, highly homologous (Parks et al. 2018). However, we also discovered intra-individual sequence variation across the rRNA genes, some of which occurred in regions of functional relevance and were associated with population groups. rRNA variants were also found to be conserved between human and mouse and to exhibit tissue-specific expression. These discoveries raise the possibility that sequence variation in the rRNA component of the ribosome may be of beneficial functional significance. These observations led us to ask whether the expression of rRNA sequence variants can affect gene expression and phenotype.
Based on the rationale that beneficial impacts of sequence variation within the rRNA genes on ribosome function, if present, would likely be an evolutionarily conserved phenomenon, we examined whether rRNA sequence variation contributes to physiological programs in laboratory strains of Escherichia coli. We discovered that rRNA alleles are differentially expressed in response to nutrient limitation-induced stress and that the rRNA allele most upregulated on a relative basis is distinguished by conserved sequence variants clustered in the small subunit head domain of the assembled ribosome (Kurylo et al. 2018). These findings revealed for the first time that the rRNA allele composition of the actively translating ribosome pool is indeed regulated in response to physiological stimuli. Remarkably, we further showed that ribosomes bearing these sequence variations causally affected stress-response gene expression and phenotype, including biofilm formation, cell motility, and antibiotic sensitivity. Consistent with the sequence variants modulating the so-called ribo-interactome (Simsek et al. 2017), we further identified rRNA allele-dependent genetic interactions with stress-related proteins that transiently interact with the ribosome in the proximity of the sequence variants during protein synthesis. The varied expression of this operon alters the mechanisms of ribosome-interacting factor engagement during translation to affect gene expression and cell physiology during stress. Interestingly, the exact same sequence variations were found to be conserved in many Enterobacteriaceae, including Salmonella enterica, which diverged from E. coli more than 120 million years ago. These findings provide compelling evidence that classes of bacteria encode what may be considered an evolutionarily conserved “stress-response ribosome.” As the theory of concerted evolution has been extended to prokaryotes with multiple copies of rRNA (Liao 2000), these results point towards a new dimension of rRNA gene evolution. We note, however, that context-specific benefits for the expression of rRNA genes with sequence variation within pre-rRNA or the rRNA genes have yet to be definitively shown in eukaryotes.
In light of previous literature and the more recent discoveries described herein, we posit that concerted evolution should be expanded to account for the possibility that positive selective pressures favor the maintenance of a plurality of rRNA alleles in the genome (Fig. 1). This may include pressures that favorably modulate ribosome function in certain environmental conditions or cellular states. For instance, the rRNA sequence variants that we observed to be conserved in Enterobacteriaceae appear to modulate ribosome function in a manner that influences gene expression and cell physiology to promote survival. Analogously, extremophilic or extremotolerant microorganisms seem to possess unusually high levels of intragenomic rRNA sequence variation (Sun et al. 2013), possibly contributing to their ability to navigate harsh environmental conditions (Lauro et al. 2007; López-López et al. 2007; Johansen et al. 2017).
In addition to uncovering a novel mechanism of gene expression regulation, our recent findings raise important questions about concerted evolution and rRNA. For instance, how do sequence variants in rDNA operons arise in the first place and how are they maintained? Do sequence variations within the rRNA genes correlate with variations within the non-coding regions of the pre-rRNA transcript? How do the potentially distinct constraints on sequence variation within the genes encoding the functional rRNA genes and the non-coding regions of rDNA operon influence and impact the mechanism and occurrence of rDNA recombination events? How is an optimal plurality of rRNA alleles achieved within an individual and is this plurality maintained over the course of an organism’s lifetime? Do the positions of the rDNA operons within the acrocentric arms of chromosomes alter the efficiency of recombination events in a meaningful way? We must also seek to understand how ribosome biogenesis and translation factors coevolve to maintain functional interactions with rRNAs of distinct sequence. Do variations in rRNA sequence drive the evolution of ribosome biogenesis and translation factors or do variations in ribosome biogenesis and translation factors drive the evolution of rRNA sequence? How does rRNA sequence variation relate to the observation that mammalian organisms typically encode multiple distinct isoforms of the core translation factors (Genuth and Barna 2018)?
To begin to answer these questions and to provide insights into the role of rRNA expression in concerted evolution, future investigations must be aimed at examining whether diverse eukaryotic species exhibit rRNA allele-specific expression levels under distinct physiological conditions. In this context, efforts must also be given to establishing a robust means of determining the complete primary sequence of individual rDNA operons within a single cell or organism. As it is difficult to achieve this goal with short-read length whole genome sequencing data, such efforts will doubtless benefit from the implementation of single-chromosome or single-molecule, long-read length technologies (Pollard et al. 2018). Equipped with this knowledge, one could endeavor to genetically or pharmacologically regulate the levels at which specific variant rDNA operons are expressed. In doing so, one could assess the impact of ribosomes composed of certain rRNA alleles on gene expression and phenotype and track rRNA genotypes over time. Such knowledge will inform targeted knock-in and knock-out studies to solidify the contribution of rRNA sequence variation in gene expression and physiology. It will also provide a much-needed foundation for genome-wide association studies, which may identify rRNA alleles to be implicated in disease conditions. These important first steps may be critical to motivating the necessary tool developments, genomics approaches, and research collaborations needed to navigate and chart this important research frontier.
As we touch upon herein, rRNA may be subject to distinct evolutionary processes and pressures. Understanding how rRNA alleles are maintained in the genome and regulated in expression will be of extraordinary importance given the centrality of the ribosome to cellular physiology. In light of the evidence showing that rRNA sequence variation has the capacity to regulate gene expression and cell physiology (Kurylo et al. 2018), as well as evidence showing a connection between genomic instability of rDNA and disease (Stults et al. 2009), these areas of investigation appear both critical to pursue and distinctly uncharted.
Change history
28 June 2019
The article Implications of sequence variation on the evolution of rRNA, written by Matthew M. Parks, Chad M. Kurylo, Jake E. Batchelder, C. Theresa Vincent and Scott C. Blanchard, was originally published electronically on the publisher’s internet portal (currently SpringerLink).
Abbreviations
- mRNA:
-
Messenger RNA
- rDNA:
-
Ribosomal DNA
- rRNA:
-
Ribosomal RNA
References
Arnheim N, Krystal M, Schmickel R, Wilson G, Ryder O, Zimmer E (1980) Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc Natl Acad Sci U S A 77:7323–7327
Bernier CR, Petrov AS, Kovacs NA, Penev PI, Williams LD (2018) Translation: the universal structural core of life. Mol Biol Evol 35:2065–2076
Bierhoff H, Postepska-Igielska A, Grummt I (2014) Noisy silence: non-coding RNA and heterochromatin formation at repetitive elements. Epigenetics 9:53–61
Brown DD, Wensink PC, Jordan E (1972) A comparison of the ribosomal DNA’s of Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J Mol Biol 63:57–73
de Capoa A, Marlekaj P, Baldini A, Rocchi M, Archidiacono N (1985a) Cytologic demonstration of differential activity of rRNA gene clusters in different human tissues. Hum Genet 69:212–217
de Capoa A, Marlekaj P, Baldini A, Archidiacono N, Rocchi M (1985b) The transcriptional activity of individual ribosomal DNA gene clusters is modulated by serum concentration. J Cell Sci 74:21–35
Eickbush TH, Eickbush DG (2007) Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175:477–485
Genuth NR, Barna M (2018) Heterogeneity and specialized functions of translation machinery: from genes to organisms. Nat Rev Genet 19:431–452
Gonzalez IL, Gorski JL, Campen TJ, Dorney DJ, Erickson JM, Sylvester JE, Schmickel RD (1985) Variation among human 28S ribosomal RNA genes. Proc Natl Acad Sci U S A 82:7666–7670
Grummt I (2003) Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev 17(14):1691–702
Hastings PJ, Lupski JR, Rosenberg SM, Ira G (2009) Mechanisms of change in gene copy number. Nat Rev Genet 10:551–564
Johansen JR, Mareš J, Pietrasiak N, Bohunická M, Zima J, Štenclová L, Hauer T (2017) Highly divergent 16S rRNA sequences in ribosomal operons of Scytonema hyalinum (cyanobacteria). PLoS One 12:e0186393
Krystal M, D’Eustachio P, Ruddle FH, Arnheim N (1981) Human nucleolus organizers on nonhomologous chromosomes can share the same ribosomal gene variants. Proc Natl Acad Sci U S A 78:5744–5748
Kurylo CM et al (2018) Endogenous rRNA sequence variation can regulate stress response gene expression and phenotype. Cell Rep 25:236–248.e6
Lauro FM, Chastain RA, Blankenship LE, Yayanos AA, Bartlett DH (2007) The unique 16S rRNA genes of piezophiles reflect both phylogeny and adaptation. Appl Environ Microbiol 73:838–845
Liao D (2000) Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J Mol Evol 51:305–317
López-López A, Benlloch S, Bonfá M, Rodríguez-Valera F, Mira A (2007) Intragenomic 16S rDNA divergence in Haloarcula marismortui is an adaptation to different temperatures. J Mol Evol 65:687–696
Noller HF (2005) RNA structure: reading the ribosome. Science 309:1508–1514
Parks MM, Lawrence CE, Raphael BJ (2015) Detecting non-allelic homologous recombination from high-throughput sequencing data. Genome Biol 16:72
Parks MM et al (2018) Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv 4:eaao0665
Peña C, Hurt E, Panse VG (2017) Eukaryotic ribosome assembly, transport and quality control. Nat Struct Mol Biol 24:689–699
Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, Hud NV, Williams LD (2014) Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci U S A 111:10251–10256
Pollard MO, Gurdasani D, Mentzer AJ, Porter T, Sandhu MS (2018) Long reads: their purpose and place. Hum Mol Genet 27:R234–R241
Simsek D et al (2017) The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169:1051–1065.e18
Stults DM, Killen MW, Williamson EP, Hourigan JS, Vargas HD, Arnold SM, Moscow JA, Pierce AJ (2009) Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res 69:9096–9104
Sun D-L, Jiang X, Wu QL, Zhou N-Y (2013) Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl Environ Microbiol 79:5962–5969
Author information
Authors and Affiliations
Contributions
M.M.P. led the writing the manuscript. C.M.K., J.E.B., C.T.V., and S.C.B. refined the concepts and discussion points. All authors contributed to revising the manuscript.
Corresponding authors
Additional information
Responsible Editor: Jennifer Gerton and Lev Porokhovnik
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Parks, M.M., Kurylo, C.M., Batchelder, J.E. et al. Implications of sequence variation on the evolution of rRNA. Chromosome Res 27, 89–93 (2019). https://doi.org/10.1007/s10577-018-09602-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-018-09602-w