
Abstract
We investigate the dissolution mechanism of cellulose using molecular dynamics simulations in both water and a mixture solvent consisting of water with Na\(^+\), OH\(^-\) and urea. As a first computational study of its kind, we apply periodic external forces that mimic agitation of the suspension. Without the agitation, the bundles do not dissolve, neither in water nor solvent. In the solvent mixture the bundle swells with significant amounts of urea entering the bundle, as well as more water than in the bundles subjected to pure water. We also find that the mixture solution stabilizes cellulose sheets, while in water these immediately collapse into bundles. Under agitation the bundles dissolve more easily in the solvent mixture than in water, where sheets of cellulose remain that are bound together through hydrophobic interactions. Our findings highlight the importance of urea in the solvent, as well as the hydrophobic interactions, and are consistent with experimental results.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cellulose is becoming more and more prominent as a component used in development of new materials, cosmetics, as a food additive, biomedical applications and in fabrics. It is typically cited as being the worlds most abundant polymer (Dufresne 2012), and cellulose fibers are at forefront to replace conventional plastic for packaging and clothing applications in particular. Cellulose is a polymer composed of \(\beta\)(1\(\rightarrow\)4) linked units of d-glucopyranose residues (French 2017). This is shown with \(n-2\) monomers in Fig. 1. As every glucose residue is rotated about the molecular axis with regard to its predecessor by \(180^{\circ }\) in most cellulose crystals, the molecule is often depicted instead as a polymer of cellobiose residues. The present work shows that a two-fold screw axis is not preserved in solution, confirming that the individual glucose residue is the correct repeating unit. Cellulose is naturally produced as chains that immediately form sheets, then bundles (Dufresne 2012). As of yet, single chained cellulose has not been found in nature, and cellulose seemingly exists as bundles of 36, 24 or 18 chains, where 18 may be considered the most likely bundle size (Habibi et al. 2010; Cosgrove 2014; Jarvis 2018).
In order to better utilise cellulose for packaging and fabric applications and more, it is desirable to be able to dissolve the bundles into smaller units, especially single chains. Several efforts have been made on this topic in order to either form more elastic films for packaging applications or to be able to weave fabrics with specific properties, such as in the lyocell process (Zhang et al. 2018). A wide range of methods has been developed to dissolve cellulose into single chains, (Lindman et al. 2010; Medronho and Lindman 2014), using alkaline solvents (Yan and Gao 2008; Cai and Zhang 2005), ionic liquids (ILs) (Wang et al. 2020; Su et al. 2019) or deep eutectic solvents (DES) (Tenhunen et al. 2018). The aim of this work has been to investigate some of the unresolved questions behind the “Zhang-method” (Cai and Zhang 2005), especially with regard to the role of agitation and urea-cellulose interactions, as well as critically reviewing molecular simulations of cellulose dissolution by said method. We investigated the inclusion of external forces mimicking agitation of the solution, and how they affect the dissolution process.

The structure of cellulose. Carbon atoms are grey, oxygen atoms are red, and hydrogen atoms are white. The numbering of the atoms inside the glucose units is indicated. For clarity, we show \(n-2\) monomers of d-glucopyranose residues to indicate the \(\beta\)(1\(\rightarrow\)4) linkage
Cellulose bundle dissolution
There are different interactions at play in dissolution of cellulose, in conjunction with its complex structure. Cellulose is an amphiphilic molecule (Medronho and Lindman 2014), and its structure is depicted in Fig. 1. Hydrogen bonds connect chains in-plane, while van der Waals interactions act perpendicular to the plane of the molecules. The innate hydrophobicity of cellulose has been demonstrated by the difficulty of removing hydrophobic compounds from cellulose, even after extensive vacuum treatment (Staudinger et al. 1953). When cellulose dissolves, the cellulose chains form a single-chain suspension as opposed to a suspension of bundles.
While cellulose dissolution is a complex process, it appears to always be composed of two steps regardless of the chosen method: first disruption of hydrogen bonds and van der Waals interactions between molecules, and then stabilization of dissolved chains. Another complicating issue with disrupting cellulose bonds is that these are both hydrophilic and hydrophobic. This has been highlighted by several works, among others by Lindman et al. (2010), stating that hydrophobic cellulose interactions are “typically” overlooked as the stronger hydrophilic hydrogen bonds are assumed to be more important. However, experimental results indicate that both these bonds need to be broken and stabilized.
Initially, cellulose solvents were derivatizing, meaning that the cellulose chains were chemically modified to achieve dissolution. While derivatizing methods such as carboxymethylation do have some applications, they do not yield a suspension of native cellulose. There is a wide range of non-derivatizing cellulose solvents available, from water-based to non-aqueous systems as well as ionic liquid systems (Medronho and Lindman 2014).
Based on the established amphiphilic nature of cellulose, one would expect that a mixed polar/nonpolar solvent is an absolute requirement for dissolving cellulose. However, this is seemingly not the case. At short chain lengths (< 200 anhydrous glucose units), cellulose is readily dissolved in water and sodium hydroxide (Isogai and Atalla 1998). Moreover, cellulose may be dissolved in several molten salts (Fischer et al. 2003), but not in the corresponding aqueous salt solutions. In the latter case, water out-competes cellulose in forming hydrogen bonds with the ion, and interchain hydrogen bonds are not disrupted. In several ionic liquids, the ionic nature of the solvent is also considered the reason for cellulose dissolution. It is not yet fully understood if the cation or anion has the leading role in such solvents (Medronho and Lindman 2014).
Dissolution by alkaline solvents
Water-based alkaline solvent systems are typically employed as they require simple chemicals and are considered environmentally friendly. An overview of dissolution mechanisms for water-based alkaline solvent systems can be seen in Table 1, and the function of the solvent constituents is outlined in Table 2. In these solvents, the ions are believed to disrupt cellulose inter-chain hydrogen bonds, by direct binding and also by increasing the osmotic pressure as they penetrate the cellulose structure. It has even been suggested that partial deprotonation of cellulose hydroxyls (pKa = 13.3) may occur (Bialik et al. 2016), and while this would be a good reason to apply hydroxide salts in the dissolution process, we will in this work not include the effect of deprotonation. The main reason for including hydroxide is that it facilitates water intrusion into the bundle (Medronho and Lindman 2014).
The role of the cation is thus assumed to be disruption of hydrogen bonds. Larger ions such as K\(^+\) are not as efficient as smaller ions (Li\(^+\)), which is typically attributed to a larger hydrodynamic radius not allowing hydrated ions to penetrate the cellulose bundle. The degree of solubility seems to follow the Hofmeister series (Liu et al. 2016). One should note that the mechanism behind this series for protein solvation is still not completely understood.
One way to improve the dissolution is to use mixed solvents. Examples are solvents with thiourea/urea as the amphiphilic component added to a water-alkali-hydroxide mixture (Cai and Zhang 2005; Jin et al. 2007). In these cases, the amphiphilic urea/thioura are assumed to shield cellulose hydrophobic interactions, hindering re-agglomeration after dissolution. For instance, Xiong et al. (2014) found evidence of hydrophobic interactions between cellulose and urea. Accumulation of urea at hydrophobic sites of cellulose has also been suggested by others (Isobe et al. 2013). The alternative inclusion of polyethylene glycol (PEG) instead of thiourea/urea, shows that the amphiphilicity is important in the added solvent component. This has been discussed extensively for PEG in terms of protein denaturation (Parray et al. 2020). However, the role of amphiphilic solvent components is still debated, as they are not strictly required to dissolve cellulose (Medronho and Lindman 2014). For PEG, hydrogen bonding with cellulose and association with the cellulose chain (possibly due to hydrophobic interactions) have been identified as the functional roles (Yan and Gao 2008).
While most dissolution processes are favored by increasing temperatures, it is clear from experiments that low temperatures facilitate dissolution of cellulose in water-based alkaline systems (Cai and Zhang 2005), regardless of the exact solvent system composition. This is typically attributed to temperature-dependent changes in the cellulose structure. Cellulose bundles (and certain other polymeric systems) are known to become more hydrophobic with higher temperatures (Lindman et al. 2010). While the number of intrachain hydrogen bonds is dropping with increasing temperature, new interchain and intersheet hydrogen bonds are formed, giving rise to a three-dimensional hydrogen bonding network that stabilize the cellulose structure at high temperatures (425–550 K) (Agarwal et al. 2011). Considering that the hydroxide/urea/water system is based on ion and water intrusion into the cellulose bundle, increased hydrophilicity at lower temperatures favours dissolution.
Imperative to all dissolution methods is vigorous stirring. In this context, stirring supplies drag forces that act opposite of the cellulosic material direction of motion. These forces are believed to increase material swelling, and their nature is described amongst others by Tulus et al. (2018) and Guillard et al. (2013). The type of applied stirring is not believed to decrease polymer chain lengths to a large extent.
Molecular Dynamics (MD) simulations are applied to verify experimental results and assumption of cellulose dissolution mechanisms. MD simulations have inherent inaccuracies and a vast amount of tunable parameters (cellulose structure, the choice of force field, atom/molecule types, boundary conditions, etc.) which play a role in how these numerical experiments should be interpreted. MD simulations can, on the other hand, provide useful information that is not accessible in experiments about the location and dynamics of the molecules, ions, and atoms. Interactions between cellulose and urea has been studied in order to confirm its role as a cellulose stabilizer (Wernersson et al. 2015; Xiong et al. 2014; Bergenstråhle-Wohlert et al. 2012; Liu et al. 2020; Cai et al. 2012), but the literature seems to diverge on the subject. Wernersson et al. (2015) support that hydrophobic cellulose-urea interactions are predominant, supporting observations made by Xiong et al. (2014). Bergenstråhle-Wohlert et al. (2012) argue that urea is also amphiphilic, and competes with water for direct hydrogen bonding with cellulose. Liu et al. (2020) described that a reduction in urea concentration at oxygen atoms O4 and O5 (see Fig. 1) was necessary to achieve re-agglomeration. Studies also indicated that ions (i.e. Na\(^+\)) are most preferably located to the cellulose hydroxyl groups. However, Cai et al. (2012) suggested that urea bind cellulose hydroxyls through hydrogen bonding. They found a decrease in cellulose-urea bonding from 265 to 283 K (\(-8\) to \(10^{\circ }\)C), in agreement with low-temperature requirements for dissolution of cellulose. More pronounced cellulose-urea interactions at lower temperatures were also found by Wernersson et al. (2015).

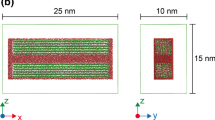
Bundle with 36 chains after energy minimization, seen from the side (a) and from the front (b). Smaller bundles are investigated in more detail, with shapes illustrated by the drawn lines: a bundle with 6 chains (blue), and a sheet with 6 chains (green)
Methodology
In this paper, we have modeled 6 and 36 chain systems with different geometries, with a basis in a maze configuration (Ding et al. 2014). The modelling aims at elucidating dissolution thermodynamics. As such, the most important feature of the models, is that the chains are aligned with in-plane hydrogen bonds and out-of-plane van der Waals interactions. Even though the exact bundle geometry is debated for different species (Jarvis 2018), there is consensus about this feature.
We simulate bundles of cellulose chains surrounded by either pure water, or a mixture solvent consisting of water, Na\(^+\), OH\(^-\), and urea. We analyze the dissolution by considering the density of the cellulose bundle in the different solvents, as well as the solvent accessible surface area. Besides investigating the effect of the solvent, we also include on agitation by external forces. Below we describe step by step how our system is set up, how the forces are added, and how we analyze the results.
Simulation setup
The cellulose molecules in our model were made with the GLYCAM carbohydrate builder (Woods and Co-workers 2019), producing chains with 8 glucose units with glycosidic angles \(\phi =22.6^{\circ }\) and \(\psi =-26.2^{\circ }\) (from Cellulose-builder: Gomes and Skaf 2012; Nishiyama et al. 2002). A bundle of 36 chains of 8 glucose units each was constructed in a maze configuration (Ding et al. 2014), with an intraplanar chain separation of 0.825 nm and an interplanar chain separation of 0.400 nm. We chose a 36 chain conformation as opposed to 18/24 chain conformation, as this provided a larger system better suited in unraveling dissolution mechanisms. On this note, it should be added that the 18-chain model is not without criticism(Jarvis 2018). The tool Intermol (v. 0.1.0) (Shirts et al. 2017) was used to convert the input files to a format compatible with LAMMPS (v. March 2018) (Plimpton 1995), which was used for all molecular dynamics simulations. Input configurations with solvents were generated with Packmol (v. 20.010) (Martínez et al. 2009), with a weight percentage of about 7% NaOH and 12% urea in the mixture solvent to match experiments (Cai and Zhang 2005).
Cellulose was modeled with the GLYCAM06 force field (Kirschner et al. 2008), water with the SPC/E model (Berendsen et al. 1987), the LJ parameters for the alkali metals were set for SPC/E (Li et al. 2015), and the standard GLYCAM06 hydroxyl LJ parameters were used for the hydroxide. Arithmetic combination rules were used for all remaining LJ parameters. The alkali metals have charge \(+1e\), in the hydroxide the oxygen had charge \(-1.02e\) and hydrogen had a charge of \(+0.02e\). The urea molecules were modeled with the improved generalized Amber force field (Özpinar et al. 2010). Non-bonded interactions between the cellulose were modeled by a switching function with an inner cutoff of 7 Å and an outer cutoff of 9 Å. For water and the other solvents, a simple cutoff of 9 Å was used. Long-range non-bonded interactions were obtained by the particle-particle particle-mesh (PPPM) solver with a relative error in forces of \(1.0\times 10^{-4}\).
The simulations started with energy minimization of the cellulose bundle without solvent by a steepest descent algorithm with about 5000 iterations, followed by 1 ns of molecular dynamics at temperature \(T=1\) K with a Langevin thermostat (Schneider and Stoll 1978). Solvent molecules were then added to the systems with Packmol (Martínez et al. 2009). The simulations were then run at \(T=1\) K with constant volume for 10 ps, before switching to a Nosé-Hoover style isothermal-isobaric simulations and linearly increasing the temperature to \(T=258\) K. We use a time step of 0.1 fs during the initialisation, and 1 fs during the rest of the simulations. These NPT simulations were initially run with a relaxation time of 10 ps for the barostat and a relaxation time of 50 fs for the thermostat while increasing the temperature to the target temperature over 0.1 ns. In the continued simulations the relaxation time for the thermostat was set to 0.1 ps and the relaxation time for the barostat was set to 1 ps. Table 3 presents an overview of the systems under investigation with the number of solvent molecules and the size of the simulation box.

Illustration of the length-controlled (left) and force-controlled (right) simulation mode. With length-controlled oscillations, the positions of the outermost carbon atoms in all the chains are controlled along the longest axis, and the chains are free to move in the axial plane. In the force-controlled case, a force is added to the outermost carbon atoms in each chain, and the chains are otherwise free to move
Agitation
We have added periodic agitation of the sample to our MD simulations. In the experiments, agitation of the system induces shear stresses on the bundle, most likely through stresses from the moving solvent. A direct implementation of this share rate in the solvent is complicated, and it is not considered to be necessary. Due to their geometry, the largest forces on the bundles will be in the direction of the long axis of the bundle.
We therefore implement agitation of our simulated bundles by periodically stretching and compressing them. This is implemented in two different ways, with length-controlled and force-controlled harmonic oscillations. These are illustrated in Fig. 3. In simulations with length-controlled oscillations, we control the position of the outermost carbon atom in each chain along the long axis, and the chains are free to move in the axial plane. In the simulations with force-controlled oscillations, a force is added to the outermost carbon atom in each chain along the longest axis, and the chains are otherwise free to move. The frequency of the oscillatory stretching and compression is 2 ns\(^{-1}\) for both modes of control. The length-controlled oscillations have an amplitude of 0.8 nm, and the force-controlled oscillations add a force with amplitude 0.35 nN per chain. The rationale behind these parameters was to apply the gentlest periodic stretching and compression that would cause the final dispersion of cellulose bundles within the time frame of the simulations, and in this way emphasize the difference in behaviour between the systems with water and those with the solvent mixture.
All chains remained intact throughout the simulations.
Modeling data analysis
The volume of the fibril was estimated by constructing convex hulls where all the atoms in the fibril were treated as point particles, using scipy.spatial.ConvexHull as implemented in SciPy (Virtanen et al. 2020). From this we directly estimated the density of the fibril, as the mass remained constant. These convex hulls were also used to determine the number of absorbed molecules: for each solvent molecule an additional convex hull was created with the center of mass of the solvent molecule as an additional node. The solvent molecule was then considered to be absorbed if the addition of the solvent molecule did not change the convex hull. Note that the absorbed molecules were not included in the mass of the bundle for the densities presented later. The solvent accessible surface area (SAS) was estimated with MDTraj (McGibbon et al. 2015), which uses the algorithm from Shrake and Rupley (1973). The probe size was set to 1.4 Å, which is approximately the radius of a water molecule.
The number of clusters was found by calculating the average distances between identical atoms in neighboring chains, if this distance is less than 5 Å they are considered to be in the same cluster.
Results
We first explore the system without agitation, shown in Fig. 2 after energy minimization. This system will serve as our reference. To gain further insight into the role of the solvents, we have also considered smaller bundles in more detail since this allows us to reach longer time scales. The configurations of these smaller bundles are illustrated in Fig. 2 with the (blue and green) outlines. We then show results with agitation.
Bundles with 36 chains
36 chains of 8 glucose units each were placed in a maze configuration, which is shown in Fig. 2 after energy minimization. The bundle was then solvated in water or in the mixture and allowed to equilibrate for 10 ns as described above. In the time span of our simulations (40–45 ns), the bundles do not dissolve in water nor in the mixture solvent. The density of the cellulose in water remains stable around 1.5 g/cm\(^3\), while the density in the mixture solvent stabilizes at approximately 1.4 g/cm\(^3\) (see Fig. 4 and Table 4).
To better understand what role the different molecules in the solvent play, we investigate how much of the different species is absorbed by the bundle. This is shown as a function of time in Fig. 5 for the mixture solvent. We see that urea is absorbed the most, with a concentration in the bundle that exceeds that of the bulk. Some water molecules are absorbed as well, and the concentration in the bundle is approximately half that of the bulk. The fraction of absorbed water in the bundle in the water-only solvent was significantly lower than that in the mixture solvent. The Na\(^+\) and OH\(^-\) ions are not absorbed into the bundle. The fact that smaller ions penetrate less deeply into the bundle could be considered counter-intuitive. Most likely this is related to the geometry of the molecules and placement of charges.
Bundles of cellulose are known to be very stable without agitation, a picture we have largely confirmed in our simulations described above. Nevertheless, we can already see that cellulose behaves differently in the mixture solvent than in pure water. The lower density of the cellulose in the mixture solvent is almost certainly due to the high concentration of urea in the bundle shown in Fig. 5, which increases the separation between the cellulose molecules. In these bundles without any agitation, the lower density of cellulose and high penetration of urea into the bundle do not yet lead to dissolution. Nevertheless, this may provide an indication as to why cellulose is more easily dissolved in the mixture solvent, and gives insight on the role of urea in dissolution.

Density of cellulose as a function of time for a bundle of 36 chains at \(T=258\) K, in pure water and the mixture solvent

Concentration ratio of solvents absorbed in a bundle with 36 chains of 8 glucose units per chain at \(T=258\) K, compared to the bulk
Smaller systems: 6 chains
In order to get a better understanding of the system on a longer time-scale, we study bundles and sheets with fewer chains, as these are far less computationally expensive and allow for more detailed investigations. In contrast to the bundles, sheets of cellulose are inherently unstable, and are not found in nature. The rationale for including sheets is that they may represent an intermediate stage of dissolution. Moreover, sheet dissolution has been suggested to occur spontaneously on timescales accessible by MD simulations (Liu et al. 2020).
We study bundles and sheets composed of 6 chains of cellulose, with 8 glucose units per chain. The initial configurations of these systems are illustrated in Fig. 2 with different colour outlines. We start simulations from the initial configurations and equilibrate in water or the mixture solvent. The density and SAS after equilibration are shown in Table 4.
The density of these small bundles in both mixture and water is similar to that of 36-chain bundles. Sheets, however, have lower density. The sheets are not perfectly flat, and probably less stiff than bundles. Consequently, when they bend, there is more extra volume in the convex hull than there is for bundles.
To investigate the surfaces of the bundles and sheets in more detail, we calculate the SAS compared to that of a set of fully dissolved chains. This is included in table Table 4 and also shown in Fig. 6 for water and Fig. 7 for the mixture solvent. The SAS of the chain with the highest and lowest areas for each system is shown with dashed lines. The systems with sheets as initial configuration have the highest SAS, both for the averages and the lowest SAS. This is to be expected, since the surface of a sheet is larger than that of a bundle. Note that the system with a sheet in water has at least one chain with a very low SAS, on par with that of bundles. This indicates that the sheet is forming a bundle. We can see this in Fig. 6, where the lowest SAS clearly decreases with time from around the typical average value in a sheet to the typical lowest level for a bundle. This does not happen in the mixture solvent, shown in Fig. 7. Snapshots of the initial sheet and resulting bundle in water are also included. We have found that in simulations without any solvent, i.e. in NVT in vacuum, these sheets also rapidly reconfigure to bundle-like configurations.
For the SAS of the sheet in mixture solvent, the average value is slightly higher than in water, but in contrast to the same system in water, none of the chains have a very low SAS in this case. In Fig. 6, it can also be seen that the smallest SAS does not drop down to the level of a bundle in the mixture solvent. This indicates that the sheet remains stable as a sheet in the mixture solvent, rather than collapsing into bundle. This increased SAS and more stable open geometry could possibly aid dissolution in the mixture solvent.
It has previously been reported that similar sheets of 6 chains actually dissolve in MD simulations in a similar mixture solvent at 261 K on a timescale of \(\sim 20\) ns (Liu et al. 2020). We have therefore performed simulations with the same configuration of the sheets with the same number of solvent molecules as those authors. The simulations were repeated twice in different ensembles, two times NPT and one NVT, as Liu et al. (2020). However in our simulations the sheets always remained intact. The main difference between our simulations and those of Liu et al., that we suspect may account for this different behaviour, is the choice of force field. Liu et al. used the CHARMM36 force field, while we use GLYCAM06. In particular the partial charges of the hydroxide are different. While Liu et al. used − 1.32e for the oxygen and 0.32e for the hydrogen, in this work these atoms were attributed with charges of − 1.02e and +0.02e respectively. It is quite likely that the dissolution mechanism is very sensitive to the force field parameters and especially the partial charges on the different atoms in the cellulose, since crystalline structure is also sensitive to it (Chen et al. 2014). We illustrate this by computing the radial distribution function (RDF) between the various solvent molecules and two specific oxygen atoms in the cellulose, O4 and O5. This is shown in Fig. 8. While there are peaks, some of them in similar locations, our RDF look qualitatively different from those by Liu et al. (2020), shown there in figure 6.

SAS per chain of cellulose, 6 chains in sheet and bundle at \(T=258\) K, water. The color indicates the system, as specified in the legend. The solid lines are averages, and the dashed lines show the SAS of the chain with the lowest value for each system. The configuration after 30 ns is shown above, confirming that the sheet is turning in to a bundle

SAS per chain of cellulose, 6 chains in sheet and bundle at \(T=258\) K, mix. The color indicates the system, as specified in the legend. The solid lines are averages, and the dashed lines show the SAS of the chain with the lowest value for each system

Agitation
The mixture solvent at 258 K is known experimentally to dissolve the cellulose bundles, but in the lab it is necessary to agitate the samples for dissolution to occur (Cai and Zhang 2005). We therefore implement agitation of our simulated bundles by periodically stretching them. We have tested two types of agitation, length- and force-controlled oscillations, in both water and mixture.
The cross sections of bundles with 36 chains after 30 ns of length-controlled oscillations and 10 ns of force-controlled oscillations in the mixture solvent and water can be seen in Fig. 9. These times correspond approximately to the earliest times where the cellulose density in both the mixture and water solvents were \(\le 0.5\) g/cm\(^{3}\). In all cases, we see signs of dissolution. However, there are clear differences. Dissolution is slower and more incomplete in water.
We see a significant dependence on whether the simulations were force-controlled or length-controlled. The snapshots of the force-controlled simulations show the most disorder. In the water solvent, there still remains a crystalline-like core, while in the mixture solvent, the bundle has fully dissolved. In water, the chains are dissolving from the outside-in, one by one, and the core has not yet fully dissolved. This indicates that the dissolution in water is slower than in the mixture solvent.
The length-controlled oscillation produces sheet-like structures in both water and the mixture solvent, but in water this is much more pronounced and the sheets are larger and more ordered. The geometry of the sheets that are formed in the length-controlled simulations is shown in Fig. 10. They are held together by hydrophobic interactions, rather than hydrophilic interactions. The fact that these structures are more present in water suggests that some of the components in the mixture play a role in breaking these hydrophobic interactions. Indeed, it has been suggested (Medronho and Lindman 2014; Lindman et al. 2010) that poor breaking down of hydrophobic interactions plays a role in why water is not able to dissolve cellulose.

Cross sections of bundles with 36 chains after 30 ns of length-controlled oscillations and 10 ns of force-controlled oscillations in mixture and water. The force-controlled oscillations dissolves the bundle fast in both water and the solvent mixture. With length-controlled oscillations, the dissolution is incomplete also after 30 ns and qualitatively different in water and in the solvent mixture

Comparison of a representative bundle of vertically stacked chains formed in the length-controlled simulations in water (left), and a sheet of 4 chains extracted from the energy minimized maze configuration (right). The chains in sheet configuration interact via in-plane hydrogen bonds, while these interactions are not present between the vertically stacked chains
The difference between the length-controlled and force-controlled simulations cannot be explained by more energetic agitation. Conversely, the amplitude for the length-controlled simulations is such that the forces on the end points vary by approximately 5 nN/chain. This is an order of magnitude more than in the force-controlled simulations, where the amplitude was set to 0.35 nN/chain.
The faster dissolution in the force-controlled simulations therefore must be related to the geometry of the two different boundary conditions. In the length-controlled simulations the ends are more restricted in space than in the force-controlled ones. This means that the length-controlled boundary conditions are more representative of the edge dynamics of a section in the middle of a larger bundle, while the force-controlled boundary conditions are more representative of an actual end of a bundle. This indicates that there may be some dissolution mechanism that involves chains loosening at the ends of the bundles. This is consistent with the fact that in experiments, shorter bundles are known to dissolve more easily.
With length-controlled oscillations, the bundle is twisting during compression, as the chains are free to move in the axial planes. This induces shear stress on the bundle, and the mechanics are reminiscent of shear stresses induced by an agitated solvent.

Time evolution of density (a) and number of clusters (b) for bundles of 36 chains of cellulose at \(T=258\) K with oscillating length and force. The difference in behaviour between the bundle in water and the bundle in the solvent mixture is most clear in the figure for the number of clusters, (b), where the horizontal line represents complete dissolution
In Fig. 11(a) we show the density as a function of the time since the start of the oscillations. In all four cases, the density immediately starts to decrease as the bundle dissolves. However, the decrease is more rapid for the systems subjected to force-controlled oscillations than those subjected to length-controlled oscillations.
We note that the density here is an indication of the maximum spreading out of the cellulose molecules, which happens by diffusion once they have detached from the bundle. While the density in water decreases at a similar rate to the density in the solvent mixture, this does not necessarily mean that the bundles dissolve at a similar rate, merely that the size of the particles diffusing is the same (i.e. single chains). In the case of length-controlled simulations, the decrease in density is slower, due to the fact that the units diffusing away from the bundle are larger and heavier sheet-like structures.
The difference in the dissolution process in water and in the solvent mixture is shown more clear in Fig. 11(b), displaying the time evolution of the number of clusters in each system. While the two systems with oscillating force both reach full dissolution, the bundle in the mixture solvent is dissolving significantly faster. The difference is largest around 10 ns, which is the time frame of the trajectories displayed in Fig. 9.
To investigate whether the dissolved states were thermodynamically stable, the simulations that reached dissolution with the presence of periodic external forces to mimic stirring or agitation was continued with the external force switched off. The density of the dissolved bundles did not change significantly even after 30 ns, indicating that the solutions achieved by agitation indeed are thermodynamically stable.
Conclusions
We have performed a series of molecular-dynamics simulations investigating the dissolution of 36-chain cellulose bundles in both water and a mixture solvent consisting of water with Na\(^+\), OH\(^-\) and urea. We find that without additional external forces to mimic stirring, cellulose bundles of 36 chains do not dissolve in our simulations. This is consistent with experimental results, not yielding dissolution in practical time frames without stirring. We do however find that in the mixture solvent, the urea intercalates into the bundles, increasing their volume significantly. This is in line with e.g. Liu et al. (2020), which found that urea had to be removed from cellulose chains in order for them to agglomerate. Moreover, a diverse range of different cellulose-urea interactions have been found in literature. For smaller bundles, we find similar results regarding the stability in the two different solvents. We also find that sheets of cellulose are stable in the mixture solvent, but in pure water they immediately collapse into bundles. This appears to be caused by shielding of cellulose hydrophobic interactions by urea in the mixture solvent.
When we add periodic external forces to mimic stirring or agitation of the solvent-cellulose mixture, we see that the bundles are being dissolved. We find that the cellulose dissolves more readily in the mixture solvent than in pure water, which is consistent with experimental results. In the water solvent, the dissolution is incomplete. Under boundary conditions that are likely more representative of longer cellulose bundles, they tend to leave sheets of cellulose that are bound together with hydrophobic interactions. This is further evidence of the urea stabilizing effect. It is difficult to compare our applied forces quantitatively to specific levels of stirring, and therefore to compare directly to dissolution in experiments. Our agitation may be significantly more energetic than what is achievable by stirring in experiments. Nevertheless, qualitatively, our findings are consistent with the experimental results that the NaOH and urea solvent is able to completely dissolve cellulose under experimental conditions when water only partially dissolves cellulose. Partial dissolution of cellulose in water, should be interpreted together with the fact that NaOH and water is indeed experimentally capable of dissolving cellulose with less than 200 glucose units (Kamide et al. 1984). It is therefore not surprising that partial dissolution occurs in a small (eight glucose units) modeled system with only water. The different behaviour of water and solvent is likely related to cellulose-cellulose hydrophobic interactions, which appear to be more stable in pure water than in the mixture solvent, where urea is absorbed into the bundles.
Data availability
Input scripts and data files are deposited in the following repository: https://doi.org/10.5281/zenodo.5592299
References
Agarwal V, Huber GW, Conner WC, Auerbach SM (2011) Simulating infrared spectra and hydrogen bonding in cellulose I\(\beta\) at elevated temperatures. J Chem Phys 135(13):13. https://doi.org/10.1063/1.3646306
Berendsen HJ, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Phys Chem 91(24):6269–6271. https://doi.org/10.1021/j100308a038
Bergenstråhle-Wohlert M, Berglund LA, Brady JW, Larsson PT, Westlund PO, Wohlert J (2012) Concentration enrichment of urea at cellulose surfaces: Results from molecular dynamics simulations and NMR spectroscopy. Cellulose 19(1):1–12. https://doi.org/10.1007/s10570-011-9616-x
Bialik E, Stenqvist B, Fang Y, Östlund Å, Furó I, Lindman B, Lund M, Bernin D (2016) Ionization of Cellobiose in Aqueous Alkali and the Mechanism of Cellulose Dissolution. J Phys Chem Lett 7(24):5044–5048. https://doi.org/10.1021/acs.jpclett.6b02346
Cai J, Zhang L (2005) Rapid dissolution of cellulose in LiOH/urea and NaOH/urea aqueous solutions. Macromol Biosci 5(6):539–548. https://doi.org/10.1002/mabi.200400222
Cai L, Liu Y, Liang H (2012) Impact of hydrogen bonding on inclusion layer of urea to cellulose: study of molecular dynamics simulation. Polymer 53(5):1124–1130. https://doi.org/10.1016/j.polymer.2012.01.008
Chen P, Nishiyama Y, Mazeau K (2014) Atomic partial charges and one Lennard-Jones parameter crucial to model cellulose allomorphs. Cellulose 21(4):2207–2217. https://doi.org/10.1007/s10570-014-0279-2
Cosgrove DJ (2014) Re-constructing our models of cellulose and primary cell wall assembly. Curr Opin Plant Biol 22:122–131. https://doi.org/10.1016/j.pbi.2014.11.001
Ding SY, Zhao S, Zeng Y (2014) Size, shape, and arrangement of native cellulose fibrils in maize cell walls. Cellulose 21(2):863–871. https://doi.org/10.1007/s10570-013-0147-5
Dufresne A (2012) Nanocellulose: from nature to high performance tailored materials. De Gruyter. https://doi.org/10.1515/9783110254600
Fischer S, Leipner H, Thümmler K, Brendler E, Peters J (2003) Inorganic molten salts as solvents for cellulose. Cellulose 10(3):227–236. https://doi.org/10.1023/A:1025128028462
French AD (2017) Glucose, not cellobiose, is the repeating unit of cellulose and why that is important. Cellulose 24(11):4605–4609. https://doi.org/10.1007/s10570-017-1450-3
Gomes TC, Skaf MS (2012) Cellulose-builder: A toolkit for building crystalline structures of cellulose. J Comput Chem 33(14):1338–1346. https://doi.org/10.1002/jcc.22959
Guillard F, Forterre Y, Pouliquen O (2013) Depth-Independent Drag Force Induced by Stirring in Granular Media. Phys Rev Lett 110(13):138303. https://doi.org/10.1103/PhysRevLett.110.138303
Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 110:3479–3500. https://doi.org/10.1021/cr900339w
Isobe N, Noguchi K, Nishiyama Y, Kimura S, Wada M, Kuga S (2013) Role of urea in alkaline dissolution of cellulose. Cellulose 20(1):97–103. https://doi.org/10.1007/s10570-012-9800-7
Isogai A, Atalla RH (1998) Dissolution of cellulose in aqueous NaOH solutions. Cellulose 5(4):309–319. https://doi.org/10.1023/A:1009272632367
Jarvis MC (2018) Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos Trans R Soc A Math Phys Eng Sci. https://doi.org/10.1098/rsta.2017.0045
Jin H, Zha C, Gu L (2007) Direct dissolution of cellulose in NaOH/thiourea/urea aqueous solution. Carbohydr Res 342(6):851–858. https://doi.org/10.1016/j.carres.2006.12.023
Kamide K, Okajima K, Matsui T, Kowsaka K (1984) Study on the solubility of cellulose in aqueous alkali solution by deuteration IR and 13 C NMR. Polym J 16(12):857–866. https://doi.org/10.1295/polymj.16.857
Kirschner KN, Yongye AB, Tschampel SM, Daniels CR, Foley BL, Woods R (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydr J Comput Chem 29:622–655. https://doi.org/10.1002/jcc.20820
Li P, Song LF, Merz KM (2015) Systematic parameterization of monovalent ions employing the nonbonded model. J Chem Theory Comput 11(4):1645–1657. https://doi.org/10.1021/ct500918t
Lindman B, Karlström G, Stigsson L (2010) On the mechanism of dissolution of cellulose. J Mol Liq 156(1):76–81. https://doi.org/10.1016/j.molliq.2010.04.016
Liu G, Li W, Chen L, Zhang X, Niu D, Chen Y, Yuan S, Bei Y, Zhu Q (2020) Molecular dynamics studies on the aggregating behaviors of cellulose molecules in NaOH/urea aqueous solution. Colloids Surfaces A Physicochem Eng Asp 594:124663. https://doi.org/10.1016/j.colsurfa.2020.124663
Liu Z, Zhang C, Liu R, Zhang W, Kang H, Li P, Huang Y (2016) Dissolution of cellobiose in the aqueous solutions of chloride salts: Hofmeister series consideration. Cellulose 23(1):295–305. https://doi.org/10.1007/s10570-015-0827-4
Martínez L, Andrade R, Birgin EG, Martíne JM (2009) Packmol: a package for building initial configurations for molecular dynamics simulations. J Comput Chem 30(13):2157–2164. https://doi.org/10.1002/jcc
McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, Pande VS (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J 109(8):1528–1532. https://doi.org/10.1016/j.bpj.2015.08.015
Medronho B, Lindman B (2014) Competing forces during cellulose dissolution: From solvents to mechanisms. Curr Opin Colloid Interface Sci 19(1):32–40. https://doi.org/10.1016/j.cocis.2013.12.001
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose I\(\beta\) from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124(31):9074–9082. https://doi.org/10.1021/ja0257319
Özpinar GA, Peukert W, Clark T (2010) An improved generalized AMBER force field (GAFF) for urea. J Mol Model 16(9):1427–1440. https://doi.org/10.1007/s00894-010-0650-7
Parray ZA, Hassan MI, Ahmad F, Islam A (2020) Amphiphilic nature of polyethylene glycols and their role in medical research. Polym Test 82:106316. https://doi.org/10.1016/j.polymertesting.2019.106316
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117(1):1–19. https://doi.org/10.1006/jcph.1995.1039
Schneider T, Stoll E (1978) Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys Rev B 17(3):1302–1322. https://doi.org/10.1103/PhysRevB.17.1302
Shirts MR, Klein C, Swails JM, Yin J, Gilson MK, Mobley DL, Case DA, Zhong ED (2017) Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J Comput Mol Des 31(1):147–161. https://doi.org/10.1007/s10822-016-9977-1
Shrake A, Rupley JA (1973) Environment and exposure to solvent of protein atoms Lysozyme and insulin. J Mol Biol 79(2):351–371. https://doi.org/10.1016/0022-2836(73)90011-9
Staudinger VH, In den Birken KH, Staudinger M (1953) Über den micellaren oder makromolekularen bau der cellulosen. Die Makromol Chemie 9(1):148–187. https://doi.org/10.1002/macp.1953.020090109
Su H, Bi Z, Ni Y, Yan L (2019) One-pot degradation of cellulose into carbon dots and organic acids in its homogeneous aqueous solution. Green Energy Environ 4(4):391–399. https://doi.org/10.1016/j.gee.2019.01.009
Tenhunen TM, Lewandowska AE, Orelma H, Johansson LS, Virtanen T, Harlin A, Österberg M, Eichhorn SJ, Tammelin T (2018) Understanding the interactions of cellulose fibres and deep eutectic solvent of choline chloride and urea. Cellulose 25(1):137–150. https://doi.org/10.1007/s10570-017-1587-0
Tulus, Mardiningsih, Sawaluddin, Sitompul OS, Ihsan AK (2018) Shear rate analysis of water dynamic in the continuous stirred tank. In: IOP conference series: materials science and engineering vol. 308(1). https://doi.org/10.1088/1757-899X/308/1/012048
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, van der Walt SJ, Brett M, Wilson J, Jarrod Millman K, Mayorov N, Nelson AR, Jones E, Kern R, Larson E, Carey CJ, Polat L, Feng Y, Moore EW, Vand erPlas J, Laxalde D, Perktold J, Cimrman R, Henriksen I, Quintero E, Harris CR, Archibald AM, Ribeiro AH, Pedregosa F, van Mulbregt P, Contributors S (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17(3):261–272. https://doi.org/10.1038/s41592-019-0686-2
Wang J, Wang Y, Ma Z, Yan L (2020) Dissolution of highly molecular weight cellulose isolated from wheat straw in deep eutectic solvent of Choline/L-Lysine hydrochloride. Green Energy Environ 5(2):232–239. https://doi.org/10.1016/j.gee.2020.03.010
Wernersson E, Stenqvist B, Lund M (2015) The mechanism of cellulose solubilization by urea studied by molecular simulation. Cellulose 22(2):991–1001. https://doi.org/10.1007/s10570-015-0548-8
Woods R, Co-workers (2019) GLYCAM Web. Complex carbohydrate research center, University of Georgia, Athens, GA. http://glycam.org
Xiong B, Zhao P, Hu K, Zhang L, Cheng G (2014) Dissolution of cellulose in aqueous NaOH/urea solution: role of urea. Cellulose 21(3):1183–1192. https://doi.org/10.1007/s10570-014-0221-7
Yamashiki T, Kamide K, Okajima K, Kowsaka K, Matsui T, Fukase H (1988) Some characteristic features of dilute aqueous alkali solutions of specific alkali concentration (2.5 mol p1) which possess maximum solubility power against cellulose. Polym J 20(6):447–457. https://doi.org/10.1295/polymj.20.447
Yan L, Gao Z (2008) Dissolving of cellulose in PEG/NaOH aqueous solution. Cellulose 15(6):789–796. https://doi.org/10.1007/s10570-008-9233-5
Zhang S, Chen C, Duan C, Hu H, Li H, Li J, Liu Y, Ma X, Stavik J, Ni Y (2018) Regenerated cellulose by the lyocell process, a brief review of the process and properties. BioResources 13(2):1–16. https://doi.org/10.15376/biores.13.2.Zhang
Acknowledgments
The authors are grateful to the Research Council of Norway for funding through project no. 250158 and project no. 262644 (PoreLab Centre of Excellence). The simulations were performed on resources provided by UNINETT Sigma2 - the National Infrastructure for High Performance Computing and Data Storage in Norway and the Idun cluster provided by the NTNU High Performance Computing Group.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital). The authors received funding by the Research Council of Norway through project no. 250158 and project no. 262644.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
The authors have followed the guidelines of the Committee on Publication Ethics.
Consent to participate
Not applicable.
Consent for publication
All authors have read and agreed to publish the latest version of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bering, E., Torstensen, J.Ø., Lervik, A. et al. Computational study of the dissolution of cellulose into single chains: the role of the solvent and agitation. Cellulose 29, 1365–1380 (2022). https://doi.org/10.1007/s10570-021-04382-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-04382-9