
Abstract
In the cellulose scientific community, hydrogen bonding is often used as the explanation for a large variety of phenomena and properties related to cellulose and cellulose based materials. Yet, hydrogen bonding is just one of several molecular interactions and furthermore is both relatively weak and sensitive to the environment. In this review we present a comprehensive examination of the scientific literature in the area, with focus on theory and molecular simulation, and conclude that the relative importance of hydrogen bonding has been, and still is, frequently exaggerated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and background
Consider a wet nanocellulose film that is rolled up and left to air dry, resulting in a nanopaper roll that is sufficiently stiff and wet-stable to be used as a drinking straw. No adhesives are used to seal the roll, but it nevertheless holds together. This mechanism is attributed to the formation of hydrogen bonds (H-bonds) between the nanopaper surfaces (Wang et al. 2020). But can this really be the full story?
The year 2020 marked the 100-year anniversary of the H-bond concept (Gibb 2020; Pauling 1939), which has, since then, been central for explaining structure-property relationships in biological matter (Jeffrey and Saenger 1994), including cellulose. Starting with the discovery of nanocellulose and promises of a bright future as sustainable load-bearing component in high performance materials (Berglund and Peijs 2010; Benítez and Walther 2017), the last decade has seen an exponential growth of the interest in cellulose research. Rapid development in cellulose chemistry, processing and characterization has led to a property range of cellulose-based materials that expanded beyond imagination and to new areas of application that are continuously discovered. But where do all these intriguing and, indeed, extraordinary properties originate from? Although justified in some cases, there is a tendency in the cellulose field to invoke H-bonding as an almost magical explanation. For example, unique characteristics of cellulose such as high axial modulus and strength of fibrils, strong fiber-fiber bonding, or forming of paper from fibers are commonly explained based on “hydrogen bonding effects”. Sometimes this explanation is wrong (high modulus and strength of fibrils) or very often incomplete (fiber-fiber bonding and forming of paper), and in most cases the effect of moisture is neglected. However, simplistic explanations to complex problems are convenient and thereby tend to survive.
Thus, there is a need from time to time to re-examine the claims made with respect to H-bonding in cellulose and cellulose-based materials, and this is the purpose of this review. To this end, we discuss H-bonds in the context of the research on cellulose and their role at different length scales (Fig. 1) including:
-
Molecular level (conformation of a cellulose molecule).
-
Intermolecular level (how cellulose molecules interact with each other).
-
Fibril level (how cellulose is arranged into crystals).
-
Interfacial level (how cellulose fibrils interact with other molecules).
-
Interfibril level (how fibrils aggregate into larger structures).
-
Fiber and interfiber level (how fibril aggregates are assembled in fibers and how fibers form joints in paper).
Based on a critical survey of suitable literature, both old and recent, our aim is to present a more nuanced description of the role of H-bonds in cellulose research. Molecular modeling such as molecular dynamics (MD) simulations has played an important role over the years for the understanding of molecular-scale phenomena in cellulose (Zhou et al. 2020) since they offer both a level of detail that surpasses what can be reached by experimental methods and the ability to quantitatively extract almost any experimental parameter from the simulated ensemble. However, how simulated microscale properties are related to the macroscale is not always clear. A molecular simulation typically represents less than one millionth of a real sample that is observed during less than one microsecond, and great care must naturally be taken when transferring simulation results to larger scales. Nevertheless, simulated molecular interactions are a strong qualitative and quantitative tool to understand interaction mechanisms. Thus, results from modeling studies are central to this text. Our most important message is that H-bonding is just one of several such mechanisms, and the main reason why it stands out is because its importance is frequently exaggerated sometimes to the extent as if it was the only reason behind a variety of complex cellulose-related phenomena.

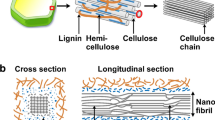
Structure of cellulose at various length scales and organizational levels. The dihedral angles \(\varphi\) and \(\psi\) are defined by the atomic sequences O5’-C1’-O4-C4 and C1’-O4-C4-C3, respectively. The figure depicts a tentative model for elementary fibrils from wood, although its exact dimensions and shape are still matters of debate
One example for this interpretational havoc is the debate concerning the physical mechanisms behind cellulose dissolution. Owing to the abundance of accessible hydroxyl groups on its surface, cellulose is rightfully considered a hydrophilic molecule with a pronounced hygroscopic character and wetting. At the same time, as distinct molecules it is completely insoluble in water at ambient conditions, which has been attributed to the formation of H-bonds between the cellulose molecules themselves leading to large and readily precipitating aggregates. However, more than a decade ago MD simulations were used to show that the contribution from H-bonding to the insolubility of cellulose in an aqueous environment was an order of magnitude smaller than hydrophobic solvation energies (Bergenstråhle et al. 2010). About the same time, in a series of papers, Björn Lindman and co-workers argued that H-bonding can hardly be the driving force for aggregation in water, and that one should treat cellulose as an amphiphilic molecule dressed with several interactions (Medronho et al. 2012; Lindman et al. 2021) which at the time was coined the “Lindman hypothesis” (Glasser et al. 2012). A few years later Nishiyama (2018) showed that London dispersion interaction is the dominating contribution to the total cohesive energy of cellulose. Today, it seems a large fraction of the cellulose community concurs with the notion that H-bonds play only a minor role for the precipitation of cellulose chains in aqueous environments, but solubility is not the only area where H-bonding effects are being exaggerated.
In this context, it is important to consider that all biological processes take place in water, the hydrogen-bonding liquid par excellence. This means that H-bonds within or between biomolecules always have to compete with H-bonds to water, and in this competition the relatively high mobility, both translational and rotational, of the water molecules with the unparalleled capacity of forming four hydrogen bonds for a puny molar mass of 18 gives the latter an advantage. This hints to that H-bonds themselves cannot be the main driving force for biomolecular assembly in water, as the total number of H-bonds in a hydrated system will remain more or less constant. In fact, we are probably lucky that H-bonds between biomolecules in water are weak, reversible, and dynamic, since the molecules of life would otherwise be strongly associated in uninteresting lumps. However, their directionality can make them into a significant steering force and thereby proficient organizers of three-dimensional structures (Jeffrey and Saenger 1994). Many processes involving cellulose are non-equilibrium processes. This applies to the biosynthesis of cellulose in the plant cell walls, to the mechanical treatments of fibers by which the elementary fibrils are liberated, and to the application of shear forces (stirring) for efficient dissolution. With that in mind, it makes sense to differentiate between the making and the breaking of H-bonds, and to realize that it may require a large activation energy to both make or break, even if the net effect in the free energy from an equilibrium point of view is zero.
A single H-bond is rather unspecific and relatively weak, and thereby can form and break on short (nanosecond) timescales, activated by thermal fluctuations alone at biological temperatures. However, consider \(N\) H-bonds defining a molecular complex. These bonds are subject to \({N}^{1/2}{k}_{B}T\) thermodynamic fluctuations. If \(N\) is large the kinetic stability of this complex is decided by the relation of that term to \(NC{k}_{B}T\), where \(C{k}_{B}T\) is the average activation energy to break an intermolecular H-bond. In other words, since the total activation energy scales with \(N\) but the random force with \({N}^{1/2}\), massively H-bonded molecular complexes are not easy to disassemble since many bonds have to break simultaneously without reforming. Once formed they can thermally be prone to remain assembled, even in the case of a favorable free energy for dissolution. Thus, their strength lies in their number, which combined with ordering in specific patterns can lead to considerable specificity. When H-bonds are broken, they can readily re-form provided that the geometry is right. This allows for flexible structures which is utilized in, e.g., spider silk (Nova et al. 2010), and can be exploited in self-healing biomaterials (Brochu et al. 2011). Thus, H-bonds is utilized by nature both to create kinetically stable, highly ordered structures, and to dissipate energy in flexible and ductile materials. Both aspects are relevant to cellulose.
The physical chemistry of hydrogen bonds
Intermolecular interactions are complex and often – even in leading textbooks – their description is heavily simplified and sometimes even incorrect (Truhlar 2019). Hydrogen bonding is a simple name for a complex situation, even in the case of two isolated molecules. Its contributing molecular elements are conventionally named as “donor” (D) and “acceptor” (A) where, provided that both A and D are sufficiently electronegative, D supposedly donates a proton that A accepts, forming the H-bond system D-H···A. For two isolated molecules, the experimental standpoint is simple: in this system there is (i) a positive charge density on H, negative charge density on A and therefore there will be an electrostatic term contributing, and (ii) a nonzero electron density between H and A as unequivocally shown by the non-zero hyperfine coupling between D and A (Grzesiek et al. 2004; Dračínský and Hodgkinson 2015). Compton scattering experiments concur (Isaacs et al. 1999). This latter feature suggests that there is charge transfer or, if one wishes, covalency over the H···A bond. In addition, induction and dispersion terms are contributing as well. There is some argument going on over the relative importance of the different contributions, and the extent of the covalent nature of the bond (Grabowski 2011). However, the clear correlation between the electron density between H and A and the bond strength suggests that the covalency cannot be entirely negligible (Shahi and Arunan 2014). Even with the difficulties of uniquely defining H-bonding (the IUPAC definition from 2011 is quite vague (Arunuan et al. 2011)) it undoubtedly exists, and its energies spread from weak (about 1 \({k}_{B}T\) in C-H···C bonds), through moderate (5-15 \({k}_{B}T\), “normal” O-H···O bonds) to very strong (>50 \({k}_{B}T\), in HF). A final complication is that H-bonding is cooperative (Mahadevi and Sastry 2016). This means that a bond which is part of an extended H-bond network may be different in strength from the isolated bond. Specifically, for the case of cellulose the intermolecular H-bond strength was shown to increase with the number of glucose units, and to plateau at a degree of polymerization of four (Qian 2008). In addition to H-bonding, ionic interaction, electrostatic multi-pole interaction and London dispersion (van der Waal’s) interaction contribute to the intermolecular forces in cellulose materials. Notably, even if the dispersion force is the weakest and most short-ranged of them when considering just a pair of atoms, their additivity makes the total contribution to the force between supra-molecular objects both considerably larger in magnitude and also significantly more long-ranged (Hamaker 1937), to the end that they can become the dominating interaction between two (uncharged) molecular surfaces.
Modeling and simulation of hydrogen bonds
In quantum chemistry (QC), some profound issues concerning hydrogen bonds remain unsettled and controversies seem to prevail about methodology, specifically the choice of base functions and its consequences for covalency, namely that some models predict significant charge transfer, whereas some predict much less (Stobe 2017; Weinhold and Glendening 2018; Stone and Szalewicz 2018). A related problem is that QC estimates of H-bond strengths remain uncertain, and so does the relative importance of the electrostatic, charge transfer, induction and dispersion terms. Moreover, QC cannot tell exactly what the angular dependence of the bond strength is, that is how directional are the H-bonds (Gilli and Gilli 2009). Probably, this depends on the exact D-H···A system.
Molecular dynamics (MD) simulations are routinely used to study molecular-scale structure and dynamics in most fields of material science, and there are several optimized parameter sets (force fields) specifically designed for simulations of carbohydrates, such as GLYCAM06 (Kirshner and Woods 2001), CHARMM 36 (Guvench et al. 2008, 2009), and GROMOS 56CARBO (Hansen and Hünenberger 2010). Most contemporary empirical biomolecular force fields do not employ any special H-bonding potential. The non-bonded interactions are usually limited to the Coulomb potential, which acts pairwise between fractional charges that are distributed over the atoms with a distance dependence of \({r}^{-1}\), and the Lennard-Jones potential, which describes repulsion with an \({r}^{-12}\), term, and dispersion attraction with a \({r}^{-6}\), term. Nevertheless, H-bond configurations form anyway as a consequence of a favorable combination of Coulomb and Lennard-Jones energies for atoms in the H-bonding geometries. Although it may seem so, this fact is in no way lending support for the non-covalent side in the controversy discussed above but is rather indicative of successful parameterization of the models. It has been noted that quantum mechanical contributions are required to capture the fine details of the structure of liquid water in simulations (Chen et al. 2003), and that inclusion of explicit polarizability affects both H-bond strength and kinetics (Xu et al. 2002), but in general empirical force fields are accurate enough to reproduce structure and dynamics of massively H-bonded systems, such as hydrated polysaccharides (Chen et al. 2018). Since H-bonds in MD do not possess a distinct and fundamental identity, they are usually identified in the post-processing of the molecular trajectories based on a set of geometrical criteria. A common choice is a donor-acceptor distance less than 0.35 nm, and a hydrogen-donor-acceptor angle below 30°. The actual H-bond definition will affect the number of detected bonds at a certain instant, but the H-bond dynamics(Luzar 2000) can be analyzed in a way such that it becomes independent of the cutoffs.
Cellulose from the inside
The cellulose polymer
Cellulose is a linear polymer composed of d-glucopyranose units linked by β-1,4-glycosidic bonds. The native polymer has high molecular weight with a degree of polymerization sometimes exceeding 10,000 (Gralén and Svedberg 1943). It is also rather inflexible with a persistence length that has been estimated to ~15 nm (or ~30 d-glucopyranose residues) from molecular modeling (Kroon-Batenburg et al. 1997). Since the sugar rings are relatively stiff, conformational freedom around the glycosidic bonds, commonly described by the \(\varphi\) and \(\psi\) torsional angles (Fig. 1), lends flexibility to cellulose, and polysaccharides in general. As a consequence of having its hydroxyl groups equatorially positioned a cellulose polymer can form intra-molecular hydrogen bonds between sequential glucose units: between the hydroxyl group on C3 and the ring oxygen (O3H3···O5) and between the hydroxyl groups on C2 and C6 respectively (O2H2···O6 or O6H6···O2). Due to the geometry of the molecule, these hydrogen bonds can only form if two consecutive glucose units attain a 21-fold (or close to) conformation, meaning that the two glucose units are twisted 180° with respect to each other around the chain axis (Fig. 1). This conformation is indeed the most dominant one in water soluble cellooligomers both in solution (Kroon-Batenburg et al. 1993) and in the solid state (Chu and Jeffrey 1968; Ham and Williams 1970). One question that arises is whether these hydrogen bonds play an active role in driving the molecular conformation, or if they are merely a consequence of favorable geometry.
The influence of trans-glycosidic H-bonding was investigated by several computer modeling studies where the H-bonding capability was modulated either by chemical substitution of the hydroxyls (French et al. 2005, 2021), by using glucose epimers having the hydroxyl groups in axial position as opposed to equatorial (e.g., mannose or allose) (Wang et al. 2013), or by looking at hemicelluloses (Berglund et al. 2016, 2019, 2020), which, due to its variation in chemical structure, can be viewed as a combination of both approaches. Indeed, both Density Functional Theory (DFT) calculations with solvent effects included and MD simulations in explicit water show that the conformation of the β-1,4 linkage is very consistent regardless of the number of H-bonds. The exceptions to this are chemical substitutions that have a relatively large effect on the steric environment (Yu et al. 2019), which can both shift the lowest energy conformation and influence the rigidity of the structure. Another example is xylan, where the hydroxymethyl group is removed altogether, which favors a twisted conformation in solution but has a relatively low free energy barrier for conversion to 21-fold (Berglund et al. 2016; Ling et al. 2020). The conclusion is that since the intra-molecular H-bonds can be effectively replaced by H-bonds to water molecules they do not contribute to the energy of the different conformations. In the gas phase on the other hand, trans-glycosidic H-bonding may indeed have profound effects on the conformation of celloligomers, as revealed by both modeling(Strati et al. 2002; French 2012) and experiments (Anggara et al. 2021). In this context, using a combination of vibrational spectroscopy and QM calculations (Cocinero et al. 2009), it was shown that the cis (untwisted) conformation produced the lowest energy conformers of phenyl \(\beta\)-cellobioside in vacuum, which were stabilized by H-bonding between O2’ and O3. Furthermore, using cellobiose analogues that lack H-bonding, French (2012) showed that the region of 2-fold conformations was the most stable one, also in vacuum. Thus, in an environment where intermolecular H-bonds are easily exchanged (e.g., in water) the propensity for the (near) 21-fold conformation in β-1,4-linked carbohydrates such as cellulose seems to be caused mainly by steric forces.
Intermolecular H-bonds and the formation of fibrils
During the biosynthesis, glucan chains coalesce into extended structures – the elementary fibrils. These fibrils are often perceived as being constituted by a crystalline core covered by more disordered surface chains and, moreover, occasional regions along the fibril where the crystalline order may be lacking. From X-ray, neutron diffraction, and NMR studies, four major crystalline allomorphs have been reported – cellulose I, II, III and IV. Cellulose I is the native form and also the most widely studied. It is present in all plant cell walls and is further divided into two sub forms, Iα (Nishiyama et al. 2003) and Ιβ (Nishiyama et al. 2002). Native cellulose is most commonly a combination of these two allomorphs (Atalla and VanderHart 1984) in proportions that depend on the source. Cellulose II is irreversibly obtained from cellulose I upon regeneration or alkali treatment (Langan et al. 1999, 2001). Cellulose III (Wada et al. 2004a, b) can be obtained from both cellulose I (in that case called cellulose IIII) and cellulose II (cellulose IIIII), by soaking in liquid ammonia or organic amine at low temperatures, whereas cellulose IV can be formed by thermal treatment of cellulose III in glycerol (Wada et al. 2004a, b). All these allomorphs are recognized by conformational differences, various packing arrangements and, importantly, their different intra- and inter-molecular hydrogen bonding patterns (Kovalenko 2010) (Fig. 2).

H-bonding system in two cellulose allomorphs, cellulose Ib and cellulose II. Based on the location of the heavy atoms, several patterns are possible. The figure depicts the one for Ib that is lowest in energy based on simulations, denoted pattern A. For cellulose II, the sole difference between the patterns is whether OH2 and OH6 act as donor or acceptor, respectively. The orientation of the hydroxymethyl groups is highlighted, denoted tg in cellulose Ib, and gt in cellulose II
The exact location of hydrogens in the crystal structure is difficult to determine experimentally due to their small X-ray scattering power. Furthermore, even if heavier atoms are ordered, the hydrogens may be disordered in the structure, as for instance seen in hexagonal ice (Peterson and Levy 1957). Consequently, the H-bonding in crystalline cellulose is not necessarily fixed to a single pattern but may be prone to fluctuations. Indeed, based on X-ray and neutron fiber diffraction several different H-bonding arrangements were concluded leading to two, mutually exclusive, H-bonding pattern models existing alongside each other in (local) equilibrium in both cellulose Ιβ (Nishiyama et al. 2002) and cellulose Iα (Nishiyama et al. 2003). Based on MD and DFT, alternate H-bonding patterns for cellulose II and IIII were also proposed (Chen et al. 2015).
From computational modeling it was shown that for Ιβ specifically, one of the two suggested patterns (denoted pattern A) was energetically more stable than the other one (pattern B) but that (presumably) static domains exhibiting pattern B still existed within real samples, possibly as disordered regions (Nishiyama et al. 2008). The preference for pattern A was also revealed by comparing DFT calculations with dichroism data from polarized IR spectroscopy of aligned cellulose samples (Lee et al. 2015). For cellulose II, however, the energy difference between the different patterns was not large enough to exclude the possibility of co-existence in dynamic equilibrium (Hayakawa et al. 2017). Interestingly, the notion that the cellulose crystal structure is not entirely static and homogeneous, but prone to structural disorder and possibly fluctuations may be coupled to the dynamical heterogeneity of the glucan chains that is manifested in broad distributions of 13 C NMR T1 relaxation times, also within the supposedly crystalline fibril core (Terenzi et al. 2015; Chen et al. 2019).
The sensitivity to small variations in the cellulose crystal structure is also seen in numerous MD simulations. Using the experimentally determined atomic coordinates as input structure, including the hydroxyl groups’ hydrogen positions, MD simulations employing common force fields typically yield structures that are stable in periodic crystal models, i.e. where the cellulose chains are covalently bonded to their own periodic image (Mazeau and Heux 2003; Bergenstråhle et al. 2007). Such structures can to reproduce crystallographic lattice parameters with reasonable deviations (within 1–8%). On the contrary, significant structural disorder including deviations from the experimental H-bonding patterns is obtained in simulations of finite fibrils, i.e. models including cellulose chain ends and interfaces to water, especially in long (microsecond) simulations (Matthews et al. 2012). Further, at high temperature a structural transition, initiated by a change in the conformation of hydroxymethyl groups from tg to gg has been observed in simulations of cellulose Iβ (Bergenstråhle et al. 2007; Matthews et al. 2011). The intermolecular H-bonds at high temperature were more dynamic and less regular than the relatively stable pattern at room temperature, but remarkably, the intramolecular H-bond O3H3···O5 was found to persist even at 500 K.
The regularity of the interchain H-bond arrangements in native cellulose suggests that H-bonds have an organizing role in creating the fibril structures. That would not be unique to cellulose since hydrogen bonds define some of the most important structures known to molecular biology: the secondary structure in proteins (Pauling and Corey 1951; Pauling et al. 1951), and the formation of base-pairs in DNA (Watson and Crick 1953). Yet, it has been argued that even for the structure of DNA, hydrophobic interactions have a strong contributing and perhaps dominant role (Lindman et al. 2021). Hydrophobic interactions are, however, not specific. This means that even if they constitute a strong thermodynamic driving force to compact moieties together to minimize the total amount of non-polar (“hydrophobic”) surface area that is exposed to water, hydrophobicity does not, for the same reason, concern the interactions between those molecular surfaces. To create crystalline order, additional interactions that are specific to the type of atoms involved are needed, such as ionic interaction, van der Waal’s interaction, or H-bonding, in combination with a regular chemical structure.
However, the cellulose I structure is not merely a result of spontaneous processes. It is well known that cellulose, when precipitated from solution, crystallizes in the form of cellulose II. This indicates that this structure is lower in energy than cellulose I, and thereby that native cellulose is in a metastable state (Stöckmann 1972). Moreover, over the scale of organisms, cellulose fibrils have, with retained interchain arrangements within the fibrils, an extreme variation in cross-sectional fibril dimensions that are regular and reproducible within a given species (Tsekos 1999). Fibril dimensions vary from thin (two to three nanometers) and isotropic (i.e. the width approx. equal to the height) in most land plants, via large (up to 20 nm) isotropic shapes in some species (e.g. Oocystis, Valonia and Hypoglossum), to the highly anisotropic ribbon-like structures produced by, e.g., Acetobacter and Erythrocladia. The cross-sectional variation is correlated with the spatial arrangement of the linear terminal complexes (TC) that secrete the glucan chains and hence the fibrils: lateral TC sizes in land plants are much smaller than those in Valonia or Oocystis and in Acetobacter the TC shape is elongated. The only possible way of having such correlation is that cellulose association into fibrils and ribbons is not random but directed by the regular arrangement of cellulose synthase units. Curiously, cyanobacteria, which seems to lack regularly arranged TCs, create fibrils that have very small cross-sectional size (Nobles et al. 2001). The organization of fibrils outside the TC has been suggested to be a self-organizing process (Emons and Mulder 2000) possibly guided by the microtubules (Paradez et al. 2006). The environment into which the glucan chains are secreted will influence the microfibril architecture. Specifically, it was shown that it is possible to influence the ratio between cellulose I\(\alpha\) and I\(\beta\) produced by Acetobacter by the addition of polymers to the culture medium (Yamamoto et al. 1996), or even to make it produce cellulose II (Shibazaki et al. 1998).

The size and spatial arrangement of the cellulose-synthesizing complexes are responsible for the large variation in fibril dimensions among different species
These observations suggest that native cellulose is not created by spontaneous self-organization, but rather that during biosynthesis the glucan chains are guided to associate into a regular intermolecular arrangement within which they remain trapped. Hence, H-bonds do not alone cause the regular intermolecular arrangement of as-biologically-produced cellulose chains within fibrils but are merely consistent (in form of a local free energy minima) with the chains being guided into a specific spatial arrangement, thereby contributing to the stabilization of the metastable state. But even in this respect, they are not working alone. An often-neglected fact in the case of cellulose is that the molecular packing of chains is to a large extent a joint result of steric repulsion and attractive dispersion interactions and their total contribution to the cohesive energy of cellulose may be as high as 70% (Nishiyama 2018).
Axial stiffness of the cellulose crystal
Cellulose has an axial elastic modulus which is often compared to that of steel or Kevlar (Moon et al. 2011). The many hydrogen bonds within and between cellulose chains have been proposed to be important for the mechanical properties of cellulose crystals and highly crystalline cellulose materials. Specifically, it has been suggested that the intramolecular trans-glycosidic H-bonds (O3HO3···O5 and O2HO2···O6 in cellulose Iβ) contributes to the axial stiffness of cellulose, since they have a large component in the chain direction. Quantum mechanics (Santiago Cintrón et al. 2011), molecular mechanics modeling(Eichhorn and Davies 2006) and MD simulations(Wohlert et al. 2012; Wu et al. 2013) are generally fairly successful in reproducing the experimental axial crystal modulus of 138 GPa (Nishino et al. 1995). Thus, computer modeling is relevant and suitable for investigating the contribution of H-bonds to the stiffness, both regarding nature and extent.
As mentioned above, the intramolecular O2H2···O6 H-bond is on average broken in glucan chains residing in surfaces exposed to water. Owing to the 21-fold symmetry, this means that surface chains have, on average, 1.5 intramolecular H-bonds per glucose unit, as opposed to 2 in the crystalline core. Thus, if H-bonds contributed significantly to the axial modulus, one would expect a lateral size dependence of the stiffness, since the proportion of surface to core becomes smaller as the fibril cross sections become larger. A nano-scale three-point-bending experiment using atomic force microscopy (AFM) on bacterial cellulose fibrils between 35 and 90 nm thick did not detect any difference (Guhados et al. 2005), but that range was probably too small to see any such effect. Moreover, the measured moduli were low, around 76 GPa, which could indicate large contributions from non-crystalline material within their samples. However, also in MD simulations using fibrils of lateral size between 2.3 and 6.8 nm (Wohlert et al. 2012), no such size dependence was found.
Cellulose II and cellulose IIII are both different from native cellulose with respect to their dominating H-bonding pattern. Due to that the hydroxymethyl group has rotated with respect to the conformation in cellulose I (from tg to gt, see Fig. 2), the trans-glycosidic hydrogen bond to the hydroxyl in position two is no longer possible. Therefore, these allomorphs have only one intramolecular H-bond per glucose (Hayakawa et al. 2017), although this conformation does permit a bifurcated O3H3···O6 H-bond as reported for Me-cellobioside (French 2012). Based on literature values, cellulose II and IIII are generally 10-40% less stiff than cellulose I, athough experimental variability makes direct comparisons difficult. This variation was reproduced in MD simulations of all three allomorphs (Djahedi et al. 2015). However, in the only experimental study that compared elastic moduli of all three allomorphs using similar conditions it was concluded that the stiffnesses of cellulose I and IIII were similar, while it was lower for cellulose II (Ishikawa et al. 1997).
Both dynamic FTIR and Raman spectroscopy (Hinterstoisser and Salmén 1999; Kong and Eichhorn 2005) have shown that intramolecular H-bonds shift their frequencies during axial deformation of cellulose, and there was also evidence that they may disassociate if the strain was sufficiently high. However, a H-bond is approximately ten times easier to stretch than a C-O-C bond angle and hundred times less stiff than an ordinary C-O covalent bond. Glucan chains in cellulose are both aligned and extended, which leads to a dominant part of the axial deformation being associated with covalent degrees of freedom (Djahedi et al. 2015). Completely removing hydrogen bonds within any cellulose crystalline allomorph (Wohlert et al. 2012; Eichhorn and Davies 2006) causes a significant reduction of its stiffness, but also a loss in structural organization of the chains. Therefore, cooperative effects between hydrogen- and covalent bonding were suggested, and also investigated spectroscopically by Altaner et al. (Altaner et al. 2014). They proposed a molecular-scale leverage mechanism by which the deformation of the O3H3···O5 H-bond was enhanced since FTIR spectroscopy showed that the O2H2···O6 bond on the opposite side of the glycosidic linkage was not deforming during chain extension (Fig. 4). This effect was analyzed in a simplified spring model with parameters based on MD data (Djahedi et al. 2016), which showed that the leverage effect was indeed present within MD simulations, although mitigated by the fact that a large part of the total deformation took place in the sugar rings. Nevertheless, this study showed a relative effect of intramolecular hydrogen bonds of about 15-20% of the total stiffness, similar to what has been seen in MD simulations of crystalline cellulose where H-bonds were artificially turned off (Wu et al. 2013). However, analysis of the respective energy contributions to the total MD potential showed that the major part of the axial stiffness comes from deformations of bonds, angles and dihedrals in combination with a large contribution from dispersion interactions (Djahedi et al. 2015). In the transverse directions on the other hand, the situation is different, since covalent interactions can be expected to contribute less. DFT calculations (Chen et al. 2021) show that dispersion interactions contribute around 50% of the stiffness in the direction perpendicular to the H-bonded planes, and about 30% in the direction parallel to the H-bonded planes (Fig. 2). In that direction, removal of inter-chain H-bonds would likely have a large effect on the stiffness. However, the specific effect of H-bonds on the stiffness, in any direction, is not easy to isolate, both because removal of the H-bonds in simulations simultaneously changes other parameters as well (chain packing, etc.) and because different contributions to the stiffness (H-bonds, dispersion interactions, covalent bonds, etc.) are not necessarily additive.

A molecular scale leverage effect, proposed by Altaner et al. (Altaner et al. 2014), amplifies the relative contribution of the O3H3···O5 H-bond to the axial stiffness of cellulose (top). MD simulations show that the O3H3···O5 bond is stretched during axial deformation, whereas the O2H2···O6 bond remains unchanged (bottom, from Djahedi et al. (2016))
At the surfaces
Accessible OH groups are abundant on cellulose crystallite surfaces. The “hydrophilic” (110) and (1-10) crystallographic planes (Fig. 1) are most likely the dominant planes exposed in native fibrils from wood (Nishiyama 2009; Daicho et al. 2018), although alternative models have larger exposure of the (010) or the “hydrophobic” (200) planes (Fernandes et al. 2011; Yang and Kubicki 2020). The two hydrophilic surfaces are similar, exposing about 5.4 OH per nm2 that are potentially accessible to the environment. However, both modeling (Heiner and Teleman 1997; Heiner et al. 1998) and experiments (Lindh et al. 2016) show that the intra-chain H-bond O3H3···O5 is so stable that, in practice, the O3H3 hydroxyl does not act as a donor. This leaves 3.6 OH groups per nm2 that are available for H-bonding with other fibrils, macromolecules, or solvent molecules. In addition, their high reactivity makes them serve as points for chemical surface modification such as acetylation (Sassi and Chanzy 1995), TEMPO-mediated oxidation (Saito et al. 2006), or polymer grafting (Rol et al. 2019), which naturally affect the fibril’s H-bonding capability.
Adsorption to cellulose
To explain adsorption, it is common to look for favorable specific interactions, such as hydrogen bonds, electrostatic attraction, or \(\pi -\pi\) interactions, and seeking explanations in the chemical structure of the surface and the adsorbing molecule. Since both cellulose and molecules that adsorb to cellulose are often decorated with polar groups, such as hydroxyl groups, it is assumed that hydrogen bonds are important. This explanation is so common in the literature that it is frequently used without scientific evidence. What is often forgotten or actually neglected is the chemical structure and adsorption energy of the solvent. This is especially true in studies from the early days of MD simulation where the solvent was often left out due to computational limitations. It is useful to view adsorption as a process in which solvent molecules close to the surface are replaced by the adsorbing species, and that adsorption will only occur if the total free energy balance of this exchange process is negative (Fleer et al. 1993). In this context, it is important to emphasize that the adsorption energy is a free energy, which besides enthalpic contributions from specific molecular interactions also contains a (often significant) contribution from entropy: \(\varDelta G=\varDelta H-T\varDelta S\). One example of this is the well-studied case of polyelectrolyte adsorption to charged surfaces. Despite the strong Coulombic interactions that are present between the adsorbing molecule and the surface, the net result is an ion-exchange process driven by the increased entropy of releasing counterions and associated water (Fu and Schlenoff 2016; Michaels 1965). This becomes relevant for most colloidally stable nanocelluloses that are decorated with charged groups.
For the adsorption of nonionic molecules, the literature is quite ambiguous concerning the role of H-bonds. Hydrogen-bonded polymer association in water is indeed a frequent description, such as hydrogen-bonded multilayer assembly or hydrogen-bonded polymer complexation (Kharlampieva et al. 2009; Tsuchida and Abe 1982). However, even in the highly cited work by Tsuchida and Abe the message is incomplete. They describe the interaction as driven by hydrogen bonds, but then show that \(\varDelta H\)and \(\varDelta S\) are both positive upon complexation with water as solvent, meaning that H-bonds alone could not be responsible for the adsorption process. However, just as oppositely charged groups pair up in polyelectrolyte association, hydrogen bonds do indeed form between the adsorbing molecule and the cellulose surface, but that does not automatically contribute favorably to the adsorption energy. Namely, in a simplified picture, breaking one water-surface H-bond and one water-solute H-bond to form one solute-surface and one water-water H-bond is a net zero process in terms of enthalpy (\(\varDelta H\)). If \(\varDelta H=0\) then a favorable change in entropy is required to drive the adsorption.
Xyloglucan (XG) is a good example in the discussion about hydrogen bond-driven adsorption. XG is a nonionic hemicellulose found in the primary cell wall of all vascular plants. It adsorbs strongly to cellulose, which is not surprising since, to put it simply, XG is a cellulose chain decorated with xylose or xylose-galactose residues. The interaction between XG and cellulose has historically been ascribed to hydrogen bonds, but this perception has recently started to change due to measurements that indicate an endothermic process (Lopez et al. 2010; Benselfelt et al. 2016). Recently, MD simulations were used to show that adsorption of XG to cellulose is driven by the increased translational entropy of releasing interfacial water (Fig. 5) from reducing the total solvent-accessible surface area and that the number of hydrogen bonds was the same before and after adsorption (Kishani et al. 2021). The interaction was endothermic at room temperature but turned exothermic as the temperature increased due to the less favorable hydration of cellulose and XG. However, such considerations are not unique to XG, and it is clear that reduction of solvated interfaces leading to the release of (relatively) constrained water molecules, i.e. hydrophobic effects, is the main driving force for all hemicellulose adsorption to cellulose in water.
The analogy with cellulose association is obvious. Although H-bonds do not drive adsorption (making) from an equilibrium thermodynamics point of view, they can still affect how easy it is to remove an adsorbed molecule (breaking), either by force or desorption, by contributing to the kinetic stability (Stuart and Fleer 1996), since the probability of many bonds being spontaneously broken at the same time becomes low. They can also contribute to specificity, namely how hemicelluloses organize on the cellulose surface. Specifically, recent experimental results in combination with MD simulation suggest that both xylan and glucomannan exhibit an abundance of structural motifs that allow them to adsorb to cellulose in conformations where they become almost seamless extensions to the cellulose crystal structure, including the H-bond network (Busse-Wicher et al. 2014, 2016; Martinez-Abad et al. 2017, 2020; Grantham et al. 2017; Simmons et al. 2016; Pereira et al. 2017). However, while the simulations generally show that hemicellulose molecules adsorbed in this manner can be remarkably stable against fluctuations, they also show that in terms of adsorption free energy, hemicelluloses prefer the essentially non-H-bonded association to hydrophobic (200) surfaces (Fig. 1), if such surfaces are present (Martinez-Abad et al. 2017; Pereira et al. 2017; Zhang et al. 2011).

Adsorption of xyloglucan (XG) to native or charged cellulose. a Schematic of the adsorption process. b Simulated entropy of a single water molecule at different distances from charged and native cellulose, and XG, which determine the free energy gain upon aggregation from decreasing the total solvent-accessible surface area. These thermodynamic principles govern the adsorption of many molecules to cellulose (Lombardo and Thielemans 2019)
Nanocelluloses are suitable substrates for experimental determination of thermodynamic quantities of adsorption using isothermal titration calorimetry (ITC). A recent review (Lombardo and Thielemans 2019) that compiles the findings from ITC studies shows that adsorption of biomacromolecules, dyes, or drugs to cellulose or partially modified cellulose, is entropy driven and endothermic in most cases. In addition, MD simulations have shown that both urea (Chen et al. 2017) and the poorly soluble drug furosemide (Lombardo et al. 2018) adsorb to cellulose driven by a combination of dispersion interactions and translational entropy when they are exchanged with surface water. The exothermic exceptions, where H-bonding is a possibility, are the adsorption of cellulose binding modules (CBMs) of carbohydrate active enzymes or small and uncharged organic molecules. The binding site of CBMs is rich in aromatic amino acids such as tyrosine or tryptophane, placed at a distance so that they can stack on top of glucose rings along the cellulose backbone (Beckham et al. 2010; Ponyi et al. 2000). This suggests that the main mechanism for the adsorption is hydrophobic association by the release of interfacial water from both CBM and cellulose. At these less hydrophilic locations, there are no polar groups for water interaction, which means that the free energy of the hydrated state is high. Reducing the exposure of these areas to solvent thus results in an exothermic response upon release of water. Aromatic groups are relatively polar and interact strongly with water, compared to for example cyclohexane, which leads to a more favorable interface to water (Raschke and Levitt 2005). In addition, exchange of O-H···O H-bonds to O-H···N which have slightly lower energy (Pandey et al. 2017) would also lead to an exothermic response. Indeed, amines or amides are found in both CBMs and aromatic-rich molecules such as methylene blue (Lombardo and Thielemans 2019) that undergo exothermic adsorption.
Fibril-fibril aggregation and hornification
Fibrils both in suspension and in the plant cell wall form self-associated structures. However, under ambient conditions fibrils in plant cell walls do not fuse completely to form larger crystallites, which makes them distinguishable as separate entities using experimental methods such as X-ray and neutron scattering (Jarvis 2018). This can in part be explained by the fibrils being partly covered by hemicelluloses, although small-angle neutron scattering show fibrils that are in direct contact in conifers (Fernandes et al. 2011), bamboo (Thomas et al. 2015), and spruce wood (Thomas et al. 2020). The presence of a structurally disordered (as compared to crystalline order) cellulose-cellulose interface was also identified by spectral fitting of the C4 region in the 13 C NMR spectrum, which also showed that these surfaces exhibited significantly different polymer dynamics based on their T1 relaxation times (Wickholm et al. 1998). This was later replicated in MD simulations of fibril aggregates (Chen et al. 2019).
Wherever the molecular structure prevents a perfect fit between fibrils, such as anti-parallel arrangement (Chen et al. 2019), fibril twist (Paajanen et al. 2019), adsorbed hemicelluloses(Thomas et al. 2020) or the presence of a chemically modified surface (Chen et al. 2020), small sub-nano-sized cavities are present, which can harbor water molecules (Fig. 6). These waters are confined to the interface between the fibrils and substantially restricted with respect to their translational and rotational degrees of freedom. Indeed, both 2 H NMR(Lindh et al. 2017) and neutron scattering(O’Neill et al. 2017) reveal a population of water in hydrated fibril systems that has significantly slower dynamics than those normally associated with surface-bound water.

The continuous interface between two crystalline domains (left) can be perturbed by molecular scale “defects” such as chemical surface modification (right), in this case surface acetylation. This leads to sub-nanometer sized cavities that can harbor water molecules. Cellulose is shown in black for carbon and yellow for oxygen, surface acetyl groups in green (carbon) and red (oxygen). Water is shown in light blue. Reproduced from Chen et al. (2020) with permission from the Royal Society of Chemistry
Computer simulation of fibril aggregates in excess water show that water molecules between fibrils tend to stay in place on MD (100 ns) timescales (Chen et al. 2019; Paajanen et al. 2019). This indicates that these water molecules are either in thermodynamic equilibrium with the water outside the aggregate or kinetically trapped due to their slow dynamics. The first case is supported by the observation that these water molecules can lower the total enthalpy of the system by saturating potential H-bonds that are lost due to defects. This will lower the total free energy provided that the entropic penalty associated with being confined is not too large. MD simulations further show that the inter-fibrillar water molecules will spontaneously leave their confinement at high (160 °C) temperature (Langan et al. 2014). This could be due to either the thermal energy becoming high enough to overcome the activation energy for diffusion, or the entropic term for the confined water becoming larger than the enthalpy gain. Interestingly, X-ray diffraction shows that hydrothermal treatment of wood does induce co-crystallization of fibrils, although not into a regular Iβ structure (Kuribayashi et al. 2016). Instead a structure was obtained that was consistent with the fusion of anti-parallel fibrils.
When cellulose is dried from air it will form large, micrometer-sized aggregates (Peng et al. 2012). Such drying-induced association is a technical problem of great significance since the dry fibrils can be difficult to redisperse. Thus, from a practical perspective, the association is described as irreversible, a phenomenon usually referred to as hornification in the pulp and paper community. This has large consequences for the industrial use of nanocellulose since fibrils have to be kept in their dispersed state, with large transportation costs as a consequence (Posada et al. 2020). The extent of hornification depends strongly on drying methods (Peng et al. 2012; Nodenström 2020) and can also be mitigated by additives such as glycerol (Moser et al. 2018), which presumably act as spacers between fibrils. It also depends on surface chemistry of the fibrils (Benselfelt et al. 2019), where for instance acetylation was shown to reduce the work of adhesion between fibrils in water (Chen et al. 2020) due to the surface acetyl groups preventing tight association leading to the interpenetration of water molecules at the interface (Fig. 6). Although hornification is often explained as irreversible H-bonding between the fibrils, H-bonding is not sufficient to explain this process. With respect to making, hornification must be driven by a force that is both sufficiently long ranged and sufficiently strong to force fibrils and fibril aggregates into close contact. The best candidate is capillary forces that arise from the water seeking to minimize its liquid/air interface (Fig. 7). The magnitude of the capillary force depends on the geometry of the problem and the solid/liquid work of adhesion, WA. The crystalline cellulose surfaces are strongly hydrophilic and their WA to water was calculated to 118 mJ m−2 from MD simulations (Karna et al. 2020). This leads to the idea that the forces can become substantial at the nanoscale and are strong enough to deform fibrils plastically (Ogawa et al. 2020). Moreover, MD simulations have shown that the capillary force can act over large distances (several nanometers) through liquid capillary bridges (Sinko and Keten 2014; Zhang et al. 2021) and thus fulfils both criteria.

Drying-induced deformation of nanofibrils driven by capillary effects. The “dry” fibril-fibril joint contains structured and immobile water, which can contribute to both total adhesion and deformation mechanisms
When two cellulose surfaces finally are in contact there will of course be H-bonds. However, with respect to breaking, since the H-bond energy is smaller than the dispersion energy even for the best possible interface with respect to H-bond formation (i.e., the crystal structure), H-bonding likely plays a minor role for the stability also here. Thus, if the surfaces are close enough to be in molecular contact (i.e., van der Waal’s and/or H-bonding) the London dispersion will dominate over the H-bond contribution to the interaction energy. However, there is undoubtedly water present even in cellulose material that is perceived as dry and this will complicate the picture further.
Even if the porosity of cellulose nanomaterials can be surprisingly low, unless fibrils are perfectly aligned, they are prohibited from fusing completely into one continuous phase. Thus, as mentioned above, sub-nanometer sized cavities are formed, for instance around fibril-fibril joints, where surface hydroxyl groups are available for H-bonding. This space can be effectively filled by water molecules which will saturate the H-bonds, as long as the entropic penalty of confinement is not too high (Fig. 7). Thus, it is not only difficult to remove the water fraction that is most tightly attached to cellulose, but it is even more difficult to prepare and maintain samples (either as a specimen to be studied or a starting point for controlled hydration) that are devoid of all water (Lindh et al. 2017), because the initial water uptake of dry cellulose from ambient air is extremely rapid and amounts to about 8 wt% (based on cellulose dry mass) at 50% relative humidity. The emerging picture is that the extent to which H-bonding contributes to the adhesion between fibrils, even in “dry” conditions, is limited and an indirect effect of the high surface tension of liquid water.
Interfacial water also affects the friction between fibrils. In completely dry conditions, which can only be realized in simulations, the traction is dominated by a stick-slip behavior, in part related to breaking and reformation of interfacial H-bonds (Zhang et al. 2021). In that respect, the small size and relatively high mobility of water molecules make them into an effective lubricant, which can significantly decrease the resistance to interfacial shear stress (Sinko and Keten 2014), and possibly also serve as a toughening mechanism since a weaker interface reduces brittleness (Hou et al. 2021).
Cellulose nanopaper films
A unique feature of cellulose nanofibrils is that “non-porous” nanopaper films can be formed by drying from colloidal dispersions of the nanofibers in water. Filtration and drying results in nanofibrils which are oriented random-in-plane, with slightly swirled conformation (Henriksson et al. 2008). The formation process can be compared to the formation of photonic crystals where capillary force through liquid bridges (Fig. 7) is the main adhesion mechanism, not hydrogen bonding (Zhou et al. 2006). Such forces can become very large, enough to deform the nanofibers plastically (Ogawa et al. 2020), which explains the low porosity of cellulose nanopaper dried from water.
The Young’s modulus of dry wood cellulose nanofibril films was recently reported to be 24.9 GPa by careful strain field measurements (Yang et al. 2021). This result is much higher than for any polymer film with comparable molecular orientation distribution, for instance 5.2 GPa for biaxially oriented PET-films (Breil 2010). From modeling work, the H-bonding between nanofibrils was erroneously stated to be critical for mechanical performance (Meng et al. 2017), and such ideas were further developed in a recent review paper (Meng and Wang 2019). Although the suggested model is interesting, the statement needs correction.
The ultimate strength of CNF nanopaper depends on cellulose molecular weight, indicating that it depends critically on cellulose nanofibril strength (Henriksson et al. 2008) and nanofibril length (Fukuzumi et al. 2013), but defects and local stress transfer mechanisms are also important. High hemicellulose content has a positive contribution to stress-strain behavior of nanopaper, which is related to interfibril bonding (Kontturi et al. 2021; Yang et al. 2021). However, in contrast to typical polymer films, even dry cellulose films (in the example above vacuum dried at 75 °C for three days) will unavoidably contain water due to the finite, however brief, time it is exposed to moist air during the actual mechanical testing (Lindh et al. 2017). The large majority of the moisture is located in the interfibril region, since the fibril center is inaccessible to water (Sakurada et al. 1962). At 50% relative humidity, the moisture content of cellulose nanopaper is 8-10% (Yang et al. 2021). There is strong dependence of both modulus and yield strength on moisture content (Benítez et al. 2013; Yang et al. 2021). The yield strength in nanopaper is probably related to interfibril shearing in the interphase region, and is lowered by increasing moisture content, a problem that has been analyzed at a molecular scale (Sinko and Keten 2014; Zhang et al. 2021). In the dry state, no apparent yielding is observed (Yang et al. 2021). The mechanisms for low strain deformation (modulus) and plastic yielding (yield strength) in practice thus depend primarily on moisture-related effects. Thus, cellulose nanopaper does not obtain its excellent properties thanks to H-bonding but rather despite the abundant H-bonding sites on the nanofibril surfaces. On one hand, they lead to formation of dense structures through capillary forces during drying, but on the other hand H-bonding sites increase moisture sorption, even at low relative humidity, with reduced properties as consequence. The main reason for the superior modulus of nanopaper to polymer films, however, is the high axial modulus of the cellulose nanofibril, which, even for random-in-plane orientation, dominates in-plane modulus for paper structures (Page 1965) and composites.
The cellulose fiber and paper making
The scientific background to the excellent mechanical properties of different paper qualities has attracted substantial research interest over the years. This is partly due to the possibilities to form numerous materials from different types of fibers via water-based processing routes, and partly since the excellent inherent mechanical properties of the fibers are utilized (Bolam 1961). A large focus has been on the contact zone between the fibers and how the properties of this contact zone can be understood and manipulated (Lindström et al. 2005; Hirn and Schennach 2017). To a large extent, the extensive work by Nissan (Nissan 1955, 1976a, b; Nissan and Batten 1997) has dominated the view among paper scientists that hydrogen-bonding between the fibers is the major interaction responsible for the mechanical properties of the paper. This is appealing at a first glance, since cellulose and hemicelluloses, the dominating components of most delignified, papermaking fibers, contain a large number of OH-groups that are known to be able to engage in H-bonds. However, the process of rough cellulose-rich fibers approaching each other during drying and consolidation of paper in the presence of water is complex and a more elaborate description of the mechanisms behind the making and the breaking of fiber-fiber joints is needed, especially considering the very short-ranged nature of H-bond interaction and formation (Santiago Cintrón et al. 2017). The development of new measuring techniques and the development of model materials mimicking the components of the fiber wall (Gustafsson et al. 2012; Li et al. 2021) provide new experimental information to separate the different molecular mechanisms responsible for fiber-fiber interactions, but still, there is no quantitative description of their relative influence on the formation of a strong fiber-fiber joint (Lindström et al. 2005; Hirn and Schennach 2017).
The making and the breaking of a fiber-fiber joint are schematically described in Fig. 8. As the water between the fibers is evaporating the capillary pressure increases, pulling the fibers together. Under wet conditions, the delignified fiber wall is highly hydrated and the water has a strong plasticizing effect. Since the wet fiber wall has a modulus (\(E\)) of around one to ten MPa (Nilsson et al. 2001) a capillary radius (\(r\)) of one micrometer (a reasonable dimension considering the dimensions of the fiber) would lead to a strain (\(\epsilon\)) of the wet fiber wall,
between 1.5 and 15% assuming a fully wetted fiber surface (\(\text{cos}\theta =1\)) and using the surface tension of pure water \(\gamma =72\) mN m−1, and as the capillaries narrow, the deformation will be even higher. This means that just as for the fibrils (see above) the fibers are pulled closely together during drying. However, unlike the elementary fibril, the fiber surface is not molecularly smooth, which means that only a part of the interface between the fibers will be in molecular contact whereas other parts will be separated. As mentioned earlier, H-bonds are specific and short-ranged, and practically require direct contact to form, whereas other forces (ionic and van der Waals) can be significantly long-ranged at this scale to act over the separation distance.
It has been shown that the migration of polymers across the interface has a significant influence on the dry adhesive properties of the fiber-fiber joints (Johansson et al. 2009) and that the thermodynamic work of adhesion is similar for cellulose-cellulose, cellulose-hemicellulose, and cellulose-lignin interactions as evaluated by contact adhesion testing (Gustafsson et al. 2012). Considering the function of these components in trees, this result is also very logical since there should be a good adhesive interaction between these components to create a strong structure.

Schematic description of the making and breaking of fiber-fiber joints. The wet fibers are pulled together by the capillary pressure and the wet fiber wall will yield in the wet zone. In the dry joint there will be areas in molecular contact and areas not in molecular contact but still close enough to allow for an interaction between the surfaces. Of the proposed mechanisms, only van der Waals and ionic interactions are significant when there is no direct molecular contact (during the making of the joints in the wet state)
As the dry joint is strained, all the separate interactions across the interface will act to resist the load and breakage can occur either through adhesive failure of the joint or cohesive failure of the fiber wall (Fig. 8). The cohesive interactions inside the fiber wall are similar to the interactions that exist between fibers, which means that in an ideal joint it is difficult to identify the actual interface. The presence of water, in the form of moist air, will have several effects. First, it will soften the fiber wall due to moisture adsorption. Further, as water adsorbs at the interface, specifically into those regions where the molecular contact between the fibers is poor, the water will weaken the dispersion interaction, enable disentanglement of molecules that might have moved across the interface and naturally also disrupt possible H-bonds.
The making and breaking of fiber-fiber joints is an interplay between the deformation of the macroscopic fiber wall due to massive capillary forces which allows close contact between dry fibers, and the development of molecular interactions at those contact points, including H-bonds. Once formed, these interactions will play different roles during the loading of the joints and also the combination of different interactions will contribute. H-bonding naturally influences the strength of these joints, as they will inevitably form wherever a favorable molecular geometry arises, but to what extent they contribute at the fiber scale in comparison to other relevant molecular forces is not possible to determine using any technique available today. However, considering their short range and directional sensitivity, contributions from other interactions are most likely dominating.
Perspective and outlook
In this paper we have reviewed the role of H-bonding in cellulose-based materials at different scales, from H-bonds within small molecular fragments up to macroscopic fiber-fiber bonds. It is clear that the role of H-bonds has in many cases been exaggerated, at all levels in the structural hierarchy of cellulose. Considering that cellulose H-bonds in most relevant systems can exchange with H-bonds to water molecules it becomes evident that their net effect on the overall energetics is relatively small compared to other factors such as dispersion interactions and hydrophobic effects. A misplaced focus on H-bonding is an oversimplification that has indeed slowed down the development of our fundamental understanding of cellulose-rich materials and impeded their use in more advanced applications. On the other hand, to ignore H-bonding completely in the analysis of cellulose is not a solution; they play an important role for defining the crystal structure of cellulose and give a surprisingly large contribution to its elastic modulus. H-bonds can also be expected to contribute to molecular adhesion in dry conditions although completely dry cellulose hardly exist except in theory, and the role of water for interactions at both fibril- and fiber level under normal conditions cannot be stressed enough. This observation also applies to drinking straws.
Molecular modeling has indeed been instrumental for our understanding of the molecular-scale properties of cellulose. But at the same time the atomistic picture that permits us to actually count H-bonds has almost been a curse. Furthermore, it is important to realize that when we discuss experimental observations, we almost exclusively limit ourselves to a material that has been taken out of its native environment and has been mechanically and/or chemically modified. This undoubtedly affects its physico-chemical properties, particularly so when comparing to computer modeling studies, which are restricted to highly idealized models.
The emerging conclusion is that H-bonding is and should be viewed as one interaction among several, and its relative contribution is highly dependent on the specific conditions and cannot easily be determined by intuition alone or, indeed, in some cases not even by careful analysis. Based on our combined experience from working with cellulose and from the review of the scientific literature in this field presented in this paper, we suggest a ranking of the relative importance of H-bonding for different physical and chemical properties of cellulose and cellulose based materials (Table 1). We hope that this work will inspire other scientists in this field to see beyond the standard explanations, and to not be content with descriptions of H-bonding as the sole defining feature for cellulose-related phenomena. This, we believe, would be of great benefit for the cellulose community, as a whole.
References
Altaner CM, Thomas LH, Fernandes AN, Jarvis MC (2014) How Cellulose Stretches: Synergism between Covalent and Hydrogen Bonding. Biomacromolecules 15:791–798
Anggara K, Zhu Y, Fittolani G, Yu Y, Tyrikos-Ergas T, Delbianco M, Rauchenbach S, Abb S, Seeberger PH, Kern K (2021) Identifying the origin of local flexibility in a carbohydrate polymer. Proc. Natl. Acad. Sci. U. S. A. 118:2102188118
Arunuan E, Desiraju GR, Klein RA, Sadle J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ (2011) Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl Chem 83:1637–1641
Atalla RH, VanderHart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Beckham GT, Matthews JF, Bomble YJ, Bu L, Adney WS, Himmel ME, Nimlos MR, Crowley MF (2010) Identification of amino acids responsible for processivity in a family 1 carbohydrate-binding module from a fungal cellulase. J Phys Chem B 114:1447–1453
Benítez AJ, Torres-Rendon J, Poutanen M, Walther A (2013) Humidity and multiscale structure govern mechanical properties and deformation modes in films of native cellulose nanofibrils. Biomacromolecules 14:4497–4506
Benítez AJ, Walther A (2017) Cellulose nanofibril nanopapers and bioinspired nanocomposites: a review to understand the mechanical property space. J Mater Chem A 5:16003–16024
Benselfelt T, Cranston ED, Ondaral S, Johansson E, Brumer H, Rutland MW, Wågberg L (2016) Adsorption of Xyloglucan onto Cellulose Surfaces of Different Morphologies: An Entropy-Driven Process. Biomacromolecules 17:2801–2811
Benselfelt T, Nordenström M, Hamedi MM, Wågberg L (2019) Ion-induced assemblies of highly anisotropic nanoparticles are governed by ion–ion correlation and specific ion effects. Nanoscale 11:3514–3520
Bergenstråhle M, Berglund LA, Mazeau K (2007) Thermal Response in Crystalline Iβ Cellulose: A Molecular Dynamics Study. J Phys Chem B 111:9138–9145
Bergenstråhle M, Wohlert J, Himmel ME, Brady JW (2010) Simulation studies of the insolubility of cellulose. Carbohydr Res 345:2060–2066
Berglund J, Angles d’Ortoli T, Vilaplana F, Widmalm G, Bergenstråhle-Wohlert M, Lawoko M, Henriksson G, Lindström M, Wohlert J (2016) A molecular dynamics study of the effect of glycosidic linkage type in the hemicellulose backbone on the molecular chain flexibility. Plant J 88:56–70
Berglund J, Azhar S, Lawoko M, Lindström M, Vilaplana F, Wohlert J, Henriksson G (2019) The structure of galactoglucomannan impacts the degradation under alkaline conditions. Cellulose 26:2155–2175
Berglund J, Kishani S, Morais de Carvalho D, Lawoko M, Wohlert J, Henriksson G, Lindström ME, Wågberg L, Vilaplana F (2020) Acetylation and sugar composition influence the (in)solubility of plant β-mannans and their interaction with cellulose surfaces. ACS Sustanable Chem Eng 8:10027–10040
Berglund LA, Peijs T (2010) Cellulose biocomposites—from bulk moldings to nanostructured systems. MRS Bull 35:201–207
Bolam FC (1961) “Fundamentals of papermaking fibres.” P. 497 in Transactions of the symposium held at Cambridge, September 1957, Technical Section of the British Paper and Board Makers’ Association
Breil J (2010) “Oriented film technology.” In: Multilayer Flexible Packaging, Wagner JR Jr (Eds), Elsevier, Armsterdam
Brochu ABW, Craig SL, Reichert WM (2011) Self-healing bomaterials. J Biomed Mater Res A 96:492–506
Busse-Wicher M, Gomes TCF, Tryfona T, Nikolovski N, Stott K, Grantham NJ, Bolam DN, Skaf MS, Dupree P (2014) The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a twofold helical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J 79:492–506
Busse-Wicher M, Li A, Silveira RL, Pereira CS, Tryfona T, Gomes TCF, Skaf MS, Dupree P (2016) Evolution of xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant Physiol 171:2418–2431
Chen B, Ivanov I, Klein ML, Parrinello M (2003) Hydrogen bonding in water. Phys Rev Lett 91:215503
Chen P, Lo Re G, Berglund LA, Wohlert J (2020) Surface modification effects on nanocellulose – molecular dynamics simulations using umbrella sampling and computational alchemy. J Mater Chem A 8:23617
Chen P, Nishiyama Y, Wohlert J (2021) Quantifying the influence of dispersion interactions on the elastic properties of crystalline cellulose.Cellulose,https://doi.org/10.1007/s10570-021-04210-0
Chen P, Nishiyama Y, Wohlert J, Lu A, Mazeau K, Ismail AE (2017) Translational Entropy and Dispersion Energy Jointly Drive the Adsorption of Urea to Cellulose. J Phys Chem B 121:2244–2251
Chen P, Ogawa Y, Nishiyama Y, Bergenstråhle-Wohlert M, Mazeau K (2015) Alternative hydrogen bond models of cellulose II and IIII based on molecular force-fields and density functional theory. Cellulose 22:1485–1493
Chen P, Terenzi C, Furó I, Berglund LA, Wohlert J (2018) Hydration-dependent dynamical modes in xyloglucan from molecular dynamics simulation of 13 C NMR relaxation times and their distribution. Biomacromoleculs 19:2567–2579
Chen P, Terenzi C, Furó I, Berglund LA, Wohlert J (2019) Quantifying localized macromolecular dynamics within hydrated cellulose fibril aggregates. Macromolecules 52:7278–7288
Chu SSC, Jeffrey GA (1968) The refinement of the crystal structures of β-D-glucose and cellobiose. Acta Cryst B 24:830–838
Cocinero EJ, Gamblin DP, Davis BG, Simons JP (2009) The building blocks of cellulose: the intrinsic conformational structures of cellobiose, its epimer, lactose and their singly hydrated complexes. J Am Chem Soc 131:11117–11123
Daicho K, Saito T, Fujisawa S, Isogai A (2018) The crystallinity of nanocellulose: dispersion-induced disordering of the grain boundary in biologically structured cellulose. ACS Appl Nano Mater 1:5774–5785
Djahedi C, Bergenstråhle-Wohlert M, Berglund LA, Wohlert J (2016) Role of hydrogen bonding in cellulose deformation: the leverage effect analyzed by molecular modeling. Cellulose 23:2315–2323
Djahedi C, Berglund LA, Wohlert J (2015) Molecular deformation mechanisms in cellulose allomorphs and the role of hydrogen bonds. Carbohydr Polym 130:175–182
Dračínský M, Hodgkinson P (2015) Solid-state NMR studies of nucleic acid components. RCS Adv 5:12300–12310
Eichhorn SJ, Davies GR (2006) Modelling the crystalline deformation of native and regenerated cellulose. Cellulose 13:291–317
Emons AMC, Mulder BM (2000) How the deposition of cellulose microfibrils builds cell wall architecture. Trends Plant Sci 5:35–40
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc. Natl. Acad. Sci. U.S.A. 108:E1195–E1203
Fleer GJ, Stuart MAC, Scheutjens J M H M, Cosgrove T, Vincent B (1993) Polymers at Interfaces. Chapman & Hall, London
French AD (2012) Combining computational chemistry and crystallography for a better understanding of the structure of cellulose. In: Horton D (ed) Advances in Carbohydrate Chemistry and Biochemistry. Academic Press, Burlington
French AD, Johnson GP, Kelterer A-M, Csonka GI (2005) Fluorinated cellobiose and maltose as stand-ins for energy surface calculations. Tetrahedron Assymetry 16:577–586
French AD, Mongomery DW, Prevost NT, Edwards JV, Woods RJ (2021) Comparison of cellooligosaccharide conformations in complexes with proteins with energy maps for cellobiose. Carbohydr Polym 264:118004
Fukuzumi H, Saito T, Isogai A (2013) Influence of TEMPO-oxidized cellulose nanofibril length on film properties. Carbohydr Polym 93:172–177
Fu J, Schlenoff JB (2016) Driving forces for oppositely charged polyion association in aqueous solutions: enthalpic, entropic, but not electrostatic. J Am Chem Soc 138:980–990
Gibb BC (2020) The centenary (maybe) of the hydrogen bond. Nat Chem 12:665–667
Gilli G, Gilli P (2009) The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory. Oxford University Press, Oxford
Glasser WG, Atalla RH, Blackwell J, Brown RM Jr, Burchard W, French AD, Klemm DO, Nishiyama Y (2012) About the structure of cellulose: debating the Lindman hypothesis. Cellulose 19:589–598
Grabowski SJ (2011) What is the covalency of hydrogen bonding? Chem Rev 111:2597–2625
Gralén N, Svedberg T (1943) Molecular weight of native cellulose. Nature 152:625
Grantham NJ, Wurman-Rodrich J, Terrett OM, Lyczakowski JJ, Stott K, Iuga D, Simmons TJ, Durand-Tardif M, Brown SP, Dupree R, Busse-Wicher M, Dupree P (2017) An even pattern of xylan substitution is critical for interaction with cellulose in plant cell walls. Nature Plants 3:859–865
Grzesiek S, Cordier F, Jaravine V, Barfield M (2004) Insights into biomolecular hydrogen bonds from hydrogen bond scalar couplings. Prog Nucl Magn Reson Spectrosc 45:275–300
Guhados G, Wan W, Hutter J (2005) Measurement of the elastic modulus of single bacterial cellulose fibers using atomic force microscopy. Langmuir 21:6642–6646
Gustafsson E, Johansson E, Wågberg L, Pettersson T (2012) Direct adhesive measurements between wood biopolymer model surfaces. Biomacromolecules 13:3046–3053
Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, MacKerell AD Jr (2008) Additive Empirical Force Field for Hexopyranose Monosaccharides. J Comput Chem 29:2543–2564
Guvench O, Hatcher E, Venable RM, Pastor RW, MacKerell AD Jr (2009) CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput 5:2353–2370
Hamaker HC (1937) The London—van der Waals attraction between spherical particles. Physica 4:1058–1072
Ham JT, Williams DG (1970) The crystal and molecular structure of methyl β-cellobioside-methanol. Acta Cryst B 26:1373–1383
Hansen HS, Hünenberger PH (2010) A Reoptimized GROMOS Force Field for Hexopyranose-Based Carbohydrates Accounting for the Relative Free Energies of Ring Conformers, Anomers, Epimers, Hydroxymethyl Rotamers, and Glycosidic Linkage Conformations. J Comput Chem 32:998–1032
Hayakawa D, Nishiyama Y, Mazeau K, Ueda K (2017) Evaluation of hydrogen bond networks in cellulose Iβ and II crystals using density functional theory and Car–Parrinello molecular dynamics. Carbohydr Res 449:103–113
Heiner AP, Kuutti L, Teleman O (1998) Comparison of the interface between water and four surfaces of native crystalline cellulose by molecular dynamics simulations. Carbohyrd Res 306:205–220
Heiner AP, Teleman O (1997) Interface between monoclinic crystalline cellulose and water: breakdown of the odd/even duplicity. Langmuir 13:511–518
Henriksson M, Berglund LA, Isaksson P, Lindström T, Nishino T (2008) Cellulose nanopaper structures of high toughness. Biomacromolecules 9:1579–1585
Hinterstoisser B, Salmén L (1999) Two-dimensional step-scan FTIR: a tool to unravel the OH-valency-range of the spectrum of Cellulose I. Cellulose 6:251–263
Hirn U, Schennach R (2017) “Fiber-fiber bond fromation and failure: Mechanisms and analytical techniques.” Pp. 839-863 in 16th Fundamental Research Symposium. Cambridge
Hou Y, Guan Q-F, Xia J, Ling Z-C, He Z, Han Z-M, Yang H-B, Gu P, Zhu Y, Yu S-H, Wu H (2021) Strengthening and toughening hierarchical nanocellulose via humidity-mediated interface. ACS Nano 15:1310–1320
Isaacs ED, Shukla A, Platzman PM, Hamann DR, Barbiellini B, Tulk CA (1999) Covalency of the hydrogen bond in ice: a direct X-ray measurement. Phys Rev Lett 82:600–603
Ishikawa A, Okano T, Sugiyama J (1997) Fine structure and tensile properties of ramie fibres in the crystalline form of cellulose I, II, III and IV. Polymer 38:463–468
Jarvis MC (2018) Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos Trans R Soc A Math Phys Eng Sci 376:20170045
Jeffrey GA, Saenger W (1994) Hydrogen Bonding in Biological Structures. Springer-Verlag, Heidelberg
Johansson E, Blomberg E, Lingström R, Wågberg L (2009) Adhesive interaction between polyelectrolyte multilayers of polyallylamine hydrochloride and polyacrylic acid studied using atomic force microscopy and surface force apparatus. Langmuir 25:2887–2894
Karna NK, Wohlert J, Lidén A, Mattsson T, Theliander H (2020) Wettabiity of cellulose surfaces under the influence of an external electric field. J Colloid Interface Sci Prepare 589:347–355
Kharlampieva E, Kozlovskaya V, Sukhishvili SA (2009) Layer-by-layer hydrogen-bonded polymer films: From fundamentals to applications. Adv Mater 21:3053–3065
Kirshner Karl N, Woods Robert J (2001) Solvent Interactions Determine Carbohydrate Conformation. Proc Natl Acad Sci 98:10541–10545
Kishani S, Benselfelt T, Wågberg L, Wohlert J (2021) Entropy drives the adsorption of xyloglucan to cellulose surfaces - A molecular dynamics study. J Colloid Interface Sci 588:485–493
Kong K, Eichhorn SJ (2005) The influence of hydrogen bonding on the deformation micromechanics of cellulose fibers. J Macromol Sci Phys 44:1123–1136
Kontturi KS, Lee K-Y, Sampson JMP, Bismarck WW, Kontturi E (2021) Influence of bilogical origin on the tensile properties of cellulose nanopapers. Cellulose 28:6619–6628
Kovalenko VI (2010) Crystalline cellulose: structure and hydrogen bonds. Russ Chem Rev 79:231
Kroon-Batenburg LMJ, Kroon J, Leeflang BR, Vliegenthart JFG (1993) Conformational analysis of methyl β-cellobioside by ROESY NMR spectroscopy and MD simulations in combination with the CROSREL method. Carbohydr Res 245:21–42
Kroon-Batenburg LMJ, Kruiskamp PH, Vliegenthart JFG, Kroon J (1997) Estimation of the persistence length of polymers by MD simulations on small fragments in solution. Application to cellulose. J Phys Chem B 101:8454–8459
Kuribayashi T, Ogawa Y, Rochas C, Matsumoto Y, Heux L, Nishiyama Y (2016) Hydrothermal Transformation of Wood Cellulose Crystals into Pseudo-Orthorombic Structure by Cocrystallization. ACS Macro Lett 5:730–734
Langan P, Nishiyama Y, Chanzy H (1999) A Revised Structure and Hydrogen-Bonding System in Cellulose II from Neutron Fiber Diffraction Analysis. J Am Chem Soc 121:9940–9946
Langan P, Nishiyama Y, Chanzy H (2001) X-ray Structure of Mercerized Cellulose at 1 Å Resolution. Biomacromolecules 2:410–416
Langan P, Petridis L, O’Neill HM, Singali SV, Foston M, Nishiyama Y, Schulz R, Lindner B, Hanson BL, Harton S, Heller WT, Urban V, Evans BR, Gnanakaran S, Ragauskas AJ, Smith JC, Davison BH (2014) Common processes drive the thermochemical pretreatment of lignocellulosic biomass. Green Chem 16:63–68
Lee CM, Kubicki JD, Fan B, Zhong L, Jarvis MC, Kim SH (2015) Hydrogen-bonding network and OH stretch vibration of cellulose: comparison of computational modeling with polarized IR and SFG spectra. J Phys Chem B 119:15138–15149
Lindh EL, Bergenstråhle-Wohlert M, Terenzi C, Salmén L, Furó I (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: The role of interaction with water. Carbohydr Res 434:136–142
Lindh EL, Terenzi C, Salmén L, Furó I (2017) Water in cellulose: evidence and identification of immobile and mobile adsorbed phases by 2H MAS NMR. Phys Chem Chem Phys 19:4360–4369
Lindman B, Medronho B, Alves L, Norgren M, Nordenskiöld L (2021) Hydrophobic interactions control the self-assembly of DNA and cellulose. Q Rev Biophys 54:1–22
Lindström T, Wågberg L, Larsson T (2005) “On the nature of joint strength in paper - A review of dry and wet strength resins used in paper manufacturing.” Pp. 457-562 in 13th Fundamental Research Symposium. Cambridge
Ling Z, Edwards JV, Nam S, Xu F, French AD (2020) Conformational analysis of xylobiose by DFT quantum mechanics. Cellulose 27:1207–1224
Li H, Roth SV, Freychet G, Zhernenkov M, Asta N, Wågberg L, Pettersson T (2021) Structure development of the interphase between drying cellulose materials revealed by in situ grazing-incidence small-angle X-ray scattering. Biomacromolecules 22:4274–4283
Lombardo S, Chen P, Larsson PA, Thielemans W, Wohlert J, Svagan AJ (2018) Toward improved understanding of the interactions between poorly soluble drugs and cellulose nanofibers. Langmuir 34:5464–5473
Lombardo S, Thielemans W (2019) Thermodynamics of adsorption on cellulose surfaces. Cellulose 26:249–279
Lopez M, Bizot H, Chambat G, Marais M-F, Zykwinska A, Ralet M-C, Driguez H, Buléon A (2010) Enthalpic Studies of Xyloglucan–Cellulose Interactions. Biomacromolecules 11:1417–1428
Luzar A (2000) Resolving the hydrogen bond dynamics conundrum. J Chem Phys 113:10663
Mahadevi AS, Sastry GN (2016) Cooperativity in noncovalent interactions. Chem Rev 116:2775–2825
Martinez-Abad A, Berglund J, Toriz G, Gatenholm P, Henriksson G, Lindström M, Wohlert J, Vilaplana F (2017) Regular motifs in xylan modulate molecular flexibility and interactions with cellulose surfaces. Plant Physiol 175:1579–1592
Martinez-Abad A, Jiménez-Quero A, Wohlert J, Vilaplana F (2020) Influence of the molecular motifs of mannan and xylan populations on their recalcitrance and organization in spruce softwoods. Green Chem 22:3956–3970
Matthews JF, Beckham GT, Bergenstråhle-Wohlert M, Brady JW, Himmel ME, Crowley MF (2012) Comparison of Cellulose Iβ Simulations with Three Carbohydrate Force Fields. J Chem Theory Comput 8:735–748
Matthews JF, Bergenstråhle M, Beckham GT, Himmel ME, Nimlos MN, Brady JW, Crowley MF (2011) High-Temperature Behavior of Cellulose I. J Phys Chem B 115:2155–2166
Mazeau K, Heux L (2003) Molecular dynamics simulations of bulk native crystalline and amorphous structures of cellulose. J Phys Chem B 107:2394–2403
Medronho B, Romano A, Miguel MG, Stigsson L, Lindman B (2012) Rationalizing Cellulose (In)solubility: Reviewing Basic Physicochemical Aspects and Role of Hydrophobic Interactions. Cellulose 19:581–587
Meng Q, Li B, Li T, FEng X-Q (2017) A multiscale crack-bridging model of cellulose nanopaper. J Mech Phys Solids 103:22–39
Meng Q, Wang TJ (2019) Mechanics of strong and tough cellulose nanopaper. Appl Mech Rev 71:040801
Michaels AS (1965) Polyelectrolyte complexes. Ind Eng Chem 57:32–40
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Moser C, Henriksson G, Lindström ME (2018) Improved dispersibility of once-dried cellulose nanofibers in the presence of glycerol. Nord Pulp Paper Res J 33:647–650
Nilsson B, Wågberg L, Gray D (2001) “Conformability of wet pulp fibres at short lentgh scales.” In: CF Baker (eds), The science of papermaking. Oxford, UK
Nishino T, Takano K, Nakamae K (1995) Elastic Modulus of the Crystalline Regions of Cellulose Polymorphs. J Polym Sci Part B, Polym Phys Ed 33:1647–1651
Nishiyama Y (2009) Structure and properties of the cellulose microfibril. J Wood Sci 55:241–249
Nishiyama Y (2018) Molecular interactions in nanocellulose assembly. Philos Trans R Soc A 376:20170047
Nishiyama Y, Johnson GP, French AD, Forsyth VT, Langan P (2008) Neutron Crystallography, Molecular Dynamics, and Quantum Mechanics Studies of the Nature of Hydrogen Bonding in Cellulose Ibeta. Biomacromolecules 9:3133–3140
Nishiyama Y, Langan P, Chanzy H (2002) Crystal Structure and Hydrogen-Bonding System in Cellulose Iβ from Synchrotron X-ray and Neutron Fiber Diffraction. J Am Chem Soc 124:9074–9082
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal Structure and Hydrogen Bonding System in Cellulose Iα from Synchrotron X-ray and Neutron Fiber Diffraction. J Am Chem Soc 125:14300–14306
Nissan AH (1955) A molecular approach to the problem of viscoelasticity. Nature 174:424
Nissan AH (1976a) Bond dissociation in hydrogen bond dominated solids. Macromolecules 9:840–850
Nissan AH (1976b) Three modes of dissociation of H bonds in hydrogen-bond dominated solids. Nature 263:759
Nissan AH, Batten GL Jr (1997) The link between the molecular and structural theories of paper elasticity. Tappi J 80:153
Nobles DR, Romanovicz DK, Brown RM (2001) Cellulose in cyanobacteria. Origin of vascular plant cellulose synthase? Plant Physiol 127:529–542
Nodenström M (2020) “Colloidal interactions and arrested dynamics of cellulose nanofibrils.” PhD Thesis, KTH Royal Institute of Technology, Stockholm, Sweden
Nova A, Keten S, Pugno NM, Redaelli A, Beuhler MJ (2010) Molecular and nanostructural mechanisms of deformation, strength and toughness of spider silk fibrils. Nano Lett 10:2626–2634
Ogawa Y, Nishiyama Y, Mazeau K (2020) Drying-induced bending deformation of cellulose nanocrystals studied by molecular dynamics simulations. Cellulose 27:9779–9786
O’Neill H, Pingali SV, Petridis L, He J, Mamontov E, Hong L, Urban V, Evans B, Langan P, Smith JC, Davison BH (2017) Dynamics of water bound to crystalline cellulose. Sci Rep 7:11840
Paajanen A, Ceccherini S, Maloney T, Ketoja JA (2019) Chirality and bound water in the hierarchical cellulose structure. Cellulose 26:5877–5892
Page DH (1965) A theory for the elastic modulus of paper. Brit J Appl Phys 16:253–258
Pandey SK, Manogaran D, Manogaran S, Shaefer HF III (2017) Quantification of hydrogen bond strength based on interaction coordinates: a new approach. J Phys Chem A 121:6090–6103
Paradez AR, Somerville CR, Ehrhardt DW (2006) Visualization of cellulose synthase demonstrate functional association with microtubules. Science 312:1491–1495
Pauling L (1939) The nature of the chemical bond. Cornell University Press, Ithaca, NY
Pauling L, Corey RB (1951) Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proc Natl Acad Sci USA 37:729–740
Pauling L, Corey RB, Branson HR (1951) The structure of proteins. Two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 37:205–211
Peng Y, Gardner DJ, Han Y (2012) Drying cellulose nanofibrils: in search of a suitable method. Cellulose 19:91–102
Pereira CS, Silveira RL, Dupree P, Skaf MS (2017) Effects of xylan side-chain substitutions on xylan–cellulose interactions and implications for thermal pretreatment of cellulosic biomass. Biomacromolecules 18:1311–1321
Peterson SW, Levy HA (1957) A single-crystal neutron diffraction study of heavy ice. Acta Cryst 10:70–76
Ponyi T, Szabó L, Nagy T, Orosz L, Simpson PJ, Williamson MP, Gilbert HJ (2000) Trp22, Trp24, and Tyr8 play a pivotal role in the binding of the family 10 cellulose-binding module from Pseudomonas xylanase A to insoluble ligands+. Biochemistry 39:985–991
Posada P, Velásquez-Cock J, Gómez-Hoyos C, Serpa Guerra AM, Lyulin SV, Kenny JM, Gañán P, Zuluaga R (2020) Drying and redispersion of plant cellulose nanofibers for industrial applications: a review. Cellulose 27:10649–10670
Qian X (2008) The effect of cooperativity on hydrogen bonding interactions in native cellulose Iβ from ab initio molecular dynamics simulations. Mol Sim 34:183–191
Raschke TM, Levitt M (2005) Nonpolar solutes enhance water structure within hydration shells while reducing interactions between them. Proc Natl Acad Sci 102:6777–6782
Rol F, Belgacem MN, Gandini A, Bras J (2019) Recent advances in surface-modified cellulose nanofibrils. Prog Polym Sci 88:241–264
Saito T, Nishiyama Y, Puteaux J-L, Vignon M, Isogai A (2006) Homogeneous suspensions of indiviualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 7:1687–1691
Sakurada I, Nukushina Y, Ito T (1962) Experimental Determination of Elastic Modulus of Crystalline Regions in Oriented Polymers. J Polym Sci 57:651–660
Santiago Cintrón M, Johnson GP, French AD (2011) Young’s modulus calculations for cellulose Iβ by MM3 and quantum mechanics. Cellulose 18:505–516
Santiago Cintrón M, Johnson GP, French AD (2017) Quantum mechanics models of the methanol dimer: O-H⋯O hydrogen bonds of β-D-glucose moieties from crystallographic data. Carbohydr Res 443–444:87–94
Sassi J-F, Chanzy H (1995) Ultrastructural aspects of the acetylation of cellulose. Cellulose 2:111–127
Shahi A, Arunan E (2014) Hydrogen bonding, halogen bonding and lithium bonding: an atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys Chem Chem Phys 16:22935–22952
Shibazaki H, Saito M, Kuga S, Okano T (1998) Native cellulose II production by Acetobacter xylinum under physical constraints. Cellulose 5:165–173
Simmons TJ, Mortimer JC, Bernardinelli OD, Pöppler A-C, Brown SP, deAzevedo E R, Dupree R, Dupree P (2016) Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nature Commun 7:13902
Sinko R, Keten S (2014) Effect of moisture on the traction-separation behavior of cellulose nanocrystal interfaces. Appl Phys Lett 105:243702
Stöckmann VE (1972) Developing a hypothesis: Native cellulose elementary fibrils are formed with metastable structure. Biopolymers 11:251–270
Stobe AJ (2017) Natural bond orbitals and the nature of the hydrogen bond. J Phys Chem A 121:1531–1534
Stone AJ, Szalewicz K (2018) Reply to “Comment on ‘Natural bond orbitals and the nature of the hydrogen bond’”. J Phys Chem A 122:733–736
Strati GL, Willet JL, Momany FA (2002) Ab initio computational study of β-cellobiose conformers using B3LYP/6-311++G**. Carbohydr Res 337:1833–1849
Stuart MC, Fleer G (1996) Adsorbed polymers in nonequilibrium situations. Annu Rev Mater Sci 26:463–500
Terenzi C, Prakobna K, Berglund LA, Furó I (2015) Nanostructural Effects on Polymer and Water Dynamics in Cellulose Biocomposites: 2H and 13 C NMR Relaxometry. Biomacromolecules 16:1506–1515
Thomas LH, Forsyth VT, Martel A, Grillo I, Altaner CM, Jarvis MC (2015) Diffraction evidence for the structure of cellulose microfibrils in bamboo, a model for grass and cereal celluloses. BMC Plant Biol 15:153
Thomas LH, Martel A, Grillo I, Jarvis MC (2020) Hemicellulose binding and the spacing of cellulose microfibrils in spruce wood. Cellulose. https://doi.org/10.1007/s10570-020-03091-z
Truhlar DG (2019) Dispersion forces: neither fluctuating nor dispersiing. J Chem Educ 96:1671–1675
Tsekos I (1999) The sites of cellulose sythesis in algae: diversity and evolution of cellulose-sythesizing enzyme complexes. J Phycol 35:635–655
Tsuchida E, Abe K (1982) Interactions between macromolecules in solution and intermacromolecular complexes. Springer, Berlin, Heidelberg
Wada M, Chanzy H, Nishiyama Y, Langan P (2004a) Celluluse IIII Crystal Structure and Hydrogen Bonding by Synchotron X-ray and Neutron Fiber Diffraction. Macromolecules 37:8548–8555
Wada M, Heux L, Sugiyama J (2004b) Polymorphism of Cellulose I Family: Reinvestigation of Cellulose IVI. Biomacromolecules 5:1385–1391
Wang D, Ámundadóttir ML, van Gunsteren WF, Hünenberger PH (2013) Intramolecular hydrogen-bonding in aqueous carbohydrates as a cause or consequence of conformational preferences: a molecular dynamics study of cellobiose stereoisomers. Eur Biophys J 42:521–537
Wang X, Pang Z, Chen C, Xia Q, Zhou Y, Jing S, Wang R, Ray U, Gan W, LI C, Chen G, Foster B, Li T, Hu L (2020) All-natural, degradable, rolled-up straws based on cellulose micro- and nano-hybrid fibers. Adv Funct Mater 30:1910417
Watson JD, Crick FHC (1953) A structure for deoxyribose nucleic acid. Nature 171:737–738
Weinhold F, Glendening ED (2018) Comment on “Natural bond orbitals and the nature of the hydrogen bond”. J Phys Chem A 122:724–732
Wickholm K, Larsson PT, Iversen T (1998) Assignment of non-crystalline forms in cellulose I by CP/MAS 13 C NMR spectroscopy. Carbohydr Res 312:123–129
Wohlert J, Bergenstråhle-Wohlert M, Berglund LA (2012) Deformation of Cellulose Nanocrystals: Entropy, Internal Energy and Temperature Dependence. Cellulose 19:1821–1836
Xiawa Wu, Robert M, Ashlie M (2013) Crystalline cellulose elastic modulus predicted by atomistic models of uniform deformation and nanoscale indentation. Cellulose 20:43–55
Xu H, Stern HA, Berne BJ (2002) Can water polarizability be ignored in hydrogen bond kinetics? J Chem Phys B 106:2054–2060
Yamamoto H, Horii F, Hirai A (1996) In situ crystallization of bacterial cellulose II. Influences of different polymeric additives on the formation of celluloses Iα and Iβ at the early stage of incubation. Cellulose 3:229–242
Yang X, Jungstedt E, Reid MS, Berglund LA (2021) Polymer films from cellulose nanofibrils - Effects from interfibrillar interphase on mechanical behavior. Macromolecules 54:4443–4452
Yang H, Kubicki JD (2020) A density functional theory study on the shape of the primary cellulose microfibril in plants: effects of C6 exocyclic group conformation and H-bonding. Cellulose 27:2389–2402
Yu Y, Tyrikos-Ergas T, Zhu Y, Fittolani G, Bordoni V, Singhal A, Fair RJ, Grafmüller A, Seeberger PH, Delbianco M (2019) Systematic hydrogen-bond manipulation to establish polysaccharide structure-property correlations. Angew Chem Int Ed 58:13127–11332
Zhang Q, Brumer H, Ågren H, Tu Y (2011) The Adsorption of Xyloglucan on Cellulose: Effects of Explicit Water and Side Chain Variation. Carbohyd Res 346:2595–2602
Zhang C, Keten S, Derome D, Carmeliet J (2021) Hydrogen bonds dominated frictional stick-slip of cellulose nanocrystals. Carbohydr Polym 258:117682
Zhou S, Jin K, Buehler MJ (2020) Understanding plant biomass via computational modeling. Adv Mater 33(28):2003206
Zhou Z, Qin L, Zhao XS (2006) Evolution of interparticle capillary forces during drying of colloidal crystals. Langmuir 22:3692–3697
Acknowledgments
The authors thank Dr. Alfred D. French and Dr. Tomas Larsson for helpful discussions during the writing of this manuscript. Benselfelt SciArt is acknowledged for the illustrations.
Funding
Open access funding provided by Royal Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wohlert, M., Benselfelt, T., Wågberg, L. et al. Cellulose and the role of hydrogen bonds: not in charge of everything. Cellulose 29, 1–23 (2022). https://doi.org/10.1007/s10570-021-04325-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-04325-4