
Abstract
Knowledge of genetic diversity and connectivity within and between populations of specialist meadow plants is crucial to developing effective conservation strategies at the landscape-scale. This study investigated levels of genetic diversity within, and gene flow between populations of a key annual species, Rhinanthus minor in protected meadows and landscape matrix sites in two contrasting regions. Possible barriers to gene flow were also analysed. Leaf material from 714 individuals in an extensively managed upland region and an intensively managed lowland region of the UK was genotyped using microsatellite markers. Genetic diversity was similar in the two regions (He = 0.48 and 0.44). FST values indicated population differentiation in both regions but the estimate was higher in the lowland (FST = 0.28) than in the upland region (FST = 0.19); evidence of global structure was revealed in a spatial principal components analysis but a maximum likelihood population effects model did not identify significant predictors of population differentiation after testing the effects of Euclidean geographic distance, land cover and elevation. Conservation strategies should aim to maintain large populations in meadows to enhance genetic diversity. At the same time the focus should be on existing and additional species-rich grassland fragments, particularly in areas of intensive land-use, if genetic connectivity is to be retained.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Habitat fragmentation is detrimental to genetic diversity as well as species diversity (Kahilainen et al. 2014; Schlaepfer et al. 2018). An increased understanding of genetic variation and connectivity in species found in habitats of high nature value is key to the development of conservation strategies for small and isolated populations (Picó and van Groenendael 2007; Mijangos et al. 2015). The broader goal of the conservation of genetic diversity is a key element of the Convention on Biological Diversity (CBD) but signatories to the CBD have not made significant progress in developing targets or actions to tackle the loss of genetic diversity, particularly in populations of wild plants and animals (Laikre et al. 2010). The need for an integrated, evidence-based approach to managing the conservation of small and genetically isolated populations has been recognised (Cook and Sgrò 2016: Ralls et al. 2018). Although the impact of fragmentation has been acknowledged in government reports and policies (Lawton et al. 2010; Reed et al. 2017), very little attention has been given to the role of genetic connectivity in such documents (Eigenbrod et al. 2016; Natural England 2016; Simeonova et al. 2017).
Investigations into the impact of fragmentation have included the examination of relationships between patch size and species richness, the analysis of the influence of past landscapes and habitats, the role played by land use and management of isolated sites, and the study of various aspects of functional connectivity (Krauss et al. 2004; Purschke et al. 2014; Auffret et al. 2015; Huber et al. 2017). Such approaches will be complemented by studies of the loss of functional connectivity through gene flow, since reductions in genetic variability within populations, and genetic divergence between populations, are likely to be associated with isolated habitat patches (Young et al. 1996). There is evidence to show that lower levels of genetic diversity have been found in small, isolated populations (Jacquemyn et al. 2010; Crichton et al. 2016) including those of species which were once common, but which now survive in small fragments as a result of processes such as agricultural intensification (Honnay and Jacquemyn 2007; Aguilar et al. 2008). Such populations are likely to be more susceptible to genetic drift, higher levels of inbreeding and reduced gene flow between populations (Ellstrand and Elam 1993) and it has been recommended that the impacts of reduced genetic diversity on fitness and the responses of populations to environmental change should not be overlooked in conservation strategies (Hooftman et al. 2003; Jump et al. 2008).
Effective landscape-scale conservation strategies will require an understanding of the connectivity provided by gene flow between protected sites but will also benefit from knowledge about such connectivity across the landscape matrix. The landscape matrix can be an important influence on plant species diversity in isolated sites both in terms of surrounding land use types and availability of other potential habitat patches such as roadside verges (Schmucki et al. 2012; Arenas et al. 2017). For example, species richness of grassland plants was found to be significantly lower in areas dominated by arable land than it was in forested landscapes (Öckinger et al. 2012). Landscape features such as roadside verges and field margins are thought to play a role in dispersal for semi-natural grassland plant species (Thiele et al. 2018) but evidence of functional connectivity between such sites is difficult to establish. An investigation of gene flow between isolated populations across different landscapes will provide more information about functional connectivity in this context.
The present study investigated genetic connectivity in species-rich hay meadows, a habitat of high nature value which has been subjected to significant losses in terms of area and numbers of sites since the middle of the twentieth century (Critchley et al. 2003; Hodgson et al. 2005; Hooftman and Bullock 2012). Hay meadows are managed using a low intensity regime and are mown once a year after seeds have been set to enable suitable conditions for a high diversity of plant species (Crofts and Jefferson 2007). Many species-rich hay meadows are protected (Jefferson 2005; Sengl et al. 2016) and their management, and the abundance of key plant species, are subject to review and monitoring, but this does not account for connectivity, particularly in relation to gene flow, to other protected and non-protected sites. This study will examine, for the first time, the degree of connectivity in two contrasting regions, between protected meadows and other grassland sites in the landscape matrix such as roadside verges, common land and non-protected, semi-natural grasslands which are known to support specialist grassland plants (Cousins 2006).
Rhinanthus minor was selected as an ideal study species for this investigation of genetic connectivity in species rich hay meadows. R. minor is an annual species and so will reflect changes in genetic patterns more readily than a long-lived perennial, and it is insect pollinated so pollen dispersal will be more limited than that of wind pollinated species (Westbury 2004). R. minor can be relatively abundant in grasslands with a low intensity management regime but is rarely found in intensively managed grasslands and is therefore an appropriate study species for an investigation of isolated grassland habitats.
This study investigated the level of genetic diversity in protected meadow sites, and analysed gene flow to examine the extent to which protected meadows and landscape matrix sites were functioning as a meta-population. A review of the impact of fragmentation on plant populations has identified a knowledge gap concerning the role of the landscape matrix (Ibáñez et al. 2014). Reference has already been made to the potential part played by linear landscape elements in plant dispersal (Arenas et al. 2017; Thiele et al. 2018) but there is still some debate about the role of these features. Other studies have found that large high-quality habitat patches are required for plant population persistence and that dispersal along linear elements in agricultural landscapes was limited (Soons et al. 2005; Liira et al. 2009). In addition to the role played by linear sites, land-use type and intensity would also be expected to influence the functional connectivity of insect pollinated species, such as R. minor, with more limited nectar and pollen sources in intensively managed arable landscapes compared with low input grasslands (Pfiffner et al. 2018; Hass et al. 2019). The present study takes a comprehensive approach to analysing genetic diversity and connectivity in a hay meadow species by including meadows and landscape matrix sites in two regions with contrasting land-use and topography, an extensively managed upland and an intensively managed lowland landscape. In doing so this study investigated: (1) genetic diversity within populations of R. minor in species-rich hay meadows and landscape matrix sites across the two study regions; (2) evidence of gene flow between populations of R. minor in meadows and landscape matrix sites within the two study areas; (3) effects of geographical distance and landscape variables on genetic connectivity in the two study regions.
Methods
Study species
Rhinanthus minor L. is an annual hemi-parasitic herb which has a widespread distribution in the UK and much of Europe and North America (Westbury 2004). It is found in range of grassland habitats but is most commonly associated with meadows (Coulson et al. 2001). More intensive grassland management, such as that which requires an early cut for silage, limits the ability of seed production so R. minor has seen a decline with the changes in agricultural practices since the middle of the 20th century (Online Atlas of the British Flora 2018). It is a diploid species (2n = 22) and is either insect- or self-pollinated with the main pollinators being Bombus spp. (Natalis and Wesselingh 2012). Seeds do not persist in the seed bank and seed dispersal is poor with most seeds located < 1.5 m from the parent plant (Bullock et al. 2003; Westbury 2004) which makes this a suitable species for research into the impacts of isolation. R. minor has a variable morphology which may be due to ecotypic variations associated with different land use types or may be associated with host type (Westbury 2004). However, Houston and Wolff (2012) did not find any evidence for distinct subspecies. The meadow sites sampled in the study are all managed in a similar way (see ‘study areas’ below) but there are more variations in the management of the landscape matrix sites.
Study areas
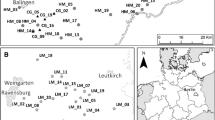
The study areas are the Forest of Bowland, North West England and Worcestershire, Central England (Fig. 1). The Forest of Bowland is an upland area (with the central moorland areas in excess of 500 m above sea level) and is situated at 53°58ʹN, 2°26ʹW with the meadow sites located in valleys. The study sites are set in an area of approximately 20 km × 20 km. The mean annual precipitation for the region is 1294 mm, mean minimum temperature is 6.1 °C and mean maximum temperature is 12.7 °C (Met Office 2018a). The Worcestershire study sites are approximately 200 km south of the Bowland sites in a lowland area situated at approximately 52°08ʹN, 2°13ʹW. This area is approximately 20 km × 30 km. The mean annual precipitation for this region is 606 mm, mean minimum temperature is 6.1 °C and mean maximum temperature is 14.5 °C (Met Office 2018b).

Maps showing location and spatial distribution of sampling sites in a the upland and b the lowland study areas. Meadow sites are represented by open circles and landscape matrix sites by open squares
The two study regions have contrasting land-use and topographical characteristics. In the Bowland (hereafter upland region) sampling areas (Ribble Valley and Lancaster administrative areas) cereals occupy < 1% of the farmed area and permanent grassland makes up > 95%. In contrast, approximately 25% of the farmed area is occupied by cereal crops and 45% by permanent grassland in the areas of Worcestershire (the lowland region) in which the sample sites are located (Worcester and Wychavon; Malvern Hills administrative areas) (Source: DEFRA 2017). There are more protected meadow sites in the lowland region (approximately 30 sites) than in the upland area (9 sites). In the lowland area 9 sites were selected for which management history was known. Sites were also chosen because their distribution had similarities to that of the upland sites with some sites close together and others were more isolated. In both regions all the meadows are managed for a field dried hay crop using a low-input regime, and are mown once a year no earlier than 7 July in the lowland area and after 15 July in the upland area. The meadows are grazed in the autumn in both regions and in the spring in the upland region. Management of the landscape matrix sites varies with some sites being managed as non-protected semi-natural grasslands through mowing and/or grazing, but features such as roadside verges and track edges are not grazed by agricultural livestock.
Sampling design
In the meadow sites leaf samples were collected from individuals spread evenly across the site and at least 5 m apart by collecting one sample from each corner of a 5 m × 5 m quadrat. 20–30 individuals were sampled. Some of the sites have multiple fields in which case each field was sampled. In some sites R. minor was infrequent or field sizes were small, so a smaller number of samples was collected.
Samples were also collected from landscape matrix sites. Landscape matrix sites were identified by surveys of the local area, information provided by local conservation groups and plant records from the Botanical Society of Britain and Ireland (https://bsbi.org/). These landscape matrix sites included other less species-rich agricultural grasslands, commons, roadside verges, a churchyard and other grassland fragments alongside footpaths and tracks. Since many of the landscape matrix sites were linear features, transects were used for sampling. Where the landscape matrix site was close to a main meadow site, the transect was located a minimum of 100 m from the main site. Samples were collected at 5 m intervals along the transect until the boundary of the site was reached and this method was applied to all landscape matrix sites. Estimates of population sizes were made and populations were grouped according to the following categories (> 100 small; 100–1000 medium; > 1000 large). Euclidean distances to the nearest protected meadow site, study site (meadow or landscape matrix site) and the nearest semi-natural grassland site were measured. Semi-natural grasslands were defined and measurements were made using maps of the UK priority grassland habitats inventory (https://magic.defra.gov.uk/MagicMap.aspx). Table 1 gives details of the sampling sites.
DNA extraction and microsatellite analysis
Leaf samples were stored in silica gel prior to DNA analysis. DNA was extracted following the crude sample PCR protocol supplied with the KAPPA3G plant PCR kit (KAPPA Biosystems 2016). An approximately 2.5 × 2.5 mm piece was cut from each leaf sample using sterile forceps and added to 50 μl of an extraction buffer containing: 50 mM Tris–HCl (pH 8.0) and 0.1 mM EDTA along with 2% ß-mercaptoethanol and 1.0 mM TCEP before heating at 95 °C for 5 min.
Six microsatellite primer pairs were used in the analysis. Initial primer testing was carried out on eight primers which were developed for R. minor specifically (Houston and Wolff 2009). Of these, only two (RM20 and RM24) were successfully amplified or found to provide consistent results so testing was also carried out on six primers which were developed for Rhinanthus angustifolius but had also been found to be effective in cross-species amplification (Ducarme et al. 2008). Four of these (RA53, RA75, RA81 and RA87) were used to amplify DNA from R. minor successfully in the present study. RA53, RA75 and RA81 were multiplexed using 6-FAM, ATTO 532 N and ATTO 565 N dyes respectively (Integrated DNA Technologies, Leuven, Belgium). RM20, RM24 and RA87 were tagged with 6-FAM (Integrated DNA Technologies) and were amplified in individual reactions.
PCR was carried out in 10 μl reactions containing: 1.0 μl template DNA, 5.0 μl Bioline My Taq Plant Kit (Bioline Reagents Ltd, London, UK), 0.1 μM of each primer pair and 0.2 μl TCEP. Forward primers were fluorescently tagged. The PCR programs followed those in Houston and Wolff (2009) for RM20 and RM24 and Ducarme et al. (2008) for RA53, RA75, RA81 and RA87.
The PCR products were diluted 1:4 using nuclease-free H2O after which 1.0 μl of the diluted PCR product was added to 8.9 μl formamide and 0.1 μl of Applied Biosystems GeneScan 500 LIZ size standard (Applied Biosystems, Warrington, UK). Fragment analysis was undertaken using capillary electrophoresis on an AB 3500 Genetic Analyzer (Applied Biosystems, Warrington, UK) and scored using Gene Mapper 5.0 software (Applied Biosystems, Warrington, UK) with manual checks carried out for scoring errors. A subset of samples was re-genotyped to test the accuracy of the procedure and the results are available in the Supplementary Information (Table A1) Allele binning was carried out using the TANDEM program (Matschiner and Salzburger 2009).
An analysis was carried out using the POWSIM software (Ryman et al. 2006) to test the power of the microsatellites used in the study to detect population differentiation. FST values were generated using an effective population size of 1000 with 2000 iterations. The tests showed that the probability of detecting FST of 0.015 or greater was 100% in both study regions (Fig A1 Supplementary information). These results are comparable with other studies which have reported POWSIM results for microsatellite power (e.g. Shohami and Nathan 2014; Landaverde-González et al. 2018).
Data analysis
Data analysis was carried out in R version 3.3.2 (R Core Team 2016).
To investigate levels of genetic diversity within populations of R. minor across the two regions several estimates of diversity were made. Raw allelic richness was calculated, along with allelic richness values corrected for sample size by rarefaction using the R package hierfstat (Goudet and Jombart 2015). A summary of the observed number of alleles and their diversity across all loci is included in Table A2 in the Supplementary Information. Estimates were made of observed heterozygosity (Ho) and expected heterozygosity (He) by site (population) and for each study region using the poppr package version 2.3.0 (Kamvar et al. 2014). Populations with less than 5 samples were excluded from these calculations because of the sensitivity of some genetic diversity measures to sample size. Finally, estimates of the levels of inbreeding were made by calculating the inbreeding coefficient (FIS) in the adegenet package (Jombart 2008). R. minor has a mixed mating system so higher values of FIS (indicating a greater degree of inbreeding) could suggest that there was more selfing, or would point to more incidences of mating between closely related individuals due to factors such as poor seed dispersal, low migration rates or a small population size.
To examine the possible drivers of genetic diversity in the studied R. minor populations, generalized linear models, assuming a gaussian distribution, were used to test the effects of population size (small, medium or large), site size (in ha), region and type (meadow or landscape matrix site) on genetic diversity (raw allelic richness). Distances to the nearest protected meadow, nearest study site and semi-natural grassland (see Table 1) were also included in the model. Tests for collinearity resulted in nearest study site and nearest semi natural grassland being eliminated from the model but nearest protected meadow was retained. There was also collinearity between type and population size so population size was retained and type was eliminated because population size provides more information about the sites. The final model tested the effects of population size, region, site size and distance to nearest protected meadow on genetic diversity. The effects were also tested using a generalized least squares model with a spatial covariance structure in order to investigate whether the differences in the spatial distribution of the sampling sites in the two study regions (see Fig. 1a, b) had an effect on genetic diversity. A Gaussian spatial correlation structure was assumed.
Patterns of gene flow between populations were investigated by analysing pairwise FST values for each population (Weir and Cockerham 1984) along with a global estimate of FST in the package hierfstat. A permutational test for significance was carried out using the adegenet package (Jombart 2008). FST is a commonly used measure of population differentiation, and is therefore useful for comparisons with other population genetics studies, but it has been suggested that that FST may over- or under-estimate population differentiation (Jost 2008; Gregorius 2010). Alternative measures have been proposed so Hedrick’s standardised GʺST was also calculated because it accounts for demographic processes such as genetic drift and migration on population structure (Meirmans and Hedrick 2011). Calculations were made in the mmod package (Winter 2012). Private alleles (alleles which are only found in one population) can be used to inform the consideration of gene flow and so were identified using the poppr package (Kamvar et al. 2014).
The investigation of patterns of gene flow was taken further by carrying out a spatial principal components analysis (sPCA) (Jombart et al. 2008) to explore spatial genetic structure of R. minor in the two regions using the package adegenet (Jombart 2008). sPCA analyses allele frequencies and their spatial autocorrelation using Moran’s I on an individual, rather than population, basis and is therefore unaffected by sample size. The analysis allows for a choice of connection networks between individuals. Some networks do not allow for connections between all available sites whilst others are more appropriate if there are data available on pollen transport distances or other dispersal information. These data were not available in the present study so the analysis was performed using inverse-distance weighting. This method assumes that all individuals can be neighbours and that individuals will be less similar as geographical distance increases. sPCA also allows for tests of global and local spatial structure in the genetic data. These tests were performed using the function spca_randtest from the adegenent package (Montano and Jombart 2017). Where there is global structure there is a high degree of spatial autocorrelation and individuals are likely to be genetically similar to their neighbours. The presence of local structure indicates negative autocorrelation and genetic dissimilarity (Warren et al. 2016). The results of the sPCA using the lagged scores were displayed using the colorplot function in the adegenet package. The lagged scores are computed from the average values of scores at neighbouring locations. This has a ‘smoothing’ effect and helps to clarify spatial structures in the data (Jombart 2017). The colorplot function translates each component into a colour from the RGB colour channel such that the different shades of the red, green and blue colour system give an indication of genetic differentiation with similar colours representing genetic similarity. An examination of the extent of variance and spatial autocorrelation displayed by the eigenvalues in the analysis can be made using the package adegenet (Jombart 2017) and this guides the decision as to how many components to retain. The colorplot function allows up to three components to be displayed in one plot but using less than three components limits the range of colours displayed in the plot.
Dispersal limitations were assessed by testing for the presence of isolation by distance (IBD) between populations. Pairwise FST was calculated between populations and these comparisons were used to generate a maximum likelihood population effects model (MLPE) with FST as the dependent variable and pairwise Euclidean geographic distance as a predictor (Clarke et al. 2002). This was fitted using the ‘gls’ function in the R package NLME (Pinheiro et al. 2017), together with the corMLPE package (provided at https://github.com/nspope/corMLPE). The latter package allows for the specification of a mixed model, accounting for the lack of independence between pairwise observations from the same populations. To that end two variables indicating the populations associated with each observation were included as random effects terms. As well as IBD, land use and topography can produce spatially-structured genetic variation by resisting gene flow between populations (e.g. Cushman et al. 2006). Bombus species, which are the main pollinators of Rhinanthus spp. (Natalis and Wesselingh 2012), are much studied in this regard and therefore landscape resistance to gene flow was assessed following Jha (2015), with some modifications. The potential drawbacks and difficulties of generating resistance surfaces that describe this phenomenon are well documented elsewhere (Spear et al. 2010; Richardson et al., 2016). Therefore, a pre-existing method already demonstrated as capable of detecting barriers to gene flow is of obvious benefit, as bee movements should be correlated with pollen dispersal in R. minor.
Briefly, then, two surfaces were generated for each area, describing both land use and elevation and their hypothesised resistances to gene flow. Raw data for these were processed in the software QGIS 2.18 and final surfaces were calculated in the R package raster 2.6-7 (QGIS Development Team 2016; Hijmans 2017). Digital terrain models (DTM) were used to calculate elevation relative to the maximum altitude within the sampled areas overall (Ordnance Survey (GB) 2018a), and vector maps were used to categorise land use as high (open water or wetland, urban areas, arable land) or low (semi-natural open areas, woodland) resistance (Ordnance Survey (GB) 2018b).These four surfaces were used to generate pairwise resistance distances (RD) in the software Circuitscape 4.0.5 (https://circuitscape.org) (McRae 2006). Resolution of all rasters was 50 m. Detection of landscape resistance to gene flow was done in the same way as for IBD, with these pairwise RD used as predictors in a MLPE model, including associated populations as random effects.
Results
Genetic diversity
Genetic diversity parameters are shown in Table 2. Values for heterozygosity (He) based on means by populations and based on overall allele frequencies were higher in the upland region (0.35 and 0.48) than the lowland region (0.29 and 0.44). Measures of genetic diversity were generally higher in the meadow sites than in the landscape matrix sites. For example, mean He in the meadows was 0.38 for the upland region and 0.34 for the lowland region but in the landscape matrix sites the estimates were 0.29 (upland) and 0.23 (lowland). Allelic richness values were also higher for the meadow sites in both regions but this measure was affected by the differences in sample sizes to some extent. Estimates of inbreeding were high in both regions with the upland region having a global FIS of 0.67 compared to 0.56 in the lowland region.
The generalized linear model revealed that (see Table 3) population size had a significant positive effect on allelic diversity (t = 3.78, P < 0.001). No other effects were found to be significant. The generalized least squares model showed very similar results after the spatial distribution of sites had been taken into account (see Table 3).
Population differentiation and spatial analysis
Global FST values were lower for the upland region than the lowland region populations but indicated that gene flow was limited in both regions, a finding that was confirmed by high values for GʺST (Table 4). FST values were significantly different from zero for both regions (P < 0.001 in both cases). Analysis of pairwise FST indicated that mean pairwise values for all sites were similar to those between meadows and landscape matrix sites in both regions (Table 4) and revealed population differentiation, although this was more pronounced in the lowland region. These results suggest that there is limited genetic connectivity between all populations and between meadows and landscape matrix sites. Sites with some identical genotypes (e.g. BM04), or very small populations (e.g. WL05) had very high pairwise FST values (> 0.4). The full matrices of pairwise FST are in Table A3 and A4 in the Supplementary Material.). Private alleles were identified in two of the meadow populations and two of the landscape matrix sites in the upland region. In the lowland sites there were more private alleles and they were found in six of the meadows and two of the landscape matrix sites, supporting the finding that there is a greater degree of population differentiation in the lowland region that the upland region.
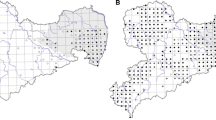
Permutation tests on the sPCA for the upland region data indicated that there was significant global structure (λ = 0.60, P = 0.002) but no local structure (λ = 0.02, P = 1). In the lowland region the permutation test also revealed significant global structure, and with a higher degree of variance explained by spatial patterns (λ = 0.84, P = 0.002) but no indication of local structure (λ = 0.009, P = 1). The colorplots (Fig. 2a, b) confirm the global structure results, although this is more pronounced in the lowland region (Fig. 2a). Here, there is little evidence of clustering of similar colours indicating a high degree of genetic dissimilarity and restricted gene flow. In the upland region (Fig. 2b) there are some clusters of similar colours, particularly in the large group of sites in the south east of the region where there appears to be genetic similarity indicating gene flow between sites. These sites include meadows and landscape matrix sites. Figure 2a is based on the components associated with the first three positive eigenvalues and Fig. 2b is based on the components associated with first two positive eigenvalues.

Spatial principal components analysis colorplots showing the lagged scores of a the first three components in the lowland area and b the first two components in the upland area. Both analyses were performed using inverse distance weighting and both revealed significant global structure (denoted by λ). Plots are overlain on a landcover map
MLPE models did not detect isolation by distance between populations in either the upland or lowland region (Fig. 3); Euclidean geographic distance between locations was not a significant predictor of genetic differentiation as FST (Table 5). Similarly, the examined landscape factors did not affect gene flow between populations, as pairwise resistance distances derived from both relative elevation and land use were not significant predictors of FST between locations (Table 5 and Fig. 4).

Graphical representation of the relationship between pairwise Euclidean geographic distances and pairwise FST. The scatterplots show results rom the MLPE analysis of isolation by distance in a the upland region and b the lowland region

Graphical representation of the relationship between pairwise resistance and pairwise FST. The scatterplots show results from the MLPE analysis of a land cover resistance in the upland region and b land cover resistance the lowland region and c elevation resistance in the upland region and d elevation resistance in the lowland region
Discussion
This study investigated genetic diversity and connectivity in populations of R. minor across two contrasting regions. Genetic diversity was similar to other studies of R. minor, and population size was found to be a significant predictor of genetic diversity. Gene flow was limited in both regions, but the analysis did not find distance, land cover or elevation to be significant predictor of population differentiation.
Genetic diversity
Global estimates of genetic diversity in the two regions (He 0.48 upland; He 0.44 lowland) were similar to a study of R. minor populations across the UK in which He ranged from 0.35 to 0.53 (Houston and Wolff 2012). They were also comparable with the He of 0.46 for annual species reported by Nybom (2004) in a wide-ranging review of genetic diversity in plants. However, the upland and lowland regions results were lower than the He of 0.60 for species with a mixed breeding system reported in the same study. Regional He estimates of an annual hemi-parasitic plant with a mixed breeding system, Melampyrum sylvaticum, were 0.20 and 0.56 with the lower value relating to samples from small isolated populations and the higher value relating to large populations (Crichton et al. 2016). The low values of observed heterozygosity could be explained by the high levels of inbreeding, a finding which was also reported in other studies of R. minor (Ducarme and Wesselingh 2013; Talve et al. 2013; Hargreaves et al. 2015) although Houston and Wolff (2012)’s UK-wide study reported lower values of inbreeding FIS 0–0.44). Inbreeding is predicted to lead to a reduction in fitness and to be more problematic in small populations where individuals are more likely to mate with others with a recent common ancestry (Lienert 2004; Young et al. 1996).
Low numbers of pollinators could explain the high rates of inbreeding, and by implication, selfing rates, but selfing has been shown to occur in R. minor when pollinators were recorded as present (Hargreaves et al. 2015). Some species of Bombus have been shown to trigger self-pollination according to how they land on, and enter, the R. minor flower (Kwak 1979; Westbury 2004). It is possible that these pollinators, which visit the flower sternotribically (i.e. landing on the upper lip of the flower with pollen being deposited on the underside of the animal), were more abundant at the sites with higher rates of selfing. Observations of pollinator behaviour and records of abundance would be required to confirm whether selfing was associated with a lack of pollinators or a higher proportion of sternotrobic species.
The analysis of possible drivers of genetic diversity found that population size had a significantly positive effect on allelic richness, a finding which has been widely reported in previous studies (Van Rossum et al. 2004; Leimu et al. 2006; Honnay and Jacquemyn 2007). Study region was not a significant predictor of allelic richness, even when the spatial structure of the populations was taken into account, despite the differences in the distribution of the study sites in the two regions. Similarly, there was no impact of distance to the nearest protected meadow on allelic richness. Reisch et al. (2017) found that the effects of the process of isolation of species rich grasslands may not yet have been realised in terms of the loss of genetic diversity though their study measured the effects of isolation on long-lived perennial plants, and it would be expected that change would be more rapid in annuals like R. minor.
Evidence of gene flow and spatial analysis
Estimates of FST revealed population differentiation in both regions but this was more marked in the lowland region (FST = 0.28) than in the upland region (FST = 0.19), and this was confirmed by high levels of GʺST. Houston and Wolff (2012) reported FST = 0.16 in their study of R. minor in populations in the UK which was comparable to the upland region estimate. Pairwise FST values indicated that levels of gene exchange were limited between landscape matrix sites and meadows (Table 3) which suggests a lack of genetic connectivity across the landscape matrix regardless of site type. This contrasts with other studies of perennial grassland plant species which included grasslands and linear landscape elements such as ditches and verges, and which reported evidence of gene flow across the different sites, even in intensively managed landscapes (Mix et al. 2006; Aavik et al. 2013). However, these studies did highlight a lack of recent migration between sites (Aavik et al. 2013) and the importance of wind dispersal in gene flow (Mix et al. 2006). The lack of connectivity was more pronounced in the lowland area where there were also higher numbers of private alleles indicating greater population differentiation, although this may have been influenced by different sampling approaches in the meadows and landscape matrix sites. The findings were reflected in the sPCA analysis which confirmed that there was significant global structure in both regions, and more particularly in the lowland region.
The examination of the effects of geographical distance elevation and land cover indicated some differences between the two regions in terms of elevation and land cover but these were not statistically significant (Table 5). Research into the effects of landscape features on gene flow has shown that altitudinal differences, even at substantially greater elevations than in the upland region, were not always a barrier to pollinators (Hargreaves et al. 2015; Kamm et al. 2010). The lowland region landscape has less varied topography, although there are other potential physical barriers including major rivers such as the River Severn which divides the north eastern and south western sampling sites. Investigations into the barriers of landscape features to bumblebee movements have found that water bodies do limit movement to some extent (Jha 2015; Lozier et al. 2013) as do artificial barriers such as roads and railways (Bhattacharya et al. 2003) but the analysis did not show such landscape features to be a significant influence in the present study.
It has been found that Bombus species (the principal pollinators of R. minor) were more abundant in landscapes with a diversity of habitats including semi-natural grasslands than they were in simple landscapes with large fields and no permanent pasture (Persson and Smith 2013). There are more protected meadows in the area included in the lowland region than in the upland area but these only account for a fraction of the land surface and the lowland region is characterised by a much more intensively farmed landscape than the upland region. It was, therefore, expected that land use would be a predictor of population differentiation, but this was not reflected in the MLPE results (Table 5 and Figs. 3, 4). Another possible factor which could affect genetic differentiation is that of ecotypic variation. Although the meadow sites in the two regions are managed in a similar way and are subject to standard agri-environment scheme management prescriptions there will be local differences in site characteristics and the management of some landscape matrix sites will differ from that of the meadows, e.g. in that there is no grazing on roadside verges. The Rhinanthus genus is known to be very variable and to have recognisable ecotypes (Zopfi 1993) which could affect patterns of genetic differentiation, possibly via pollinator behaviour related to recognition. More detailed investigations into all the possible landscape and management variables which could affect population differentiation would be valuable in further research in this area (Holderegger et al. 2010).
There was no evidence of a significant relationship between geographical distance and population differentiation across either of the two regions (Table 5 and Fig. 3). However, the colorplot for the upland region (Fig. 2b) indicates that there may be local gene flow in the cluster of the sites in the south east of the region. The mean pairwise FST for the meadows in this cluster is 0.08 and is 0.12 for all the sites in the cluster (see Supplementary Information Table A4). It may be that there is a threshold of site proximity and density which is important for conservation, but which is difficult to extract from regional analyses of isolation by distance. This pattern in the upland region may also explain why global FST and the results of the sPCA indicate relatively less population differentiation in the upland than the lowland region.
It is possible that some gene flow between populations of R. minor may be attributable to seed dispersal as well as to pollination. Reference has already been made to the fact that seed dispersal is poor and most seeds fall within 1.5 m of the parent plant (see methods: study species). However, seeds of R. minor have been shown to be dispersed on the clothing of people working in meadows (Auffret and Cousins 2013) and via animal fur (Hovstad et al. 2009). Seeds can also be dispersed by vehicles and farm machinery (Strykstra et al. 1997). It is not possible to quantify what proportion of gene flow is due to seed dispersal by any of these methods without further research but this should be taken account in conservation strategies to address limited abundance or movement of pollinators.
Further research on genetic diversity and connectivity in wind-pollinated meadow species, and in longer-lived perennial species, will be of value to investigate whether these types of species are being similarly affected by fragmentation of this important habitat. For example, studies by Münzbergová et al. (2012), Aavik et al. (2013), Takkis et al. (2013) emphasised the influence of past landscapes and connectivity patterns on current genetic connectivity in grassland perennials and a wind pollinated grassland species. Findings like this suggest that time lags in response to landscape change should be considered in conservation management for perennial meadow species and indicate that the results from the present study of an annual species, R. minor, may give a more immediate representation of the impacts of fragmentation.
Conclusions and implications for conservation
This study has shown that populations of a key meadow species, R. minor have intermediate levels of genetic diversity which are comparable with other studies of this species, and that they have relatively high levels of inbreeding. There is evidence that gene flow is limited in both regions and that there is a low level of connectivity between meadows and landscape matrix sites, with the exception of one area in the upland region where site density and proximity is higher and gene flow is less restricted. The lowland region is more intensively managed and developed, and land use was expected to limit gene flow but analyses of the predictors of population differentiation did not find any significant effects associated with distance between sites, land cover or elevation. Nevertheless, conservation should focus on maintaining large populations and enhancing and creating landscape matrix sites, along with nectar sources and habitat for pollinating species if genetic diversity and fitness are to be sustained in this key meadow species. The results of this study support recent conservation policy approaches advocating bigger and more connected habitats (e.g., Lawton et al. 2010) but more detailed research is needed to understand the barriers to gene flow at the landscape-scale, and to clarify the density and proximity of populations needed to support functional connectivity in habitats of high conservation value.
References
Aavik T, Holderegger R, Edwards PJ, Billeter R (2013) Patterns of contemporary gene flow suggest low functional connectivity of grasslands in a fragmented agricultural landscape. J Appl Ecol 50:395–403. https://doi.org/10.1111/1365-2664.12053
Aguilar R, Quesada M, Ashworth L, Herrerias-diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188. https://doi.org/10.1111/j.1365-294X.2008.03971.x
Arenas JM, Escudero A, Mola I, Casado MA (2017) Roadsides: an opportunity for biodiversity conservation. J Veg Sci 20(4):527–537. https://doi.org/10.1111/avsc.12328
Auffret AG, Cousins SAO (2013) Humans as long-distance dispersers of rural plant communities. PLoS ONE 8(5):e62763. https://doi.org/10.1371/journal.pone.0062763
Auffret AG, Plue J, Cousins SAO (2015) The spatial and temporal components of functional connectivity in fragmented landscapes. Ambio 44(Suppl. 1):S51–S59. https://doi.org/10.1007/s13280-014-0588-6
Bhattacharya M, Primacka RB, Gerweinb J (2003) Are roads and railroads barriers to bumblebee movement in a temperate suburban conservation area? Biol Conserv 109:37–45. https://doi.org/10.1016/S0006-3207(02)00130-1
Bullock JM, Moy IL, Coulson SJ, Clarke RT (2003) Habitat-specific dispersal: environmental effects on the mechanisms and patterns of seed movement in a grassland herb Rhinanthus minor. Ecography 26:692–704. https://doi.org/10.1034/j.1600-0587.2003.03525.x
Clarke RT, Rothery P, Raybould AF (2002) Confidence limits for regression relationships between distance matrices: estimating gene flow with distance. J Agric Biol Environ Stat 7:361–372. https://doi.org/10.1198/108571102320
Cook CN, Sgrò CM (2016) Aligning science and policy to achieve evolutionarily enlightened conservation. Conserv Biol 31(3):501–512. https://doi.org/10.1111/cobi.12863
Coulson SJ, Bullock JM, Stevenson MJ, Pywell RF (2001) Colonization of grassland by sown species: dispersal versus microsite limitation in responses to management. J Appl Ecol 38:204–216. https://doi.org/10.1046/j.1365-2664.2001.00585.x
Cousins SAO (2006) Plant species richness in midfield islets and road verges—the effect of landscape fragmentation. Biol Conserv 127:500–509. https://doi.org/10.1016/j.biocon.2005.09.009
Crichton RJ, Dalrymple SE, Woodin SJ, Hollingsworth PM (2016) Conservation genetics of the annual hemiparasitic plant Melampyrum sylvaticum (Orobanchaceae) in the UK and Scandinavia. Conserv Genet 17:547–556. https://doi.org/10.1007/s10592-015-0803-4
Critchley CNR, Burke MJW, Stevens DP (2003) Conservation of lowland semi-natural grasslands in the UK: a review of botanical monitoring results from agri-environment schemes. Biol Conserv 115:263–278. https://doi.org/10.1016/S0006-3207(03)00146-0
Crofts A, Jefferson RG (eds) (2007) The lowland grassland management handbook, 2nd edn. English Nature/The Wildlife Trusts. http://publications.naturalengland.org.uk/publication/35034. Accessed 22 Sept 2018
Cushman SA, McKelvey KS, Hayden J, Schwartz MK (2006) Gene flow in complex landscapes: testing multiple hypotheses with causal modeling. Am Nat 168:486–499
DEFRA (Department for the Environment, Food and Rural Affairs) (2017) Structure of the agricultural industry in England and the UK at June. https://www.gov.uk/government/statistical-data-sets/structure-of-the-agricultural-industry-in-england-and-the-uk-at-june. Accessed 22 Sept 2018
Ducarme V, Wesselingh RA (2013) Outcrossing rates in two self-compatible, hybridising Rhinanthus species: implications for hybrid formation. Plant Biol 15:541–547. https://doi.org/10.1111/j.1438-8677.2012.00668.x
Ducarme V, Risterucci AM, Wesselingh RA (2008) Development of microsatellite markers in Rhinanthus angustifolius and cross-species amplification. Mol Ecol Resour 8:384–386. https://doi.org/10.1111/j.1471-8286.2007.01966.x
Eigenbrod F, Adams W, Hill C, Macgregor N, Osborne P, Clarke D, Sandbrook L, Hodge I, Steyl I, Thompson A, Van Dijk N, Watmough G (2016) A review of large-scale conservation in England, Scotland and Wales. Natural England Joint Publication JP019. http://publications.naturalengland.org.uk/publication/5762035722223616. Accessed 25 Sept 2018
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242. https://doi.org/10.1146/annurev.es.24.110193.001245
Goudet J, Jombart T (2015) hierfstat: estimation and tests of hierarchical F-statistics. R package version 0.04-22. https://CRAN.R-project.org/package=hierfstat. Accessed: 22 Sept 2018
Gregorius H-R (2010) Linking diversity and differentiation. Diversity 2:370–394. https://doi.org/10.3390/d2030370
Hargreaves AL, Weiner JL, Eckert CG (2015) High-elevation range limit of an annual herb is neither caused nor reinforced by declining pollinator service. J Ecol 103:572–584. https://doi.org/10.1111/1365-2745.12377
Hass AL, Brachmann L, Batáry P, Clough Y, Behling H, Tscharntke T (2019) Maize-dominated landscapes reduce bumblebee colony growth through pollen diversity loss. J Appl Ecol 56:294–304. https://doi.org/10.1111/1365-2664.13296
Hijmans RJ (2017) raster: geographic data analysis and modeling https://cran.r-project.org/web/packages/raster/index.html. Accessed 26 Apr 2019
Hodgson JG, Grime JP, Wilson PJ, Thompson K, Band SR (2005) The impacts of agricultural change (1963–2003) on the grassland flora of Central England: processes and prospects. Basic Appl Ecol 6:107–118. https://doi.org/10.1016/j.baae.2005.01.009
Holderegger R, Buehler D, Gugerli F, Manel S (2010) Landscape genetics of plants. Trends Plant Sci 15(12):675–683. https://doi.org/10.1016/j.tplants.2010.09.002
Honnay O, Jacquemyn H (2007) Susceptibility of common and rare plant species to the genetic consequences of habitat fragmentation. Conserv Biol 21(3):823–831. https://doi.org/10.1111/j.1523-1739.2006.00646.x
Hooftman DAP, Bullock JM (2012) Mapping to inform conservation: a case study of changes in semi-natural habitats and their connectivity over 70 years. Biol Conserv 145:30–38. https://doi.org/10.1016/j.biocon.2011.09.015
Hooftman DAP, van Kleunen M, Diemer M (2003) Effects of habitat fragmentation on the fitness of two common wetland species, Carex davalliana and Succisa pratensis. Oecologia 134:350–359. https://doi.org/10.1007/s00442-002-1096-0
Houston K, Wolff K (2009) Eight polymorphic microsatellite markers for Rhinanthus minor. Mol Ecol Resour 9:174–176. https://doi.org/10.1111/j.1755-0998.2008.02415.x
Houston K, Wolff K (2012) Rhinanthus minor population genetic structure and subspecies: potential seed sources of a keystone species in grassland restoration projects. Perspect Plant Ecol Evol Syst 14:423–433. https://doi.org/10.1016/j.ppees.2012.08.001
Hovstad KA, Borvik S, Ohlson M (2009) Epizoochorous seed dispersal in relation to seed availability—an experiment with a red fox dummy. J Veg Sci 20:455–464. https://doi.org/10.1111/j.1654-1103.2009.01049.x
Huber S, Huber B, Stahl S, Schmid C, Reisch C (2017) Species diversity of remnant calcareous grasslands in south eastern Germany depends on litter cover and landscape structure. Act Oecol 83:48–55. https://doi.org/10.1016/j.actao.2017.06.011
Ibáñez I, Katz DSW, Peltier D, Wolf SM, Barrie BTC (2014) Assessing the integrated effects of landscape fragmentation on plants and plants communities: the challenge of multiprocess-multiresponse dynamics. J Ecol 102:882–895. https://doi.org/10.1111/1365-2745.12223
Jacquemyn H, Roldán-Ruiz I, Honnay O (2010) Evidence for demographic bottlenecks and limited gene flow leading to low genetic diversity in a rare thistle. Conserv Genet 11:1979–1987. https://doi.org/10.1007/s10592-010-0089-5
Jefferson RG (2005) The conservation management of upland hay meadows in Britain: a review. Grass Forage Sci 60:322–331. https://doi.org/10.1111/j.1365-2494.2005.00489.x
Jha S (2015) Contemporary human-altered landscapes and oceanic barriers reduce bumble bee gene flow. Mol Ecol 24:993–1006. https://doi.org/10.1111/mec.13090
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T (2017) An introduction to adegenet 2.1.0. https://github.com/thibautjombart/adegenet/wiki/Tutorials. Accessed: 22 Sept 2018
Jombart T, Devillard S, Dufour A-B, Pontier D (2008) Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101:92–103. https://doi.org/10.1038/hdy.2008.34
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Jump AS, Marchant R, Peñuelas J (2008) Environmental change and the option value of genetic diversity. Trends Plant Sci 14(1):51–58. https://doi.org/10.1016/j.tplants.2008.10.002
Kahilainen A, Puurtinen M, Kotiaho JS (2014) Conservation implications of species–genetic diversity correlations. Glob Ecol Conserv 2:315–323. https://doi.org/10.1016/j.gecco.2014.10.013
Kamm U, Gugerli F, Rotach P, Edwards P, Holderegger R (2010) Open areas in a landscape enhance pollen-mediated gene flow of a tree species: evidence from northern Switzerland. Landsc Ecol 25:903–911. https://doi.org/10.1007/s10980-010-9468-z
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
KAPPA Biosystems (2016) KAPPA 3G Plant PCR kit: technical data sheet. https://www.kapabiosystems.com/product-applications/products/pcr-2/kapa3g-plant-pcr-kits/. Accessed 22 Sept 2018
Krauss J, Klein A, Steffan-Dewenter I, Tscharntke T (2004) Effects of habitat area, isolation, and landscape diversity on plant species richness of calcareous grasslands. Biodivers Conserv 13:1427–1439. https://doi.org/10.1023/B:BIOC.0000021323.18165.58
Kwak MM (1979) Effects of bumblebee visits on the seed set of Pedicularis, Rhinanthus and Melampyrum (Scrophulariaceae) in the Netherlands. Acta Botanica Neerlandica 28(2–3):177–195. https://doi.org/10.1111/j.1438-8677.1979.tb00333.x
Laikre L, Allendorf FW, Aroner LC, Baker CS, Gregovich DP et al (2010) Neglect of genetic diversity in implementation of the convention on biological diversity. Conserv Biol 24(1):86–88
Landaverde-González P, Baltz LM, Escobedo-Kenefic N, Mérida J, Paxton RJ, Husemann M (2018) Recent low levels of differentiation in the native Bombus ephippiatus (Hymenoptera: Apidae) along two Neotropical mountain ranges in Guatemala. Biodivers Conserv 27:3513–3531. https://doi.org/10.1007/s10531-018-1612-0
Lawton JH, Brotherton PNM, Brown VK, Elphick C, Fitter AH, Forshaw J, Haddow RW, Hilborne S, Leafe RN, Mace GM, Southgate MP, Sutherland WJ, Tew TE, Varley J, Wynne GR (2010) Making space for nature: a review of England’s wildlife sites and ecological network. Report to Defra. http://webarchive.nationalarchives.gov.uk/20130402170324/http://archive.defra.gov.uk/environment/biodiversity/documents/201009space-for-nature.pdf. Accessed 22 Sept 2018
Leimu R, Mutikainen P, Koricheva J, Fischer M (2006) How general are positive relationships between plant population size, fitness and genetic variation? J Ecol 94:942–952. https://doi.org/10.1111/j.1365-2745.2006.01150.x
Lienert J (2004) Habitat fragmentation effects on fitness of plant populations—a review. J Nat Conserv 12:53–72. https://doi.org/10.1016/j.jnc.2003.07.002
Liira J, Schmidt T, Aavik T, Arens P, Augenstein I, Bailey D, Billeter R, Bukáček R, Burel F, De Blust G, De Cock R et al (2009) Plant functional group composition and large-scale species richness in European agricultural landscapes. J Veg Sci 19:3–14. https://doi.org/10.3170/2007-8-18308
Lozier JD, Strange JP, Koch JB (2013) Landscape heterogeneity predicts gene flow in a widespread polymorphic bumble bee, Bombus bifarius (Hymenoptera: Apidae). Conserv Genet 14:1099–1110. https://doi.org/10.1007/s10592-013-0498-3
Matschiner M, Salzburger W (2009) TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics 25(15):1982–1983. https://doi.org/10.1093/bioinformatics/btp303
McRae BH (2006) Isolation by resistance. Evolution 60:1551–1561
Meirmans PG, Hedrick PW (2011) Assessing population structure: F ST and related measures. Mol Ecol Resour 11:5–18. https://doi.org/10.1111/j.1755-0998.2010.02927.x
Met Office (2018a) Stonyhurst climate station https://www.metoffice.gov.uk/public/weather/climate/gcw4rg5df. Accessed 30 July 2018
Met Office (2018b) Pershore climate station https://www.metoffice.gov.uk/public/weather/climate/gcq2vqb9v. Accessed 30 July 2018
Mijangos JL, Pacioni C, Spencer PBS, Craig MD (2015) Contribution of genetics to ecological restoration. Mol Ecol 24:22–37. https://doi.org/10.1111/mec.12995
Mix C, Arens PFP, Rengelink R, Smulders MJM, Van Groenendael M, Ouborg NJ (2006) Regional gene flow and population structure of the wind-dispersed plant species Hypochaeris radicata (Asteraceae) in an agricultural landscape. Mol Ecol 15:1749–1758. https://doi.org/10.1111/j.1365-294X.2006.02887.x
Montano V, Jombart T (2017) An Eigenvalue test for spatial principal component analysis. BMC Bioinform 18:562. https://doi.org/10.1186/s12859-017-1988-y
Münzbergová Z, Cousins SAO, Herben T, Plačková I, Mildén M, Ehrlén J (2012) Historical habitat connectivity affects current genetic structure in a grassland species. Plant Biol 15:195–202. https://doi.org/10.1111/j.1438-8677.2012.00601.x
Natalis LC, Wesselingh RA (2012) Shared pollinators and pollen transfer dynamics in two hybridizing species, Rhinanthus minor and R. angustifolius. Oecologica 170:709–721. https://doi.org/10.1007/s00442-012-2346-4
Natural England (2016) Conservation 21: natural England’s conservation strategy for the 21st century https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/562046/conservation-21.pdf. Accessed 22 Sept 2018
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155. https://doi.org/10.1111/j.1365-294X.2004.02141.x
Öckinger E, Lindborg R, Sjödin NE, Bommarco R (2012) Landscape matrix modifies richness of plants and insects in grassland fragments. Ecography 35:259–267. https://doi.org/10.1111/j.1600-0587.2011.06870.x
Online Atlas of the British Flora (2018) http://www.brc.ac.uk/plantatlas/plant/rhinanthus-minor. Accessed 23 Sept 18
Ordnance Survey (GB) (2018a) OS Terrain 5 [ASC geospatial data] [WWW Document]. EDINA Digimap Ordnance Survey Service. https://digimap.edina.ac.uk. Accessed 4 Jan 19
Ordnance Survey (GB) (2018b) OS VectorMap® Local [SHAPE geospatial data] [WWW Document]. EDINA Digimap Ordnance Survey Service. https://digimap.edina.ac.uk. Accessed 4 Jan 2019)
Persson AS, Smith HG (2013) Season persistence of bumblebee populations is affected by landscape context. Agric Ecosyst Environ 165:201–209. https://doi.org/10.1016/j.agee.2012.12.008
Pfiffner L, Ostermaier M, Stoeckli S, Müller A (2018) Wild bees respond complementarily to ‘high-quality’ perennial and annual habitats of organic farms in a complex landscape. J Insect Conserv 22:551–562. https://doi.org/10.1007/s10841-018-0084-6
Picó FX, Van Groenendael J (2007) Large-scale plant conservation in European semi-natural grasslands: a population genetic perspective. Divers Distrib 13:920–926. https://doi.org/10.1111/j.1472-4642.2007.00349.x
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team (2017) nlme: linear and nonlinear mixed effects models https://cran.r-project.org/web/packages/nlme/index.html. Accessed 26 Apr 2019
Purschke O, Sykes MT, Poschlod P, Michalski SG, Römermann C, Durka W, Kühn I, Prentice HC (2014) Interactive effects of landscape history and current management on dispersal trait diversity in grassland plant communities. J Ecol 102:437–446. https://doi.org/10.1111/1365-2745.12199
QGIS Development Team (2016) QGIS Geographic Information System. https://qgis.org/en/site/. Accessed 26 Apr 2019
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Ralls K, Ballou JD, Dudash MR, Eldridge MDB, Fenster CB, Lacy RC, Sunnucks P, Frankham RF (2018) Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett 11(2):1–6
Reed J, van Vianen J, Barlow J, Sutherland T (2017) Have integrated landscape approaches reconciled societal and environmental issues in the tropics? Land Use Policy 63:481–492. https://doi.org/10.1016/j.landusepol.2017.02.021
Reisch C, Schmidkonz S, Meier K, Schöpplein Q, Meyer C, Hums C, Putz C, Schmid C (2017) Genetic diversity of calcareous grassland plant species depends on historical landscape configuration. BMC Ecol 17:19. https://doi.org/10.1186/s12898-017-0129-9
Richardson JL, Brady SP, Wang IJ, Spear SF (2016) Navigating the pitfalls and promise of landscape genetics. Mol Ecol 25:849–863. https://doi.org/10.1111/mec.13527
Ryman N, Palm S, André C, Carvalho GR, Dahlgren TG, Jorde PE, Laikre L, Larsson LC, Palmé A, Ruzzante DE (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:2031–2045
Schlaepfer DR, Braschler B, Rusterholz HP, Baur B (2018) Genetic effects of anthropogenic habitat fragmentation on remnant animal and plant populations: a meta-analysis. Ecosphere 9(10):02488. https://doi.org/10.1002/ecs2.2488
Schmucki R, Reimark J, Lindborg R, Cousins SAO (2012) Landscape context and management regime structure plant diversity in grassland communities. J Ecol 100:1164–1173. https://doi.org/10.1111/j.1365-2745.2012.01988.x
Sengl P, Magnes M, Wagner V, Erdős L, Berg C (2016) Only large and highly-connected semi-dry grasslands achieve plant conservation targets in an agricultural matrix. Tuxenia 36:167–190
Shohami D, Nathan R (2014) Fire-induced population reduction and landscape opening increases gene flow via pollen dispersal in Pinus halepensis. Mol Ecol 23:70–81
Simeonova V, Bouwma I, van der Grift E, Sunyer C, Manteiga L, Külvik M, Suškevičs M, Dimitrov S, Dimitrova A (2017) Natura 2000 and spatial planning. Final report for the European Commission (DG ENV) (Project 07.0202/2015/716477/ETU/ENV. B.3)
Soons MB, Messelink JH, Jongejans E, Heil GW (2005) Habitat fragmentation reduces grassland connectivity for both short-distance and long-distance wind-dispersed forbs. J Ecol 93:1214–1225
Spear SF, Balkenhol N, Fortin M-J, McRae BH, Scribner K (2010) Use of resistance surfaces for landscape genetic studies: considerations for parameterization and analysis. Mol Ecol 19:3576–3591. https://doi.org/10.1111/j.1365-294X.2010.04657.x
Strykstra RJ, Verweij GL, Bakker JP (1997) Seed dispersal by mowing machinery in a Dutch brook valley system. Acta Bot Neerl 46:387–401. https://doi.org/10.1111/plb.1997.46.4.387
Takkis K, Pärtel M, Saar L, Helm A (2013) Extinction debt in a common grassland species: immediate and delayed responses of plant and population fitness. Plant Ecol 214:953–963. https://doi.org/10.1007/s11258-013-0221-y
Talve T, McGlaughlin ME, Helenurm K, Wallace LE, Oja T (2013) Population genetic diversity and species relationships in the genus Rhinanthus L. based on microsatellite markers. Plant Biol 16:495–502. https://doi.org/10.1111/plb.12057
Thiele J, Schirmel J, Buchholz S (2018) Effectiveness of corridors varies among phytosociological plant groups and dispersal syndromes. PLoS ONE 13(7):e0199980. https://doi.org/10.1371/journal.pone.0199980
Van Rossum S, Campos de Souza S, Triest L (2004) Genetic consequences of habitat fragmentation in an agricultural landscape on the common Primula veris, and comparison with its rare congener, P. vulgaris. Conserv Genet 5:231–245
Warren MJ, Wallin D, Beausoleil RA, Warheit KI (2016) Forest cover mediates genetic connectivity of northwestern cougars. Conserv Genet 17:1011–1024. https://doi.org/10.1007/s10592-016-0840-7
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Westbury DB (2004) Rhinanthus minor L. J Ecol 92:906–927. https://doi.org/10.1111/j.0022-0477.2004.00929.x
Winter DJ (2012) MMOD: an R library for the calculation of population differentiation statistics. Mol Ecol Resour 12:1158–1160. https://doi.org/10.1111/j.1755-0998.2012.03174.x
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418. https://doi.org/10.1016/0169-5347(96)10045-8
Acknowledgements
The authors would like to thank the farmers and landowners who kindly provided access to the study sites. They would also like to acknowledge the advice and support given by the Forest of Bowland Haytime Team and the Worcestershire Wildlife Trust. Thanks also go to field and lab assistants, particularly Ashley Tuffin, Heather Wickson, Katherine Judson and Mark Ashton, and to Sue Jones for help with mapping. The project was made possible by funding from Edge Hill University, Natural England, the Botanical Society of Britain and Ireland and the Natural History Museum’s Botanical Research Fund. Thanks to three anonymous reviewers for suggestions which significantly improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by David Hawksworth.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sullivan, E.R., Barker, C., Powell, I. et al. Genetic diversity and connectivity in fragmented populations of Rhinanthus minor in two regions with contrasting land-use. Biodivers Conserv 28, 3159–3181 (2019). https://doi.org/10.1007/s10531-019-01811-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-019-01811-x