
Abstract
Background
This phase I study investigated the safety and the maximum tolerated dose (MTD) of the oral fluoropyrimidine S-1 when combined with epirubicin and oxaliplatin (EOS).
Methods
Patients aged ≥18 years with advanced or metastatic solid tumors were enrolled in a 3 + 3 design with S-1 dose escalation (two planned cohorts) performed according to the occurrence of dose-limiting toxicity (DLT). On day 1 of each 21-day cycle, patients received epirubicin 50 mg/m2 followed by oxaliplatin 130 mg/m2 (maximum 8 cycles) and then S-1 [20 mg/m2 (cohort 1) or 25 mg/m2 (cohort 2), twice daily]: first dose, evening of day 1; subsequent administration on days 2–14, twice daily; last dose, morning of day 15 (unlimited number of S-1 cycles). After protocol amendment, enrollment in a third cohort was restricted to patients with chemotherapy-naïve advanced or metastatic esophagogastric cancer.
Results
DLT was reported for two of the five patients in cohort 2, defining 20 mg/m2 twice daily as the MTD of S-1 combined with epirubicin and oxaliplatin in heavily pretreated patients. Thirteen patients with chemotherapy-naïve advanced or metastatic esophagogastric cancer were subsequently enrolled and treated at an S-1 dose level of 25 mg/m2 twice daily; no DLTs were reported; median overall survival was 13.1 months. Of the 11 evaluable patients, three (27 %) had partial responses and seven (64 %) had stable disease. The safety profile was in line with expectations.
Conclusions
The promising activity of EOS (S-1 dose level, 25 mg/m2 twice daily) and acceptable safety profile support further clinical development of this combination for the first-line treatment of patients with advanced or metastatic esophagogastric cancer.
Similar content being viewed by others

Introduction
For patients with advanced gastric cancer, meta-analysis has confirmed that palliative chemotherapy improves overall survival compared with best supportive care, and that combination chemotherapy improves overall survival compared with single-agent 5-fluorouracil (5-FU) chemotherapy [1]. Proximal gastric tumors involving the cardia, tumors of the gastroesophageal junction, and lower esophageal tumors are generally characterized by the same histology (adenocarcinoma) and biological behavior. Consequently, most clinical trials enroll patients with esophagogastric adenocarcinomas irrespective of the anatomical site of the primary in the stomach or esophagus, and patients with such tumors are also treated with identical regimens in clinical practice. While a number of different first-line chemotherapy regimens have been validated for use in this setting, there is as yet no consensus recommendation for the most effective combination of agents for this patient group [2]. Most commonly, eligible patients with advanced esophagogastric cancer will receive combination chemotherapy regimens based on a platinum–fluoropyrimidine doublet [3]. The addition of an anthracycline or docetaxel to such doublets has been shown to improve overall survival, albeit at the cost of increased toxicity [2, 4–6]. The integration of targeted agents into systemic chemotherapy regimens used in this setting has so far been limited compared to their use for other gastrointestinal cancers [7].
In terms of the platinum component of combination chemotherapy regimens used in the first-line treatment of advanced esophagogastric cancer, both cisplatin and oxaliplatin appear to be equally effective, with oxaliplatin associated with reduced incidence of certain toxicities, including thromboembolic events [8–10]. The fluoropyrimidine component may be administered as infusional 5-FU. Alternatively, randomized studies have shown that capecitabine, an oral fluorouracil prodrug, is at least as effective as 5-FU when combined with a platinum agent in this setting [9, 11]. The anthracycline component added to platinum–fluoropyrimidine doublets in triplet regimens is commonly epirubicin, a derivative of doxorubicin [12], with epirubicin, cisplatin, and 5-FU (ECF) established as a standard first-line treatment for patients with advanced esophagogastric cancer [13].
S-1 represents a further development of oral fluoropyrimidine therapy in that it comprises tegafur, a fluorouracil prodrug, and two modulators of 5-FU metabolism, 5-chloro-2,4-dihydroxypyridine (CDHP) and oteracil potassium. The CDHP component of S-1, which binds dihydropyrimidine dehydrogenase (DPD), serves as an inhibitor of 5-FU catabolism, with oteracil potassium—an inhibitor of 5-FU activation by orotate phosphoribosyl transferase—preferentially localizing to the gut and thereby potentially decreasing off-target activation and collateral toxicity at this site [14]. Early phase II studies of single-agent therapy indicated that S-1 might be particularly effective in the first-line treatment of advanced gastric cancer [15, 16]. This was confirmed in the randomized phase III FLAGS study, which showed the noninferiority of cisplatin plus S-1 to cisplatin plus 5-FU, coupled with a significantly reduced rate of severe adverse events for cisplatin plus S-1 [17, 18]. This led in 2011 to regulatory approval by the European Medicines Agency (EMA) of S-1 (25 mg/m2 twice daily) combined with cisplatin for the treatment of advanced gastric cancer [19]. Subsequently, it has been shown in this setting that oxaliplatin plus S-1 is as effective as cisplatin plus S-1, with a favorable safety profile [20]. A meta-analysis has also shown that S-1-based regimens are effective and tolerable in both Western and Asian populations as first-line treatments for advanced gastric cancer [21].
As a prelude to randomized studies in patients with advanced or metastatic esophagogastric cancer, this phase I study was designed to determine the maximum tolerated dose (MTD) of S-1 when combined with epirubicin and oxaliplatin (EOS). Initially, enrollment in this study was permitted for patients with any advanced or metastatic solid tumor. Following a protocol amendment, a second phase was initiated in which enrollment was restricted to patients with advanced or metastatic esophagogastric cancer. In parallel, another phase I study (NCT01928524) aimed to establish recommended docetaxel and S-1 doses for docetaxel, oxaliplatin, and S-1 regimens in European patients.
Patients and methods
Study design
This was a multicenter, open-label, phase I dose-escalation study which evaluated the safety and antitumor activity of two doses of S-1 administered with fixed doses of oxaliplatin and epirubicin. The first two cohorts (1 and 2) enrolled patients with an advanced or metastatic solid tumor, regardless of the number of prior therapies received for this tumor. Following a protocol modification, enrollment to cohort 3 was restricted to patients with previously untreated advanced or metastatic esophagogastric cancer.
Based on the MTD of S-1 established in Western patients [22, 23], and corresponding to the EMA-approved dose level, a maximum S-1 dose of 25 mg/m2 twice daily was planned. To minimize patient risk, a lower starting dose of 20 mg/m2 twice daily was to be investigated initially. Patients were to be enrolled in a 3 + 3 design, with dose escalation of S-1 permitted according to the incidence of dose-limiting toxicity (DLT), as assessed during the first 21-day treatment cycle of the first cohort. Only toxicities that were deemed to be related to the study drug were considered as DLTs; these were ≥grade 3 nonhematological toxicity (excluding nausea/vomiting, diarrhea), ≥grade 3 nausea/vomiting uncontrolled by aggressive antiemetic support, ≥grade 3 diarrhea lasting more than 24 h despite antidiarrheal treatment, febrile neutropenia (absolute neutrophil count <1.0 × 109/L with a single temperature of >38.3 °C or a sustained temperature of >38 °C for more than 1 h), grade 4 neutropenia lasting ≥5 days, grade 4 thrombocytopenia associated with dose interruption or hemorrhage, any drug-related toxicity that resulted in a >1 week delay in the initiation of cycle 2, any drug-related toxicity that resulted in the administration of <80 % of the total planned S-1 dose.
If none of the initial three patients treated at the 20 mg/m2 dose level experienced a DLT during the first treatment cycle, escalation to the second planned S-1 dose level was to be permitted. If a DLT was observed in one of the first three patients treated, then a maximum of three additional patients were to be enrolled at the same dose level. If no additional DLTs were seen after all six patients completed the first cycle of treatment, dose escalation was to be permitted. If a DLT was observed in one of three patients of any cohort, the investigator had to wait until all three patients had completed the DLT evaluation period before enrolling additional patients at the same dose level, and had to wait until 30 days after all patients at a particular dose level had completed the DLT evaluation period before escalating the dose. Intrapatient dose escalation was not allowed. If two or more patients treated at a particular dose level developed a DLT, then dose escalation ceased and the previous dose was considered to be the MTD. At least six patients in total were to be treated at a particular dose level before it was defined as the MTD, with no more than one of the six patients experiencing a DLT. Once the MTD was established, up to six additional patients (a total of 12 patients) were to be treated at the MTD level to comprehensively explore tolerability before recommending it as the dose to be further evaluated in clinical studies.
On day 1 of each 21-day treatment cycle, patients were to receive epirubicin 50 mg/m2 via an intravenous (IV) bolus immediately prior to oxaliplatin 130 mg/m2 via a 2-h IV infusion, both of which were administered according to the institution’s standard practice. Treatment with epirubicin and oxaliplatin was limited to a maximum of 8 cycles. The first S-1 dose (orally: 20 or 25 mg/m2) was to be taken during the evening of day 1. S-1 was subsequently to be administered orally on days 2–14 twice daily. The last dose of S-1 per cycle was to be administered on the morning of day 15. There was no limit on the number of cycles for S-1. Patients were to receive study treatment until disease progression, the occurrence of intolerable side effects, pregnancy, removal by the investigator, or withdrawal of consent. A patient was considered discontinued from study treatment when S-1 therapy was discontinued. Study treatment continued until all patients had discontinued from treatment or until 12 months from the date of the first day of treatment with S-1, whichever occurred first. At that point, S-1 therapy could be continued at the discretion of the investigator and with the agreement of the sponsor. Each patient was followed for up to 12 months after their first dose of study medication to determine survival status.
The study was conducted in accordance with the Declaration of Helsinki and applicable local and national laws and regulations. The study was approved by the local ethics committee of each study center, and written informed consent was obtained from each patient before any screening procedures were performed.
Eligibility
For cohorts 1 and 2 (planned S-1 dose levels of 20 and 25 mg/m2, respectively), patients aged ≥18 years of age with an advanced or metastatic solid tumor for which no established curative therapy was available, who could take oral medication, and who had an Eastern Cooperative Oncology Group performance status at baseline of 0 or 1, a life expectancy of at least 3 months, and a left ventricular ejection fraction greater than the lower limit of normal were eligible. Patients were also required to have serum troponin T and CPK-MB values ≤upper limit of normal (ULN) for the institution and adequate organ function, as defined by aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤2.5 × ULN; or if liver function abnormalities were due to underlying liver metastasis, AST and ALT ≤ 5 × ULN; total serum bilirubin of ≤1.5 × ULN; absolute neutrophil count of ≥1500/mm3; platelet count ≥100,000/mm3; and a hemoglobin value of ≥9.0 g/dL and creatinine clearance ≥60 mL/min based on calculated creatinine clearance or 24-h urine collection. Enrollment in cohort 3 (planned S-1 dose level of 25 mg/m2) was additionally restricted to patients who had advanced or metastatic esophagogastric adenocarcinoma and who had received no previous chemotherapy for advanced or metastatic disease.
Key exclusion criteria included major surgery or radiotherapy in the 4 weeks prior to enrollment, receipt of any investigational agent concurrently or within the previous 30 days, and current enrollment in another interventional trial. For cohorts 1 and 2, patients should not have had >25 % of marrow-bearing bone irradiated, received any chemotherapy within the prior 3 weeks, have previously received oxaliplatin or S-1, have previously received epirubicin at a cumulative dose >350 mg/m2, have extensive prior exposure to other anthracycline or anthracenedione agents, or have received trastuzumab within the prior 24 weeks. Patients were excluded from all cohorts if they had another serious illness or medical condition, were receiving concomitant treatment with particular drugs which might interact with S-1 or epirubicin, if they were pregnant, or if they had a known hypersensitivity to 5-FU, epirubicin, oxaliplatin, or other platinum compounds.
Study objectives
The primary objective was to investigate the safety and determine the MTD of S-1, either 20 mg/m2 or 25 mg/m2, when combined with epirubicin and oxaliplatin in patients with advanced or metastatic solid tumors (pre-amendment) and in patients with advanced or metastatic esophagogastric cancer treated in the first-line setting (post-amendment). The secondary objective was to document any antitumor activity observed with S-1 administered in this combination regimen.
Assessments
Adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA; version 14.0 or higher), and severity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v4.03. The safety assessment period was from the time of signing of the informed consent form through to 30 days after the last dose of study medication, or until the initiation of a new anticancer therapy, whichever came first. The assessment of response was based on an investigator review of radiological images, and followed RECIST (version 1.1) guidelines [24].
Statistical considerations
The safety population included all patients who received at least one dose of the study medication. DLT was assessed in the DLT population, which included all patients in the safety population who completed at least one cycle of the study medication with at least 80 % treatment compliance, unless treatment was interrupted due to a DLT. The efficacy population included all patients in the safety population who completed at least one cycle of the study medication and had radiological or clinical progression assessments performed. The primary efficacy variable was overall survival, defined as the time from the first dose of S-1 to the date of death, as assessed in the safety population. Surviving patients were censored at the date of the last contact. Best overall response and progression-free survival (PFS)—defined as the time from the first dose of S-1 until the date of investigator-assessed radiological disease progression, clinical progression, or death due to any cause—were both assessed in the efficacy population. For the PFS analysis, patients who were alive at the cutoff date and showed no evidence of disease progression were censored at the date of the last tumor assessment.
All data analyses were carried out using SAS version 9 or higher. The study is registered with EudraCT (no: 2011‐003471‐11).
Results
Patients
The study was conducted at seven sites: three in Germany, three in the Czech Republic, and one in the UK. Of 37 patients screened, 12 were deemed to be ineligible and 25 were enrolled and treated. The first patient was enrolled on January 31, 2012. The data cutoff date for our analysis was August 4, 2015. At the cutoff date, all 25 of the enrolled patients had discontinued from the study: 14 (56 %) due to disease progression, 5 (20 %) due to an adverse event, and 6 due to other reasons. Patient characteristics for the safety population at baseline are summarized in Table 1.
Of the 25 treated patients, two were not evaluable for DLT assessment: one patient in cohort 2 due to S-1 treatment interruption in cycle 1 (compliance of <80 % caused by replacement of a stent in the ductus hepatocholedochus) and one patient in cohort 3 when it was discovered post-screening that eligibility criteria had been violated (this patient was consequently assessed as not eligible for DLT assessment). Four patients were not evaluable for efficacy as they did not complete at least one cycle of the study medication, and radiological or clinical progression assessments were not performed.
Evaluation of DLT
No DLTs were observed in the first three patients enrolled in cohort 1 (S-1 dose level 20 mg/m2); therefore, the S-1 dose was escalated to 25 mg/m2 twice daily in a second cohort. DLT was reported for two of five patients in cohort 2. The first patient developed grade 3 elevated gamma-glutamyl transferase on day 8 of cycle 1. This was assessed by the investigator as study drug related and was therefore reported as a DLT. This patient had a diagnosis of pancreatic cancer and had previously received two lines of anticancer treatment. The second patient, who had a history of cholangiocarcinoma, was hospitalized on day 9 of cycle 1 with life-threatening serious adverse events (SAEs) of grade 4 cholangitis, grade 4 sepsis, grade 4 thrombocytopenia, and grade 4 febrile neutropenia. This later event was assessed by the investigator as study drug related and therefore as a DLT. On day 13 of cycle 1, an X-ray of the thorax revealed pneumonia, which was treated with antibiotics; on day 15, the patient’s condition deteriorated and the patient died due to pneumonia. This patient had also received two prior lines of chemotherapy. These DLTs led to the closure of cohort 2. Consequently, in order to allow confirmation of the MTD, cohort 1 was expanded by enrolling three additional patients; no further DLTs were reported. Therefore, a dose of 20 mg/m2 twice daily was established as the MTD of S-1 when combined with epirubicin and oxaliplatin (EOS) in heavily pretreated patients with solid tumors.
It was noted that the DLTs in cohort 2 had occurred in patients with non-esophagogastric tumors who had received the study treatment as third-line therapy. However, as the envisaged clinical population for EOS treatment comprised chemotherapy-naïve patients, it was decided to further test the EOS triplet in this specific context. The study protocol was therefore amended (Amendment 3; April 16, 2013) to include assessment of the 25 mg/m2 twice daily S-1 dose level in patients with previously untreated advanced or metastatic esophagogastric cancer who were eligible for first-line therapy (cohort 3). This dose level was to be defined as the MTD if no more than one of the six patients experienced DLT; otherwise, the dose level of 20 mg/m2 was to be considered the MTD in this second patient population (as previously defined in patients with advanced or metastatic solid tumors).
Thirteen patients were subsequently enrolled and treated in cohort 3 (S-1 dose level 25 mg/m2). No DLTs were seen in the first six evaluable patients; one enrolled patient did experience a grade 3 rise in AST, which was assessed as a DLT. However, it was found post-screening that eligibility criteria had been violated in the case of this patient with respect to a history of alcohol abuse, which was not known at study entry. This was considered a major protocol deviation, and once discovered, the patient was deemed by the investigator and medical monitor not to be eligible for DLT assessment. Six further evaluable patients were subsequently enrolled and assessed for DLT at this dose level, and again, no DLTs were reported. Therefore, the MTD of S-1 when combined with epirubicin and oxaliplatin in chemotherapy-naïve patients with advanced or metastatic esophagogastric cancer was defined as 25 mg/m2 twice daily.
Exposure
The median number of treatment cycles initiated was 4 (range 1–18) in the overall safety population and 7 (range 1–18) in cohort 3. In cohorts 1 and 3, the median actual dose intensity for S-1 was close to the planned dose intensity, with a median relative dose intensity (ratio of actual dose intensity to planned dose intensity) of 0.9 (range 0.7–1.0 and 0.3–1.0, respectively). In cohort 2, the median actual dose intensity for S-1 was somewhat lower than the planned dose intensity, with a median relative dose intensity of 0.8 (range 0.5–0.9). The median relative dose intensities in all three cohorts for epirubicin and oxaliplatin were 0.9 and 0.9, respectively.
Safety
For all patients, across all cycles, the most frequently reported adverse events, as summarized in the Electronic supplementary material (ESM) according to MedDRA preferred terms, were: nausea, neutropenia, fatigue, paresthesia, and vomiting. Grade 3 or higher adverse events were reported for 22 (88 %) of 25 patients, the most common of which were neutropenia, thrombocytopenia, and anemia (Table 2). There were 31 treatment-emergent SAEs that occurred in 14 patients. Eight of the SAEs that occurred in three patients were S-1 related: neutropenia in one patient, febrile neutropenia, thrombocytopenia, cholangitis, pneumonia, and sepsis in one patient, and tachycardia and sepsis in one patient.
Five patients discontinued study treatment due to adverse events, which included grade 3 acute renal failure (subsequently worsening to grade 4), grade 3 hyperbilirubinemia, grade 4 febrile neutropenia, grade 3 thrombocytopenia, and grade 4 neutropenia (one patient each). The hyperbilirubinemia was considered to be unrelated to the study treatment. The febrile neutropenia and neutropenia were assessed as related to S-1. The renal failure was assessed as related to oxaliplatin; the worsening from grade 3 to grade 4 was suspected to be due to delayed oxaliplatin toxicity, with infection considered to be a likely contributor. The thrombocytopenia was assessed as related to oxaliplatin and epirubicin.
At the cutoff date, deaths had been reported for 15 (60 %) of the 25 patients: five patients in cohort 1, four patients in cohort 2; and six patients in cohort 3. Fourteen deaths (12 due to documented disease progression; two for reasons unknown) were reported to have occurred after the 30-day safety follow-up period. One patient in cohort 2 experienced a fatal adverse event of pneumonia and died on day 15 of cycle 1; this event was considered to be related to S-1.
Efficacy
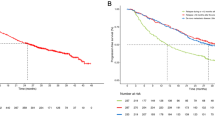
Efficacy data are summarized in Table 3. At the time of final analysis, at least 12 months of follow-up data were available for all patients. Median overall survival was 4.7 months for cohort 1, 10.4 months for cohort 2, and 13.1 months for cohort 3 (Fig. 1a). Median PFS was also longer for patients in cohort 3 (6.9 months; Fig. 1b) compared with cohorts 1 and 2 (1.9 and 5.1 months, respectively). Three of 11 (27 %) evaluable patients in cohort 3 had a partial response to treatment, and a further seven (64 %) were deemed to have stable disease, including one patient with an unconfirmed partial response (Fig. 2).

a Overall survival in cohort 3 (safety population), b progression-free survival in cohort 3 (efficacy population)

Waterfall plot of target lesion shrinkage and best overall response in cohort 3 (efficacy population). Asterisk: one partial response was not confirmed and was reported as stable disease. One patient with stable disease was excluded from the plot as they did not have measurable disease
Discussion
In the first part of this study we showed that, in cohorts of Caucasian patients with solid tumors and 0, 1, or 2 lines of prior anticancer therapy, the MTD of S-1 when combined with epirubicin and oxaliplatin was 20 mg/m2 twice daily. The DLTs seen in two of five patients at the 25 mg/m2 twice daily dose level were in patients who had received the study treatment as third-line therapy for tumors other than esophagogastric cancer. However, as our intention in this study was to determine a suitable S-1 dose for future trials exploring the EOS triplet in patients with chemotherapy-naïve advanced or metastatic esophagogastric cancer, we revised the study design through a protocol amendment to further explore safety in this specific setting. In a third cohort of 12 such predominantly Caucasian evaluable patients treated with EOS at an S-1 dose level of 25 mg/m2 twice daily, no DLTs were seen, confirming that this dose could be recommended for future studies in untreated patients with advanced disease. This recommended dose is lower than that generally administered to East Asian patients, but corresponds to the S-1 dose commonly recommended for Caucasian patients [25–28]. The biological basis for this difference in the MTD of S-1 between different ethnic groups may, at least in part, be due to population differences in the genetic polymorphism status of the CYP2A6 gene, the product of which is a principal enzyme in relation to the bioactivation of tegafur to 5-FU [25, 29, 30].
Suggestive of a high potential for tumor control, partial responses to EOS were seen in three (27 %) of 11 evaluable patients in cohort 3, and a further seven patients (64 %) had stable disease, including one patient with an unconfirmed partial response; only one patient (9 %) had progressive disease. The median PFS for this cohort was 6.9 months, and the PFS rate at 9 months was 48 %. In addition, median overall survival was 13.1 months, which, although based on a small number of patients, compares favorably with the median overall survival times reported for non-Asian patient populations in phase III studies conducted in this setting [9, 25].
The most frequently reported grade 3 or higher adverse events were neutropenia (52 %), thrombocytopenia (24 %), and anemia (12 %). Corresponding incidence rates for grade 3 or higher hematological adverse events in the 5-FU plus cisplatin arm of the FLAGS study were 64 % for neutropenia, 14 % for thrombocytopenia, and 21 % for anemia [18], and the corresponding incidence rates for the ECF regimen in the REAL-2 study were 42, 5, and 13 %, respectively [9]. Note that there was no indication in our study of a high incidence of grade 3 or higher lethargy, a treatment-related adverse event noted for 25 % of patients in the epirubicin, oxaliplatin, and capecitabine treatment group of REAL-2 [9]. The data in the current study do not therefore suggest any relevant change to the established safety profile of S-1, with no new safety concerns apparent for the EOS combination. Highlighting the potential of S-1 in this setting, a recent meta-analysis which compared S-1 based with non-S-1-based chemotherapy in the first-line treatment of advanced gastric cancer showed that S-1-based regimens were associated with higher objective response rates, longer overall survival, longer time-to-treatment failure, and lower risks of febrile neutropenia and stomatitis [31].
In summary, the encouraging activity of the EOS regimen observed in this study, along with an acceptable safety profile, supports further clinical development of this combination. The MTD of S-1 in patients with heavily pretreated solid tumors was 20 mg/m2 twice daily. The study further confirmed that an S-1 dose of 25 mg/m2 twice daily, combined with oxaliplatin 130 mg/m2 and epirubicin 50 mg/m2, is an appropriate first-line regimen for future trials in patients with advanced or metastatic esophagogastric cancer.
References
Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2010;3:CD004064.
Lordick F, Allum W, Carneiro F, Mitry E, Tabernero J, Tan P, et al. Unmet needs and challenges in gastric cancer: the way forward. Cancer Treat Rev. 2014;40(6):692–700.
Waddell T, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D, et al. Gastric cancer: ESMO-ESSO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 Suppl 6:vi57–63.
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol. 2006;24(18):2903–9.
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24(31):4991–7.
Wagner AD, Moehler M, Grothe W, Haerting J, Unverzagt S. Novel chemotherapy combinations for metastatic gastric adenocarcinoma (mAGC): an updated meta-analysis. J Clin Oncol. 2015;33(suppl 3; abstr 125).
Moehler M, Schwarz S, Wagner AD. Esophagogastric cancer: integration of targeted therapies into systemic chemotherapy. Curr Cancer Drug Targets. 2011;11(6):681–7.
Al-Batran SE, Hartmann JT, Probst S, Schmalenberg H, Hollerbach S, Hofheinz R, et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: a study of the Arbeitsgemeinschaft Internistische Onkologie. J Clin Oncol. 2008;26(9):1435–42.
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358(1):36–46.
Starling N, Rao S, Cunningham D, Iveson T, Nicolson M, Coxon F, et al. Thromboembolism in patients with advanced gastroesophageal cancer treated with anthracycline, platinum, and fluoropyrimidine combination chemotherapy: a report from the UK National Cancer Research Institute Upper Gastrointestinal Clinical Studies Group. J Clin Oncol. 2009;27(23):3786–93.
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol. 2009;20(4):666–73.
Cersosimo RJ, Hong WK. Epirubicin: a review of the pharmacology, clinical activity, and adverse effects of an adriamycin analogue. J Clin Oncol. 1986;4(3):425–39.
Webb A, Cunningham D, Scarffe JH, Harper P, Norman A, Joffe JK, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol. 1997;15(1):261–7.
Maehara Y. S-1 in gastric cancer: a comprehensive review. Gastric Cancer. 2003;6(Suppl 1):2–8.
Koizumi W, Kurihara M, Nakano S, Hasegawa K. Phase II study of S-1, a novel oral derivative of 5-fluorouracil, in advanced gastric cancer. For the S-1 Cooperative Gastric Cancer Study Group. Oncology. 2000;58(3):191–7.
Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T. Late phase II study of novel oral fluoropyrimidine anticancer drug S-1 (1 M tegafur-0.4 M gimestat-1 M otastat potassium) in advanced gastric cancer patients. Eur J Cancer. 1998;34(11):1715–20.
Ajani JA, Buyse M, Lichinitser M, Gorbunova V, Bodoky G, Douillard JY, et al. Combination of cisplatin/S-1 in the treatment of patients with advanced gastric or gastroesophageal adenocarcinoma: results of noninferiority and safety analyses compared with cisplatin/5-fluorouracil in the First-Line Advanced Gastric Cancer Study. Eur J Cancer. 2013;49(17):3616–24.
Ajani JA, Rodriguez W, Bodoky G, Moiseyenko V, Lichinitser M, Gorbunova V, et al. Multicenter phase III comparison of cisplatin/S-1 with cisplatin/infusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol. 2010;28(9):1547–53.
Matt P, van Zwieten-Boot B, Calvo Rojas G, Ter Hofstede H, Garcia-Carbonero R, Camarero J, et al. The European Medicines Agency review of tegafur/gimeracil/oteracil (Teysuno) for the treatment of advanced gastric cancer when given in combination with cisplatin: summary of the Scientific Assessment of the Committee for medicinal products for human use (CHMP). Oncologist. 2011;16(10):1451–7.
Yamada Y, Higuchi K, Nishikawa K, Gotoh M, Fuse N, Sugimoto N, et al. Phase III study comparing oxaliplatin plus S-1 with cisplatin plus S-1 in chemotherapy-naive patients with advanced gastric cancer. Ann Oncol. 2015;26(1):141–8.
Ter Veer E, Mohammad NH, Lodder P, Ngai LL, Samaan M, van Oijen MG, et al. The efficacy and safety of S-1-based regimens in the first-line treatment of advanced gastric cancer: a systematic review and meta-analysis. Gastric Cancer. 2016 (Epub).
Chung KY, Saito K, Zergebel C, Hollywood E, Segal M, Saltz LB. Phase I study of two schedules of oral S-1 in combination with fixed doses of oxaliplatin and bevacizumab in patients with advanced solid tumors. Oncology. 2011;81(2):65–72.
Zhu AX, Clark JW, Ryan DP, Meyerhardt JA, Enzinger PC, Earle CC, et al. Phase I and pharmacokinetic study of S-1 administered for 14 days in a 21-day cycle in patients with advanced upper gastrointestinal cancer. Cancer Chemother Pharmacol. 2007;59(3):285–93.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Ajani JA, Faust J, Ikeda K, Yao JC, Anbe H, Carr KL, et al. Phase I pharmacokinetic study of S-1 plus cisplatin in patients with advanced gastric carcinoma. J Clin Oncol. 2005;23(28):6957–65.
Kobayakawa M, Kojima Y. Tegafur/gimeracil/oteracil (S-1) approved for the treatment of advanced gastric cancer in adults when given in combination with cisplatin: a review comparing it with other fluoropyrimidine-based therapies. Onco Targets Ther. 2011;4:193–201.
Ryu MH, Baba E, Lee KH, Park YI, Boku N, Hyodo I, et al. Comparison of two different S-1 plus cisplatin dosing schedules as first-line chemotherapy for metastatic and/or recurrent gastric cancer: a multicenter, randomized phase III trial (SOS). Ann Oncol. 2015;26(10):2097–101.
Pfeiffer P, Qvortrup C, Krogh M, Schoennemann K, Vestermark L, Jensen H, et al. S-1 in combination with docetaxel and oxaliplatin every 2 weeks or 3 weeks in patients with advanced gastro-esophageal adenocarcinoma: final results of two parallel phase I studies. Gastric Cancer (submitted).
Ikeda K, Yoshisue K, Matsushima E, Nagayama S, Kobayashi K, Tyson CA, et al. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome P-450 2A6 in human liver microsomes in vitro. Clin Cancer Res. 2000;6(11):4409–15.
Yamamiya I, Yoshisue K, Ishii Y, Yamada H, Chiba M. Effect of CYP2A6 genetic polymorphism on the metabolic conversion of tegafur to 5-fluorouracil and its enantioselectivity. Drug Metab Dispos. 2014;42(9):1485–92.
Yang J, Zhou Y, Min K, Yao Q, Xu CN. S-1-based vs non-S-1-based chemotherapy in advanced gastric cancer: a meta-analysis. World J Gastroenterol. 2014;20(33):11886–93.
Acknowledgments
This study was funded by Nordic Pharma SAS. Medical writing support was provided by Jim Heighway PhD of Cancer Communications and Consultancy Ltd (Knutsford, UK), and was funded by Nordic Pharma SAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Markus Moehler reports receiving personal fees and non-financial support from Taiho, personal fees and non-financial support from Nordic Pharma, personal fees from Sanofi-Aventis, and personal fees from Medac, in each case outside the submitted work; Volker Heinemann reports receiving grants, personal fees, and non-financial support from Merck, Roche, and Bayer, grants and personal fees from Amgen and Sirtex, and grants from Sanofi, Taiho, Celgene, and Boehringer Ingelheim, in each case outside the submitted work; Bohuslav Melichar reports personal fees in relation to lectures and advisory boards from Roche, AstraZeneca, Novartis, Pfizer, Bristol-Myers Squibb, Astellas, Merck, MSD, Janssen, Bayer, and Amgen, and travel support from Roche, Novartis, Bristol-Myers Squibb, and Merck, in each case outside the submitted work; Fabienne Hédouin-Biville was an employee of Nordic Pharma. The other authors declare that they have no conflict of interest.
Human rights statement and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent was obtained from all patients before they were included in the study.
Additional information
M. Moehler and R. Mahlberg contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moehler, M., Mahlberg, R., Heinemann, V. et al. Phase I study of orally administered S-1 in combination with epirubicin and oxaliplatin in patients with advanced solid tumors and chemotherapy-naïve advanced or metastatic esophagogastric cancer. Gastric Cancer 20, 358–367 (2017). https://doi.org/10.1007/s10120-016-0618-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-016-0618-0