
Abstract
Diketene (4-methylideneoxetan-2-one) is a precursor to the formation of either two molecules of ketene, or allene and CO2 using pyrolysis techniques. It is not known experimentally which of these pathways is followed, or indeed if both are, during the dissociation process. We use computational methods to show that the formation of ketene has a lower barrier than formation of allene and CO2 under standard conditions (by 12 kJ/mol). According to CCSD(T)/CBS, CBS-QB3 and M06-2X/cc-pVTZ calculations the formation of allene and CO2 is favoured thermodynamically under standard conditions of temperature and pressure; however, kinetically the formation of ketene is favoured from transition state theory calculations at standard and elevated temperatures.
Graphical abstract

Similar content being viewed by others

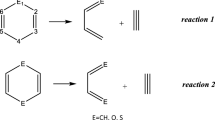
Introduction
Ketenes are a reactive class of organic oxo compounds that are useful in organic synthesis and industrial chemistry [1,2,3,4,5]. For instance, a series of organic compounds were synthesized using ketene as an intermediate in the preparation route [1]. The alkylketene dimer has been used for the preparation of hydrophobic paper sizing agents [2, 4], as well as a hydrophobic starch microcellular foam [5]. Methoxycarbonylketene can be used for the synthesis of the functionalized malonates for agrochemicals and pharmaceuticals [3].
Ketene was first synthesized by Staudinger by reaction of α-chlorodiphenylacetyl chloride with zinc at 452–453 K [6]. Staudinger was seeking to obtain the radical Ph2ĊCOCl, inspired by Gomberg who prepared a stable triphenylmethyl radical [7], but the result was the unforeseen discovery of ketene. Ketene has also been prepared by pyrolysis of acetic anhydride using hot platinum wire [8]. This new class of compound dimerized rapidly at room temperature yielding diketene [9].
Diketene is a reactive compound that is useful in synthetic and structural chemistry [10, 11]. Chick and Wilsmore made the first known diketene, as ‘acetylketene’, in 1908 [9]. On standing the liquid or gaseous ketene at room temperature, the new substance [9] was formed as a pungent smelling brown liquid with the possible formula CH3COCHCO. Five different isomeric molecular structures were proposed for this compound (one acyclic and four cyclic conformers as shown in Fig. 1) [11].

Structure of diketene (c) and its other cyclic (a, b and d) and acyclic (e) isomers
Boese [6] prepared ketene via two different methods, firstly refluxing diketene over hot metal such as platinum or resistant metal and secondly by passing diketene vapour through a hot tube at 823–873 K. The pyrolysis chamber was fitted with a reflux condenser extended to an ice bath to collect the ketene so that unreacted diketene could be observed [11]. The process [11] was about 50% efficient. In 1965 Andreades and Carlson [12] synthesized ketene by pyrolysis of diketene with a flow of nitrogen, which yielded 46–55% ketene. Ketene was prepared from pyrolysis of diluted diketene with ultra-pure argon by thermal decomposition at a temperature of 510–603 K and a constant pressure of 800 Torr [13]. The impurities, such as unpyrolysed diketene and carbon dioxide, were observed in the products by Fourier Transform Infrared spectroscopy (FTIR) investigation, but the quantity was not measured. Previous work from our group [14, 15] generated ketene from pyrolysis of three sources: acetic anhydride, Meldrum’s acid and acetone by using flash vacuum pyrolysis coupled with gas electron diffraction (FVP-GED).
In this work, we use computational methods to study the pyrolysis decomposition of diketene with two possible pathways, (I) and (II). (I) leads to the formation of two equivalent molecules of ketene, and (II) yields allene and CO2. Our work provides insight into the underlying mechanism for the pyrolysis decomposition of diketene as shown in Fig. 2 and helps to explain the experimental observations at elevated temperatures.

Potential pathways for pyrolytic decomposition of diketene to form (I) ketene and (II) allene and CO2
Computational details
All calculations were performed using either second order Møller–Plesset perturbation theory (MP2) [16], with the 6-31G*, 6-311G*, 6-311+G*, 6-311++G**basis sets [17,18,19,20] and the Minnesota hybrid meta exchange-correlation functional (M06-2X) [21] using the aug-cc-pVTZ basis set [22] with the Gaussian 09 (Revision B.01) [23] and NWChem [24] programs. The results were visualized using GaussView [25]. NWChem [24] calculations were carried out using the resources of the New Zealand e-Science Infrastructure (NeSI). All MP2 methods were frozen core [MP2(FC)]. Geometric optimization of molecules was started at the HF level with a 6-31G* basis set, and frequency calculations were undertaken at MP2/6-311++G** to verify the nature of the stationary points. Thermodynamic parameters and transition state structures with reaction pathways were calculated using the synchronous transit-guided quasi-Newton (STQN) method [26] using CCSD(T)/CBS and composite CBS-QB3 [27] method as described by Curtiss et al. [28, 29]. This method predicts thermochemical parameters with chemical accuracy in the range of mean absolute deviation less than 5.27 kJ/mol [30]. For the CCSD(T)/CBS method, the method is based on extrapolation of the energy to complete basis limit (CBS) using the power function extrapolation scheme suggested by Helgaker et al. [31] (Eq. 1) with the augmented correlation consistent basis sets (aug-cc-pVnZ) of Dunning [32], where n = D, T and Q have been used.
In Eq. 1: X is two for double-zeta basis sets, three for triple-zeta basis sets, etc. E∞ is energy at the basis set limit, and ‘β’ and ‘α’ are fitting parameters.
The energy profile diagram was plotted from the optimizations at the M06-2X/cc-pVTZ level [21]. The input coordinates were taken from the optimized transition state (TS) structures at the B97D/6-31++G(d,p) level [33]. To ascertain the identity of the relevant transition structures, intrinsic reaction coordinate [34] (IRC) calculations were undertaken at the same level of theory. After IRC calculations followed by structural optimization of the species, it was confirmed that the TS correctly connects the reactant and product(s). Global minima on the potential energy surfaces were identified by the absence of any imaginary vibrational frequencies, and all TS were identified by the presence of one imaginary vibrational frequency.
Results and discussion
Quantum chemical calculations
Theoretical calculations revealed the ground state structure of diketene has Cs symmetry and its potential decomposition products ketene, CO2 and allene have C2v, D∞h and D2d symmetry respectively at both the MP2 and M06-2X levels of theory with different basis sets. The structural parameters for each molecule for the various calculations are given in the supporting information (SI) Table S1. Calculated coordinates for each structure in Table S1 are given in the SI, Tables S2–S6. The structures with atom numbering are shown in Fig. 3.

Lowest energy ground state structures (at MP2/6-311++G**) level of (L – R) diketene (Cs), ketene (C2v), allene (D2d) and CO2 (D∞h) with atom numbering
The gas-phase and solid-state structure of diketene was previously investigated using gas electron diffraction [35] and single crystal X-ray diffraction [36, 37] as well as 1H NMR spectroscopy [38] and IR spectroscopy [39, 40]. The previous GED structure [35] differs from the computed structure at MP2/6-311++G** level for various parameters such as rC=C, rC–C and rC–C (for ∠C–C=O) by 2.0 pm, 1.4 pm and 0.9 pm respectively. The bond angles differ by a range of 0.5 to 6.5°, particularly for ∠O–C=O, ∠C–C=O and ∠C=C–C (6.5°, 5.6° and 3.8° respectively). The most recent single crystal X-ray diffraction structure of diketene [37] is in very good agreement with the computed structure. We compared the computed structures of diketene and ketene using MP2 level of theory with previous experimental and theoretical results as shown in Tables 1 and 2.
Calculated parameters from MP2 level of theory for ketene are in good agreement with the experimental structures from microwave spectroscopy and GED. There were some small differences in the bond distances and bond angles between our theoretical calculation at MP2/6-311++G** and the previous GED-2 study [14], such as the bond distances of rC=O (2.0 pm), rC–H (0.4 pm) and ∠H–C–H (0.7°).
Thermochemical calculations
Thermochemical properties such as the Gibbs energy (ΔG°), enthalpy (ΔH°) and entropy (ΔS°) changes were calculated at the CBS-QB3 and CCSD(T)/CBS level of theory (Table 3). The calculated energies and thermochemical parameters are given in SI (Table S7).
The decomposition of diketene to form allene and CO2 was found to be exothermic and spontaneous under standard thermodynamic conditions of temperature and pressure. We expected the calculated formation of ketene to be spontaneous; however, the calculations indicated that it was not under standard conditions. Given this unexpected result, thermochemical parameters were also predicted at elevated temperatures. The calculations gave different thermochemical correction parameters, such as Hcorr and Gcorr, which were then used to obtain the thermochemistry of the two reaction pathways for the decomposition of diketene at that temperature. The same energies from the CCSD(T)/CBS calculations, as shown in the SI (Table S7), were used. It was found that the change in temperature affects the thermochemical parameters of both reactions quite dramatically as shown in Table 4. The calculations were performed at three different temperatures, the lowest possible decomposition temperature was taken as 653 K and the highest possible temperature was assumed to be 823 K, along with standard room temperature 298.15 K for comparison. From this it was observed that the formation of ketene was favoured at elevated temperatures with −ΔG°.
Kinetic calculations
The proposed pyrolysis decomposition pathways of diketene were plotted as shown in Fig. 4 with the corresponding TS structures (TS1 and TS2) shown in Fig. 5. Of the two pathways, the formation of ketene (pathway I) was favoured kinetically (Ea ~236 kJ/mol) compared to that for the formation of allene and CO2 (pathway II; Ea ~248 kJ/mol) at standard temperature (298.15 K). Relative energies (at the CCSD(T)/CBS level of theory) of all species in the pathway reactions are given in the SI (Tables S7 and S8). Calculated coordinates of TS structures are given in SI (Tables S9 and S10).

Energy profile diagram for pathways I and II (TS1 and TS2 respectively) for decomposition of diketene at 298.15 K using CCSD(T)/CBS level of theory. Relative energies are given in kJ/mol

Predicted TS structures, optimized at the M06-2X/cc-pVTZ level with atom numbering. TS1 leads to 2 equivalents of ketene and TS2 leads to allene and CO2
A previous experimental kinetic study [44] found the relative activation energy of ketene formation by pyrolysis of diketene was 209.2 kJ/mol. The reverse dimerization process was found to have an activation energy of 129.7 kJ/mol by extrapolating the Arrhenius plot from the experimental reaction flow system. In our work, the calculations at CBS-QB3 and CCSD(T)/CBS level return energies that are 235.8 kJ/mol and 98.8 kJ/mol & 193.7 kJ/mol and 111.3 kJ/mol respectively for the decomposition and dimerization of ketene at 298.15 K.
Previous computational work predicted that pathway I is favoured kinetically using both G2M and BAC-G3B3 levels of theory (Ea for pathway I ~190.0 kJ/mol and pathway II ~200.0 kJ/mol) [13]. As discussed earlier, we have predicted the thermochemical parameters by using a computationally demanding and accurate level of theory, CCSD(T)/CBS, which has not been done before. This finding suggested that the decomposition of diketene still favours pathway II to form allene and CO2 at elevated temperatures, but the formation of ketene also becomes energetically more favourable. The kinetic calculations showed that the small difference in Ea facilitates the formation of ketene at all temperatures, although we predict that allene and CO2 should still be formed in observable quantities. This observation explains the early experimental observations [13, 44] of significant levels of allene in the product stream.
Thermal equilibrium constants and transition state theory (TST) rate coefficients at different temperatures were calculated using the modular program Kinetic and Statistical Thermodynamic Package (KiSThelP) version 2021 [45] in Java runtime environment using the Gaussian output files of reactant, TSs and products (M06-2X/cc-pVTZ level of theory) for reactions (I) and (II). The calculated values are given in the SI (Table S11). The ratios of equilibrium constants of reaction (I) to reaction (II) (taken from output file of the program KiSThelP [45]) were 9.36×10−17, 1.50×10−7 and 6.12×10−6 respectively at 298.15, 653 and 823 K. Both reactions (I) and (II) are predicted to occur spontaneously at 653 and 823 K with ΔG < 0, ΔS > 0 and K > 1. By applying transition state theory, KiSThelP [45] was used to determine the rate constants at 298.15 K. These values for reaction (I) and (II) were calculated as 2.34×10−21 and 4.81×10−23 s−1. Their branching ratios were 3.44 at 653 K and 2.25 at 823 K.
Mechanism of decomposition
Figure 2 (above) shows two possible pathways for the decomposition of diketene, both following a concerted single-step mechanism. The concerted nature of both TS1 and TS2 were confirmed by the internal reaction coordinate calculations. It would be expected that, due to the high electron affinity of the oxygen atom in the diketene ring, formation of ketene would be favoured via a (2+2) retro-Diels-Alder mechanism [46, 47]; however, this is not what we observed from our calculations or indeed what was observed experimentally. The transition states are very close in energy meaning that both pathways are likely to accessed leading to formation of allene and CO2 as well as ketene. Ketene is known to dimerize rapidly to diketene helping explain why it is difficult to observe experimentally. The process is kinetically (Ea = 111.3 kJ/mol at M06-2X/cc-pVTZ) and thermodynamically (ΔG = −38.6 kJ/mol at CCSD(T)/CBS) favourable at room temperature, which was not explained in the previous study [14].
Conclusions
Our work reveals that the thermal decomposition of diketene occurs via a single-step concerted mechanism, supported by the prediction of the transition state structures, TS1 and TS2. Kinetically, pathway I to form ketene (Ea = 235.8 kJ/mol at M06-2X/cc-pVTZ) is favoured. The formation of allene and CO2 is thermodynamically feasible with ΔG = −21.5, −23.0 and −59.6 kJ/mol respectively at CCSD(T)/CBS, CBS-QB3 and M06-2X/cc-pVTZ level of theory under standard conditions of temperature and pressure (298.15 K and 1.0 atm). At elevated temperatures (653 and 823 K) both reactions satisfy the condition of spontaneity (ΔG < 0, ΔS > 0 and K > 1). Transition state theory was applied to calculate the ratios of rate constants for reaction (I) to reaction (II) as 48.60, 3.44 and 2.25 respectively at 298.15, 653 and 823 K. It was observed that the formation of ketene dominates at all calculated temperatures and the presence of allene and CO2 can be explained from the thermodynamic analysis.
Data availability
Available on request.
References
Brady TW (1981) Synthetic applications involving halogenated ketenes. Tetrahedron 37:2949–2966
Bildik AE, Hubbe MA, Gürboy KB (2016) Alkyl ketene dimer (AKD) sizing of paper under simplified treatment conditions. Tappi J 15:545–552
Ramachary DB, Venkaiah C, Reddy YV, Kishor M (2009) Multi-catalysis cascade reactions based on the methoxycarbonylketene platform: diversity-oriented synthesis of functionalized non-symmetrical malonates for agrochemicals and pharmaceuticals. Org and Biomol Chem 7:2053–2062
Kumar S, Chauhan VS, Chakrabarti SK (2016) Separation and analysis techniques for bound and unbound alkyl ketene dimer (AKD) in paper: a review. Arab J Chem 9:S1636–S1642
El-Tahlawy K, Venditti R, Pawlak J (2008) Effect of alkyl ketene dimer reacted starch on the properties of starch microcellular foam using a solvent exchange technique. Carbohydr Polym 73:133–142
Staudinger H (1905) Ketene, Eine Neue Korperklasse. Ber Dtsch Chem Ges 38:1735–1739
Tidwell TT (2005) Ketenes II2nd edn. John Wiley & Sons, Inc, Hoboken, New Jersey
Wilsmore NTM (1907) CLXXXVIII-Ketene. J Chem Soc 91:1938–1941
Chick F, Wilsmore NTM (1908) LXXXIX. - Acetylketen: a polymeride of keten. J Chem Soc Trans 93:946–950
Clemens RJ (1986) Diketene. Chem Rev 86:241–318
Boese AB (1940) Diketene: a new industrial chemical. Ind Eng Chem 32:16–22
Andreades S, Carlson HD (1965) Ketene. Org Syn 45:50–52
Bui B, Tsay TJ, Lin MC, Melius CF (2007) Theoretical and experimental studies of the diketene system: product branching decomposition rate constants and energetics of isomers. Int J Chem Kinet 39:580–590
Noble-eddy R (2009) Gas-phase electron diffraction studies of unstable molecules(Ph.D. Thesis). University of Edinburgh
Atkinson SJ, Noble-Eddy R, Masters SL (2016) Gas-phase structures of ketene and acetic acid from acetic anhydride using very-high-temperature gas electron diffraction. J Phys Chem A 120:2041–2048
Frisch MJ, Head-gordon M, Pople JA (1990) A direct MP2 gradient method. Chem Phys Lett 166:275–280
Hehre WJ, Ditchfield R, Pople JA (1972) Self-consistent molecular orbital methods. XII. further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules published by the AIP publishing articles you may be interested in self-consistent molecular orbital. J Chem Phys 56:2257–2261
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650–654
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and 12 other functionals. Theor Chem Acc 119:525
Woon DE, Dunning TH (1995) Gaussian basis sets for use in correlated molecular calculations. V. core-valence basis sets for boron through neon. J Chem Phys 103:4572–4585
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE et al (2010) Gaussian 09, Revision B.01. Gaussian, Inc, Wallingford, CT
Valiev M, Bylaska EJ, Govind N, Kowalski K, Straatsma TP, van Dam HJJ, Wang D, Nieplocha J, Apra E, Windus TL, de Jong WA (2010) NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun 181:1477–1489
Frisch E, Hratchian HP, Dinnington II RD, Keith TA, Millam J, Nielsen AB, Holderand AJ, Hiscocks J (2009) GaussView 5 Reference. Wallingford, CT
Peng C, Bernhard Schlegel H (1993) Combining synchronous transit and Quasi-Newton methods to find transition states. Isr J Chem 33:449–454
Gaussian 09 User’s Reference Gaussian I, Petersson GA (2005) Complete basis set models for chemical reactivity: from the helium atom to enzyme kinetics. Quantum-mechanical prediction of thermochemical data. Springer, Netherlands, pp 99–130
Poudel P, Ja'o AM, Masters SL (2022) Should pyrolysis of diazotetranoic acid produce methylene ketene? A theoretical structural, thermochemical and kinetic study. Chem Phys Lett 802:139770
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (1997) Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J Chem Phys 106:1063–1079
Matus MH, Anderson KD, Autrey ST CDM, Dixon DA (2007) Reliable predictions of the thermochemistry of boron-nitrogen hydrogen storage compounds: BxNxHy, x = 2, 3. J Phys Chem A 111:4411–4421
Helgaker T, Klopper W, Koch H, Noga J (1997) Basis-set convergence of correlated calculations on water. J Chem Phys 106:9639–9646
Dunning TH (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Com Chem 27:1787–1799
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Bregman J, Bauer SH (1955) An electron diffraction study of ketene dimer, methylketene dimer and β-propiolactone. J Am Chem Soc 77:1955–1965
Kay MI, Katz LA (1958) Refinement of the crystal structure of ketene dimer. Acta Crystall 11:897–898
Orr VL, Esselman BJ, Dorman PM, Amberger BK, Guzei IA, Woods RC, McMahon RJ (2016) Millimeter-wave spectroscopy, X-ray crystal structure, and quantum chemical studies of diketene: resolving ambiguities concerning the structure of the ketene dimer. J Phys Chem A 120:7753–7763
Bader AR, Gutowsky HS, Williams GA, Yankwich PE (1956) The proton magnetic resonance spectrum and structure of diketene. J Am Chem Soc 78:2385–2387
Miller FA, Koch SD, Miller FA (1948) Diketene: infrared spectrum and structure. J Am Chem Soc 70:1890–1894
Mille FA, Carlson GL (1957) Diketene: infrared spectrum and structure II. J Am Chem Soc 79:3995–3997
Katz L, Lipscomb WN (1952) The crystal and molecular structure of diketene. Acta Crystallogr 5:313–118
Taylor Broun T, Livingston RL (1952) An electron diffraction investigation of the molecular structures of ketene, carbonyl fluoride and tetrafluoroethylene. J Am Chem Soc 74:6084–6091
Johnson HR, Strandberg MWP (1952) The microwave spectrum of ketene. J Chem Phys 120:687–695
Chickos JS, Sherwood DE, Jug K (1978) Mechanism of thermolysis of diketene in the gas phase. J Org Chem 43:1146–1150
Canneaux S, Bohr F, Hénon E (2014) KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results. J Comp Chem 35:82–93
Bruce R (1998) The Retro-Diels-Alder reaction part II. Dienophiles with one or more heteroatom. Org React 53:223–629
Bruce R (1998) The Retro-Diels-Alder reaction part I. C=C dienophiles. Org React 52:1–393
Acknowledgements
We are thankful to Dr Ricardo Lopez Gonzalez and Mr Hector Mancilla Diaz (University of Canterbury) for valuable discussion about the mechanism of organic reactions. P.P. and S.L.M. thank the New Zealand eScience Infrastructure (NeSI) for supercomputing resources. P.P. also thanks the School of Physical and Chemical Science for funding to write the paper.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This work was supported by the School of Physical and Chemical Sciences, University of Canterbury.
Author information
Authors and Affiliations
Contributions
S.L.M. conceived and directed the project. P.P. ran the QM calculations. P.P. and S.L.M. drafted the manuscript, and both authors contributed significantly to the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1:
Optimized structural parameters for all molecules involved in reactions (I) and (II). Calculated Cartesian coordinates at different levels of theory for all molecules including transition states. Energies and corrections for thermochemical and kinetic calculations. Calculated equilibrium constants and rate constants and their ratios for diketene decomposition reactions (I) and (II) at different temperatures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poudel, P., Masters, S.L. Structural, thermochemical and kinetic insights on the pyrolysis of diketene to produce ketene. J Mol Model 29, 168 (2023). https://doi.org/10.1007/s00894-023-05572-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05572-x