
Abstract
N-Methylated derivatives of α-tocopheramine, which have preliminarily been shown to have good performance as stabilizers of cellulose solutions in ionic liquids for production of cellulosic manmade fibers, have not been accessible in sufficient amounts by green syntheses. In this study, the N-methyl-, N,N-dimethyl-, and N,N,N-trimethylammonium derivatives of α-tocopheramine were synthesized and fully analytically characterized. The procedures used dimethyl carbonate as solvent and methylating agent as well as aluminum oxide as the reusable catalyst. Care was taken to ensure that the procedures conformed to green chemistry principles and were easily upscalable.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
α-Tocopheramine (1) has been described as early as 1942. It is distinguished from its more popular brother – α-tocopherol (2), the main component in vitamin E by an amino group instead of tocopherol´s phenolic OH group [1, 2]. Tocopheramines are biocompatible [2, 3], have been proposed as food and feed additives [4, 5] and studied for their promising anticancer and proapoptotic activities [6,7,8,9,10]. Similar to tocopherols, which made the move from medical, physiological uses and usage as major food/feed additives to all kinds of technological applications as antioxidants, also tocopheramines have been tested with good success with respect to general uses as antioxidant stabilizers in technical applications, such as surfactants [11], polymer stabilizers [12], and stabilizers for Lyocell spinning dopes and the resulting cellulosic fibers [13, 14]. However, large scale application had been hampered by two facts: first, the limited availability: α-tocopheramine is usually synthesized according to a rather complex multi-step approach which has not yet left lab scale [15]. Second, the nature of the byproducts produced from α-tocopheramine especially with regard to chromophoric degradation products has remained unclear until recently. α-Tocopherol, by contrast, is produced on a large industrial scale and its oxidation chemistry and reaction products in all kinds of different setups and reaction systems have been extensively described [16, 17]. At least the obstacle of unknown degradation products has been removed recently, with the structural elucidation and analytical characterization of all monomeric N-oxidation [18] and dimeric N–N coupling products of α-tocopheramine [19]. It can also be expected that the problem of limited availability will be tackled with new vigor as recent results have shown tocopheramines to be excellent stabilizers for cellulosic fiber spinning from ionic liquid solutions [13] which should result in much greater demand to be met.
α-Tocopherylquinone (3) [17, 20] is the main product of the oxidation of α-tocopheramine under aqueous conditions or in the presence of water, accompanied by small amounts of monomeric N-oxidized derivatives (4–6), usually around 2–5% (Scheme 1). Under non-aqueous conditions, dimeric, N,N-coupled oxidation products, namely N,N-bis(tocopheryl)hydrazine (7), azo-tocopherol (8), and azoxytocopherol (9), dominate [21,22,23,24,25] and no tocopherylquinone (3) is formed. Compared to α-tocopherol, which forms 3 in aqueous media and the transient 5a-ortho-quinone methide intermediate (10) [26,27,28] that dimerizes into the α-tocopherol spiro-dimer (11) [29, 30] in organic media (Scheme 1), α-tocopheramine gives rise to a wider spectrum of oxidation products, due to the different possible oxidation and coupling products of its amino function.

Tocopheramine derivatives have recently been employed as stabilizers for cellulose solutions in N,N-dialkylimidazolium ionic liquids, to hold autoxidation reactions at bay that otherwise would oxidatively damage the polymer, decrease the molecular weight of the celluloses and cause discoloration of the spinning dope and the resulting fibers [13]. Interestingly, the N-alkyl-protected derivatives, available only in milligram amounts, performed better than the parent α-tocopheramine, both with regard to the antioxidative effect and with regard to the minimization of chromophoric compounds. This might seem logical insofar as certain UV-active oxidative byproducts, such as the nitroso, nitro, azo, and azoxy derivatives (5, 6, 8, 9), can readily be formed starting from of α-tocopheramine itself, but not from N-alkylated variants. Unfortunately, the N-alkylated derivatives – with the N-methylated compounds as the structurally simplest representatives – are not available, nor are synthesis procedures published.
This provided the impetus to the present study: development of a synthetic approach to N-methylated tocopheramines and, as reported in follow-up accounts, identification of their oxidation products. As side conditions, the synthesis approach was supposed to employ green chemistry principles as strictly as possible, to be compatible with the sustainable cellulose fiber spinning approaches in which the antioxidants are to be used. It can reasonably be hypothesized that N-methylation of the amino function in tocopheramine derivatives simplifies its oxidation chemistry in a way that the number of possible byproducts is decreased and that these byproducts are less chromophoric compared to α-tocopheramine (1). In this study, we consequently report the synthesis of the N-methyl, N,N-dimethyl, and N,N,N-trimethylammonium derivative of α-tocopheramine (1) in facile one-pot, multigram approaches, along with their comprehensive analytical data.
Results and discussion
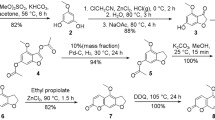
α-Tocopheramine (1) was the obvious starting material for our synthesis attempts. While chemical literature is full of procedures to methylate or permethylate amines, most N-alkylation approaches employ methyl halides, and reduction procedures use the (para)formaldehyde / formic acid couple (Leuckart–Wallach type reactions) [31, 32]. Both options seemed less optimal to us from the viewpoint of sustainable chemistry. Nevertheless, we tested different methylations with methyl iodide, and the variant in acetone in the presence of pulverized anhydrous K2CO3 worked best, giving convincing yields of 82%. Apparently, the initially formed N-methyl-α-tocopheramine (12) was a better nucleophile than the starting amine 1, because even with a 1.4-fold molar excess of alkylating agent, some starting amine remained unmethylated. With a 1.5-fold molar excess, the starting amine was completely consumed, but half of the starting amine was converted to the N,N-dialkylated product N,N-dimethyl-α-tocopheramine (13) besides the targeted N-monomethylated 12, so that chromatographic purification was required (46% and 49%, respectively). The alternative Leuckart–Wallach approaches provided always a mixture of mono- and demethylated products when using less than 2.5 molar HCHO-equivalents. Even at small ratios of HCHO and amine of 0.5 and below, the tertiary amine (dimethylated product) dominated over the secondary (monomethylated) one: at a HCHO/1 ratio of 0.3 and 0.5, the ratio between 12 and 13 was 1:2.4 and 1:2.8, respectively. At a molar HCHO/1 ratio larger than 2.5, yields of 13 were convincingly high (92%), slightly reduced only by a chromatographic purification step that turned out to be necessary to remove the dark brown discoloration of the crude product.
The major objection against these two approaches was the use of the cancerogenic reagents, methyl halide or formaldehyde, which were largely incompatible with our intention to adhere to green chemistry principles. After some additional trials, we resorted to a procedure that used dimethyl carbonate, one of the recommended sustainable and very cheap solvents, as the methylating agent. Under appropriate conditions, i.e., in the absence of water and the presence of a basic catalyst, dimethyl carbonate is able to methylate amino groups in excellent yields and in a “traceless” manner, generating only water, CO2 and some methanol as the byproducts. In our previous studies, dimethyl carbonate had been used in this way for methylation of amino functions, in the synthesis of isotopically labeled variants of the cellulose solvents N,N-dimethylacetamide, N-methylmorpholine-N-oxide and 1-alkyl-3-methylimidazolium acetate [33, 34].
Based on these experiences, we used dimethyl carbonate as solvent and reagent, in combination with aluminum oxide (alox) as a water trap and catalyst. Being used most commonly as bulk stationary phase for chromatography similar to silica gel, aluminum oxide is non-toxic, cost-effective, easy to remove by filtration, recyclable by calcination, and very effective at adsorbing polymeric and chromophoric byproducts. It is commercially available in different degrees of hydration (Brockmann grades I–V) and has either Lewis-acidic or Lewis-basic character; neutral alox is actually a mixture of both variants.
The reaction setup for the methylation was intriguingly simple, the conversion proceeds in a pressure reactor (autoclave) under stirring, in the presence of excess dimethyl carbonate in which aluminum oxide is suspended. From the optimization experiments, it was evident that a higher hydration state (larger Brockmann numbers) was detrimental: the yield dropped to 35% when using grade II instead of grade I, and down to 8% for Brockmann grade III. Interestingly, basic aluminum oxide gave rise to the tertiary amine only, not even low yields of quaternization products (ammonium salts) were formed. With acidic alox, by contrast, only the quaternary ammonium salt was produced, and no non-quaternized amines remained. This offered a very convenient way to switch between permethylation to the tertiary amine and quaternization to the ammonium salt simply by using another type of aluminum oxide as the solid catalyst.
Using the reaction conditions of previous work (350 °C for 3 h), we observed complete consumption of the starting tocopheramine (1) in the presence of basic alox, a yield of 72% of the targeted N,N-dimethyl-α-tocopheramine (13), absence of the quaternized product, and formation of byproducts as black, viscous tar. As higher reaction temperature and longer reaction times promote side reactions, in particular chromophore formation, we tried to reduce temperature and reaction time as much as possible, while in return increasing the amount of alox to keep the conversion complete. The conditions were optimized in a design-of-experiment approach using temperature, reaction time, and mass of catalyst as the influencing factors. The optimized conditions used a temperature as low as 120 °C for 30 min with a fivefold mass of alox (Brockmann grade I, relative to 1) and 50 cm3 of dimethyl carbonate up to 5 g of tocopheramine (see Scheme 2). Under these conditions, conversion of 1 stayed complete, but chromophore formation was almost completely suppressed, and the reaction mixture appeared only slightly yellow. By adding another five mass-equivalents of alox after cooling the reaction mixture to room temperature, the colored byproducts were completely adsorbed to the solid and were removed together with the solids simply by filtration. No other byproducts were detectable in the colorless filtrate, eliminating any need for chromatographic purification. With 91%, the yield of 13, a colorless oil, was quite satisfactory.

The yield of 13 can even be increased by another 6% by washing the solid alox remainder with additional solvents. However, even very apolar solvents (n-hexane, n-heptane, or petroleum ether) eluted not only residual adhering product, but also some of the colored byproducts, so that the minor yield gain would come at the expense of an additional chromatographic purification step with much higher overall solvent usage, which has to be critically considered with regard to economy and sustainability. It should be noted that the absolute amount of chromophores as usual in chromophore chemistry [35] was very low, in the sub-milligram range, which might not seem to be a critical issue. However, as the compounds are to be applied as stabilizers of spinning dopes for cellulose fibers, the starting discoloration should be as low as possible and any intake of chromophores needs to be minimized.
Under otherwise identical reaction conditions, with the only exception of using acidic aluminum oxide (Brockmann grade I) instead of basic one, the starting amine 1 was neatly quaternized to N,N,N-trimethyl-α-tocopherol ammonium, obtained initially as a mixture of carbonate and methyl carbonate as the counter anions (14-I). By dissolving the crude product in acetic acid, trituration with active charcoal and filtration through celite to remove chromophores, and evaporation under reduced pressure, N,N,N-trimethyl-α-tocopherol ammonium acetate (14) was obtained as a colorless waxy solid (Scheme 2). Alternatively, the same compound was also obtained by adding the acidic aluminum oxide to the reaction mixture containing the tertiary amine 13, to afford its quaternization under otherwise identical conditions.
The two methylation approaches afforded either the tertiary amine 13 or the ammonium salt 14, but did not offer access to the N-monomethylated, secondary amine, N-methyl-α-tocopheramine (12). To synthesize this compound, we used a previously developed approach for quantitative N-mono-demethylation of tertiary N-methylamines according to a facile chromatography-like setup [36]. The amine to be mono-demethylated passes slowly through a column with different reaction zones, the first one oxidizing the tertiary amine to the corresponding amine N-oxide, the second one effecting deoxygenative demethylation into secondary amine and HCHO, and the third one being the purification zone which removes the byproducts. The deoxygenative demethylation is based on the autocatalytic degradation of amine N-oxides by carbenium-iminium ions (Mannich intermediates) [14]. Based on this mechanism, demethylation occurs strictly just once in N,N-dimethylamino structures. This demethylation method, which has been reported to afford excellent yields for structurally diverse tertiary N-methylamines [36], worked also very well in the present case, giving a 94% yield of 12. The solvent used in the original demethylation protocol, chloroform, was replaced by the more sustainable dihydrolevoglucosenone (cyrene®) [37] without yield penalty. Preliminary experiments indicated that also oxidation of the tertiary amine 13 by the laccase mediator system [38] caused N-demethylation to give N-methyl-α-tocopheramine (12) in good yields above 70%, but this approach was not further followed.
Table 1 summarizes the 1H and 13C NMR data of the N-methylated compounds 12–14 in comparison to parent amine 1. The effects of N-methylation on the chemical shifts of the chroman system in α-tocopheramine are rather small, and no influence of methylation whatsoever on the chemical shifts of the isoprenoid side chain was seen. With increasing number of N-methyl groups, their 1H NMR shift is down-field shifted from 2.44 to 2.52 and 3.84 ppm in 12, 13, and 14, respectively. There was a peculiar down-field shift by 0.14 ppm for the H-3 methylene protons in the ammonium derivative 14. N-Methylation caused a down-field shift of carbon resonances in the aromatic system, affecting in particular the ortho-carbon atoms C-5 and C-7 (9–14 ppm) and the ipso- and para-carbon atoms (4–6 ppm). These shift effects increased over the monomethylated compound 12 to the dimethylated derivative 13, and are reduced again by quaternization (14). Quaternization causes a slight down-field shift also of the meta-carbon resonances, C-4a and C-8a (3 – 4 ppm), which was not seen in the case of 12 and 13.
Stored in an inert gas atmosphere (Ar) in the dark, all three derivatives 12–14 were completely stable at room temperature. No discoloration was detectable even after storage over more than 6 weeks under these conditions. A comparison sample stored under air in the dark showed a slight yellow discoloration after 2 weeks which, however, did not become more pronounced upon longer storage. All three derivatives 12–14 are soluble in common organic solvents, are tolerant towards acids (1 M H2SO4, glacial acetic acid) and alkali (1 M NaOH). No instabilities or rearrangements, as for instance in the case of the nitroso-derivative 6 upon alkali treatment [18], were observed.
As mentioned introductorily, smaller amounts of the N-methyl derivative 12 and N,N-dimethyl derivative 13 have been tested preliminarily as stabilizers of cellulose spinning dopes in imidazolium ionic liquids against thermal and antioxidative degradation [13, 39]. By extraction of the aqueous spinning baths with petroleum ether, non-reacted tocopheramines and their reaction products were separated, being readily retrievable because of their strongly lipophilic nature due to the isoprenoid side chain. In addition to these tocopherol derivatives, the extract contained smaller amounts (11% of extracted mass) of benzoid/furanoid compounds, derived from thermal aging and oxidation of the polysaccharide components, cellulose and hemicelluloses [40], via furfural and 5-hydroxymethylfurfural intermediates [41]. In contrast to α-tocopheramine (1) with its rather extensive range of possible oxidation products (cf. Scheme 1), only two products were formed from N-methyl-α-tocopheramine (12): hydroxylamine 15 and N,N-dimethyl-N,N-bis(tocopheryl)hydrazine (16), and only one product from N,N-dimethyltocopheramine (14), namely N,N-dimethyl-α-tocopheramine N-oxide (17), see Scheme 3. Structural elucidation of these compounds was based on NMR (see Table 2) and MS data (experimental part) as well as the comparison with authentic, independently synthesized samples.

The tetrasubstituted hydrazine 16 was synthesized by methylation of the N,N-bis(tocopher)hydrazine 7, which was available from previous work [18], see Scheme 3. The protocol was analogous to the ones used in Scheme 2. The amine N-oxide 17 was obtained by oxidation of the tertiary amine 13 with 30% hydrogen peroxide at room temperature in dioxane. Treatment of secondary amine 12 with 10% H2O2 under otherwise identical conditions, followed by treatment with aqueous sodium bisulfite solution, afforded hydroxylamine 15. The reaction appeared to proceed via an intermediate, dark-red nitroxyl radical [22], which immediately gave the colorless hydroxylamine 15 upon reduction with bisulfite. Under inert conditions, however, the radical intermediate seemed to be persistent and unreactive towards common EMPO-type radical traps [42], maintaining its red color even after weeks.
The 1H and 13C NMR data of the oxidation products 15, 16, and 17 are listed in Table 2, along with starting amine 1 for convenient comparison. The shift differences between N-methyl derivative 12 and its N–N-coupling product, hydrazine 16, are very minor both in the 1H (< 0.05 ppm) and 13C domains (< 1 ppm); only the 1H resonances of the N-methyl groups (2.44 in 12 vs. 2.28 in 16) can be used for reliable distinction. In the N-hydroxy (15) and N-oxide (17) derivatives, the 13C down-field shift effects on the ortho-, ipso- and para-carbon atoms were similar as discussed above (cf. Table 1). Note the down-field N-methyl shifts of 46.2 ppm in 15 and 52.9 ppm in 17 (relative to 40.8 ppm in N-methyl-α-tocopheramine 12).
The tocopheramine derivatives 12–17 are distinguished only by different substituents and oxidation stages of the nitrogen. To differentiate them only based on their 1H and 13C NMR data can thus be rather challenging. 15 N NMR can provide a much better distinction [18, 19]: therefore, we used the 15N-isotopically labeled model compound 1a, in which the isoprenoid side chain is replaced by a methyl group, available from previous work [43], to synthesize the corresponding 15N-isotopically labeled model compounds (12a-17a). By analogy, many earlier studies on compounds related to vitamin E (α-tocopherol) had relied on the corresponding truncated model compound 2,2,5,7,8-pentamethylchroman-6-ol [44, 45]. The 15N chemical shifts of the N-methylated, isotopically labeled compounds (12a-17a) compounds fell in the expected ranges (Scheme 4), thus being in full support of the 1H and 13C NMR data. Given for comparison, the 15N atom in 1a resonates at -331.3 ppm (see Scheme 4). Note the large down-field shift (more positive value) of the nitrogen in the amine N-oxide 17a.

Conclusion
Synthesis approaches towards N-methylated derivatives of α-tocopheramine (12–14) were developed, allowing the compounds to be prepared in facile one-pot procedures with yields of over 90% and in compliance with green chemistry principles. Any toxic reagents and solvents as well as the need for chromatographic purification are eliminated, and the procedures can be easily expanded to a scale of several 100 g and beyond. With these synthesis approaches at hand, sufficient amounts of the compounds can now be provided for comprehensive testing as stabilizers for cellulose solutions in ionic liquids in the area of fiber spinning. The performance of these compounds under different spinning conditions and their reactivity differences will be the topics of an upcoming account.
Experimental
All chemicals were commercial products, of the highest purity available and used without further purification. HPLC-grade solvents were used for all extractions and workup procedures. Bidistilled water was used for all aqueous extractions and for all aqueous solutions. Petroleum ether had a boiling range of 50—70 °C. 1,4-Dioxane, ethyl acetate, and toluene used in chromatography were distilled before use. α-Tocopheramine (1) was of the [R,R,R]-type, maintenance of stereochemical integrity over the reactions performed was not further checked, however.
All reactions involving non-aqueous conditions were conducted in oven-dried (140 °C, overnight) glassware under an argon atmosphere. TLC was performed using Merck silica get 60 F254 pre-coated plates, and flash chromatography on Baker silica gel (40 µm particle size). All products were purified to homogeneity by TLC / GC analysis; yields refer to isolated, pure products with satisfying elemental analysis data (± 0.2). Elemental analyses were performed at the Microanalytical Laboratory of the University of Vienna. Melting points were determined on a Kofler-type micro hot stage with Reichert-Biovar microscope.
1H NMR spectra were recorded at 300.13 MHz for 1H and at 75.47 MHz for 13C NMR in CDCl3 if not otherwise stated. Chemical shifts, relative to TMS as internal standard, are given in δ values, coupling constants in Hz. 13C peaks were assigned by means of APT, HMQC, and HMBC spectra.
The nomenclature and atom numbering of tocopherols and chromanols as recommended by IUPAC was used throughout [46, 47]. 1H and 13C NMR resonances of the isoprenoid side chain of tocopherols are only insignificantly influenced (Δ < 0.05 ppm) by modifications of the chroman ring [48, 49], and are thus listed only once: 19.7 (C-4aʹ), 19.8 (C-8aʹ), 21.2 (C-2ʹ), 22.7 (C-13ʹ), 22.8 (C-12aʹ), 24.6 (C-6ʹ), 24.8 (C-10ʹ), 28.0 (C-12ʹ), 32.6 (C-8ʹ), 32.8 (C-4ʹ), 37.3 (C-7ʹ), 37.4 (C-9ʹ), 37.5 (C-5ʹ), 37.5 (C-3ʹ), 39.3 (C-11ʹ), 39.9 (C-1ʹ) ppm. Analytical data for α-tocopheramine (1) agreed with the literature: [2] for methods apart from NMR, and [7] for NMR data, and so did the data for the 15N-labeled model compound 1a [43].
N,N-Dimethyl-[(R)-2-[(4R,8R)-4,8,12-trimethyltridecyl]-2,5,7,8-tetramethyl-6-chromanyl]-amine (6-desoxy-6-dimethylamino-α-tocopherol, N,N-dimethyl-α-tocopheramine, 13, C31H55NO)
In a stainless-steel autoclave with Teflon coating, 0.43 g α-tocopheramine (1, 1.00 mmol) was dissolved in 53.50 g freshly distilled dimethyl carbonate (50.00 cm3). Basic aluminum oxide (2.15 g, Brockmann grade I) was added. The vessel was flushed with argon, sealed, heated to 120 °C (20°/min) and stirred for 30 min. Efficient agitation of the alumina was crucial to complete the reaction within the time used. After cooling to r.t., the vessel was opened, another aliquot of basic aluminum oxide (2.15 g, Brockmann grade I) was added, the mixture stirred for 5 min and filtered. The solid was washed with 20 cm3 ice-cold dimethyl carbonate. The solvent of the combined organic phases was removed in vacuo at room temperature to provide 0.42 g (91%) of 13 as a colorless, viscous oil. Rf (toluene) = 0.50; 1H NMR and 13C NMR, see Table 1; EI-MS (70 eV): m/z = 459 (MH+, 55), 458 (36), 232 (40), 191 (100), 57 (25), 44 (70), 43 (10); [α]D20 = + 4.4° cm2 g−1 (c = 1, ethanol).
N,N,N-Trimethyl-[(R)-2-[(4R,8R)-4,8,12-trimethyltridecyl]-2,5,7,8-tetramethyl-6-chromanyl]ammonium acetate (N,N,N-trimethyl-α-tocopherammonium acetate, 14, C34H61NO3)
In a stainless-steel autoclave with Teflon coating, 0.43 g α-tocopheramine (1, 1.00 mmol) was dissolved in 53.50 g freshly distilled dimethyl carbonate (50.00 cm3). Acidic aluminum oxide (2.15 g, Brockmann grade I) was added. The vessel was flushed with argon, sealed, heated to 120 °C (20 °C/min) and stirred for 30 min. Efficient agitation of the alumina was crucial to complete the reaction within the time used. After cooling to r.t., the vessel was opened, 0.50 g active charcoal was added, the mixture stirred for 5 min and filtered through a 2 cm layer of celite. The solid was washed with 20 cm3 ice-cold dimethyl carbonate. The solvent was evaporated in vacuo to a volume of about 10 cm3, and 2 cm3 glacial acetic acid was added, upon which the clear liquid turned cloudy due to evolved gases. After stirring for additional 30 min, vacuum (10–3 Torr) was applied for another 30 min. A waxy, colorless solid was obtained which was recrystallized from petroleum ether to afforded 0.48 g (91%) of 14 as colorless, waxy plates. Rf (toluene) = 0.18; 1H NMR and 13C NMR, see Table 1. EI-MS (70 eV): m/z = 473 ([M-Ac]+, 85), 247 (50), 206 (100), 60 (30), 59 (40), 43 (15); [α]D20 = − 22.4° cm2 g−1 (c = 1, ethanol).
N-Methyl-[(R)-2-[(4R,8R)-4,8,12-trimethyltridecyl]-2,5,7,8-tetramethyl-6-chromanyl]amine (6-desoxy-6-aminomethyl-α-tocopherol, N-methyl-α-tocopheramine, 12, C30H53NO)
The procedure used the previously published demethylation procedure [36], starting with 0.92 g N,N-dimethyl-α-tocopheramine (13, 2.00 mmol) and replacing chloroform by dihydrolevoglucosenone (cyrene®). After evaporation of the solvent, 0.83 g (94%) of 12 was obtained as colorless, viscous oil. Rf (toluene) = 0.42; 1H NMR and 13C NMR, see Table 1. EI-MS (70 eV): m/z = MS (70 eV): m/z = 445 (MH+, 80), 444 (45), 218 (15), 177 (100), 57 (35), 43 (10); [α]D20 = + 62.4° cm2 g−1 (c = 1, ethanol).
N-Methyl-(2,2,5,7,8-pentamethyl-6-chromanyl)amine (12a, C15H23 15NO)
The procedure follows the above protocol for the preparation of 12, employing model compound 13a (1 mmol, 0.25 g) instead of N,N-dimethyl-α-tocopheramine (13). Evaporation of the solvent in vacuo afforded a solid that was crystallized twice from n-heptane to afford 0.22 g (95%) of 12a. M.p.: 80–81 °C; Rf (n-heptane / ethyl acetate, v/v = 10:1) = 0.44; 1H NMR: δ = 2.63 (2H, t, 3 J = 7.2 Hz, 4-CH2), 2.44 (3H, s, N-Me), 2.18 (s, 3H, 5a-CH3), 2.16 (s, 3H, 7a-CH3), 2.12 (s, 3H, 8b-CH3) 1.80 (t, 2H, 3 J = 7.2 Hz, 3-CH2), 1.35 (s, 6H, 2a-CH3) ppm; 13C NMR: δ = 149.2 (C-8a), 138.6 (C-6, d, C-6, JC,N = 24.0 Hz), 126.6 (C-7), 126.3 (C-5), 122.9 (C-4a), 120.0 (C-8), 74.0 (C-2), 40.2 (N-Me), 32.4 (C-3), 27.5 (C-2a), 22.4 (C-4), 12.9 (C-7a), 12.7 (C-5a), 12.0 (C-8b) ppm; 15N NMR: δ = – 336.4 ppm.
N,N-Dimethyl-(2,2,5,7,8-pentamethyl-6-chromanyl)amine (13a, C16H25 15NO)
The procedure follows the above protocol for the preparation of 13, applying model compound 1a (1 mmol) instead of α-tocopheramine (1). Evaporation of the solvent in vacuo and flash chromatography (n-heptane / ethyl acetate, v/v = 10:1) afforded 0.24 g (96%) of 13a. M.p.: 42–44 °C; Rf (n-heptane / ethyl acetate, v/v = 10:1) = 0.69; 1H NMR: δ = 2.63 (2H, t, 3 J = 7.0 Hz, 4-CH2), 2.50 (6H, s, N-Me), 2.17 (s, 3H, 5a-CH3), 2.16 (s, 3H, 7a-CH3), 2.12 (s, 3H, 8b-CH3), 1.79 (t, 2H, 3 J = 7.0 Hz, 3-CH2), 1.36 (s, 6H, 2a-CH3) ppm; 13C NMR: δ = 150.0 (C-8a), 141.1 (d, C-6, JC,N = 18.6 Hz), 130.9 (C-5), 129.8 (C-7), 123.4 (C-4a), 121.4 (C-8), 74.5 (C-2), 40.9 (N-Me), 33.2 (C-3), 27.0 (C-2a), 21.2 (C-4), 17.2 (C-7a), 13.8 (C-5a), 11.8 (C-8b) ppm; 15N NMR: δ = − 341.5 ppm.
N,N,N-Trimethyl-(2,2,5,7,8-pentamethyl-6-chromanyl)-ammonium acetate (14a, C19H31 15NO3)
The procedure follows the above protocol for the preparation of 14, employing model compound 1a (1 mmol) instead of α-tocopheramine (1). Evaporation of the solvent in vacuo and flash chromatography (n-heptane / ethyl acetate, v/v = 10:1) afforded 0.26 g (82%) of 14a. M.p.: 133–136 °C (decomp.); Rf (n-heptane / ethyl acetate, v/v = 10:1) = 0.12; 1H NMR: δ = 3.86 (s, 9H, N-Me), 2.58 (2H, t, 3 J = 6.4 Hz, 4-CH2), 2.22 (s, 3H, 5a-CH3), 2.18 (s, 3H, 7a-CH3), 2.12 (s, 3H, 8b-CH3) 1.93 (t, 2H, 3 J = 6.8 Hz, 3-CH2), 1.36 (s, 6H, 2a-CH3) ppm; 13C NMR: δ = 150.4 (C-8a), 136.1 (d, C-6, JC,N = 26.6 Hz), 129.3 (C-5), 128.4 (C-7), 126.4 (C-4a), 123.0 (C-8), 74.5 (C-2), 52.2 (N-Me), 30.5 (C-3), 29.0 (C-2a), 20.2 (C-4), 19.2 (C-7a), 18.8 (C-5a), 13.8 (C-8b) ppm; 15N NMR: δ = – 304.9 ppm.
References
Smith LI, Renfrow WB, Opie JW (1942) J Am Chem Soc 64:1082
Mayer H, Isler O (1971) Tocopheramines and tocopherthiols. In: Colowick SP, Kaplan NO (eds) Methods in enzymology, vol 18, part C. Academic Press, New York, pp 275–334
Blomstrand R, Forsgren L (1968) Int J Vit Res 38:328
Schlegel W, Schwieter U, Tamm R (1969) Non-toxic antioxidants, based on chromane derivatives US. Pat. 3458637. Chem Abstr 69:21909
Søndergaard E, Dam H (1970) Z Ernähr 10:71
Tomic-Vatic A, Eytina J, Chapman J, Mahdavian E, Neuzil J, Salvatore BA (2005) Int J Cancer 117:188
Gille L, Stamberg W, Patel A, Böhmdorfer S, Rosenau T (2012) Free Rad Biol Med 53:S100
Zingg JM (2007) Mini Rev Med Chem 7:545
Neuzil J, Tomasetti M, Zhao Y, Dong LF, Birringer M, Wang XF, Low P, Wu K, Salvatore BA, Ralph SJ (2007) Mol Pharmacol 71:1185
Gruber J, Staniek K, Krewenka C, Moldzio R, Patel A, Böhmdorfer S, Rosenau T, Gille L (2014) Bioorg Med Chem 22:684
Lambert KJ, Lal M (2002) Preparation of aminobenzopyran derivatives as surfactants. WO Pat. 2002076937. Chem Abstr 137:263202
Tokuwame M (1991) Synergistic antioxidant-heat stabilizer systems for polyolefins JP. Pat. 03043458. Chem Abstr 115:73015
Hettegger H, Zhang J, Koide M, Rinner U, Potthast A, Gotoh Y, Rosenau T (2022) Fibers 10:50
Rosenau T, Potthast A, Kosma P, Chen CL, Gratzl JS (1999) J Org Chem 64:2166
Mazzini F, Netscher T, Salvadori P (2009) Eur J Org Chem 13:2063
Preedy VR, Watson RR (2007) Encyclopedia of vitamin E. CABI Publishing, Oxford
Machlin LJ (1980) Vitamin E: a comprehensive treatise. Marcel Dekker Inc, New York
Patel A, Hofinger A, Rosenau T (2021) Monatsh Chem 152:959
Patel A, Rosenau T (2021) Monatsh Chem 152:1231
Rüegg R, Mayer H, Schudel P, Schwieter U, Tamm R, Isler R (1967) Wiss Veröff Dtsch Ges Ernähr 16:22
Bamonti L, Hosoya T, Pirker K, Böhmdorfer S, Mazzini F, Galli F, Netscher T, Rosenau T, Gille L (2013) Bioorg Med Chem 21:5039
Murphy PA, Lin JS, Olcott HS, Windle JJ (1976) Lipids 11:296
Boguth W, Hackel R (1965) Hoppe-Seyler Z Physiol Chem 342:172
Igarashi O (1977) J Nutr Sci Vitaminol 23:169
Quaife ML (1948) J Biol Chem 175:605
Rosenau T, Potthast A, Elder T, Kosma P (2002) Org Lett 4:4285
Rosenau T, Ebner G, Stanger A, Perl S, Nuri L (2005) Chem Eur J 11:280
Rosenau T, Böhmdorfer S (2009) ortho-Quinone methides in tocopherol chemistry. In: Rokita S (ed) Wiley series on reactive intermediates in chemistry and biology, 1st edn. Wiley, New York, p 163
Schudel P, Mayer H, Metzger J, Rüegg R, Isler O (1963) Helv Chim Acta 46:636
Schröder H, Netscher T (2001) Magn Reson Chem 39:701
Leuckart R (1885) Ber Dtsch Chem Ges 18:2341
Wallach O (1893) Ann Chem 272:99
Adelwöhrer C, Yoneda Y, Nakatsubo F, Rosenau T (2008) J Label Comp Radiopharm 51:28
Adelwöhrer C, Yoneda Y, Takano T, Nakatsubo F, Rosenau T (2009) Cellulose 16:139
Rosenau T, Potthast A, Milacher W, Hofinger A, Kosma P (2004) Polymer 45:6437
Rosenau T, Hofinger A, Potthast A, Kosma P (2004) Org Lett 6:541
Sherwood J, de Bruyn M, Constantinou A, Moity L, McElroy CR, Farmer TJ, Duncan T, Raverty W, Hunt AJ, Clark JH (2014) Chem Comm 50:9650
Potthast A, Rosenau T, Fischer K (2001) Holzforschung 55:47
Liebner F, Ebner G, Becker E, Potthast A, Rosenau T (2010) Holzforschung 64:161
Korntner P, Hosoya T, Dietz T, Eibinger K, Reiter H, Spitzbart M, Röder T, Borgards A, Kreiner W, Mahler AK, Winter H, French AD, Henniges U, Potthast A, Rosenau T (2015) Cellulose 22:1053
Thoma C, Konnerth J, Sailer-Kronlachner W, Solt P, Rosenau T, van Herwijnen HWG (2020) Chemsuschem 13:3544
Stolze K, Udilova N, Rosenau T, Hofinger A, Nohl H (2003) Biol Chem 384:493
Böhmdorfer S, Gille L, Rosenau T (2010) Lett Org Chem 7:335
Rosenau T (2007) 2,2,5,7,8-Pentamethylchroman-6-ol (PMC) and related model compounds. In: Preedy VR, Watson RR (eds) Encyclopedia of vitamin E. CABI Publishing, Oxford, p 21
Patel A, Liebner F, Netscher T, Mereiter K, Rosenau T (2007) J Org Chem 72:6504
IUPAC-IUB Commission on Biochemical Nomenclature (CBN) (1982) Eur J Biochem 123:473
IUPAC-IUB Commission on Biochemical Nomenclature (CBN) (1974) Arch Biochim Biophys 165:1
Urano S, Hattori Y, Yamanoi S, Matsuo M (1980) Chem Pharm Bull 28:1992
Brownstein S, Ingold KU (1989) J Org Chem 54:560
Acknowledgements
The financial support of the Austrian Biorefinery Center Tulln (ABCT) is gratefully acknowledged.
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, A., Rosenau, T. Synthesis and analytical characterization of N-methylated derivatives of α-tocopheramine and their oxidation products. Monatsh Chem 153, 919–928 (2022). https://doi.org/10.1007/s00706-022-02970-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-02970-4