
Abstract
Predictive biomarkers are the mainstay of precision medicine. This review summarizes the advancements in tissue-based diagnostic biomarkers for gastric cancer, which is considered the leading cause of cancer-related deaths worldwide. A disease seen in the elderly, it is often diagnosed at an advanced stage, thereby limiting therapeutic options. In Western countries, neoadjuvant/perioperative (radio-)chemotherapy is administered, and adjuvant chemotherapy is administered in the East. The morpho-molecular classification of gastric cancer has opened novel avenues identifying Epstein–Barr-Virus (EBV)-positive, microsatellite instable, genomically stable and chromosomal instable gastric cancers. In chromosomal instable tumors, receptor tyrosine kinases (RKTs) (e.g., EGFR, FGFR2, HER2, and MET) are frequently overexpressed. Gastric cancers such as microsatellite instable and EBV-positive types often express immune checkpoint molecules, such as PD-L1 and VISTA. Genomically stable tumors show alterations in claudin 18.2. Next-generation sequencing is increasingly being used to search for druggable targets in advanced palliative settings. However, most tissue-based biomarkers of gastric cancer carry the risk of a sampling error due to intratumoral heterogeneity, and adequate tissue sampling is of paramount importance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric adenocarcinoma (GC) is the fifth most common cancer and the third most common cause of cancer-related deaths, accounting for almost 800.000 deaths world-wide (Smyth et al. 2020). The incidence and prevalence of GC varies geographically, and its prevalence is twice as high among men than in women. Emerging data show that East Asia, Central Europe, and Eastern Europe have the highest rates of GC, collectively accounting for 87% of all new cases registered worldwide. In Africa and North America, significantly lower rates have been observed (Smyth et al. 2020). During the past 30–50 years, the standardized incidence rates of non-cardia GC have declined worldwide. However, cancer of the proximal stomach/cardia and esophagogastric junction has either been stable or even increased. Typically, the incidence of GC is predominantly observed in the elderly, often peaking at the 7th and 8th decade of life; moreover, with a rapidly aging population, the prevalence of GC is relatively high (Smyth et al. 2020). In regions reporting low incidence of the disease, it is diagnosed at an advanced stage, thereby restricting targeted therapeutic treatment options, with patient prognosis remaining rather dismal. The disease specific 5 year survival rate for both sexes ranges between 30% and 35%.
Etiology
Gastric adenocarcinoma has a diverse etiology, and the main risk factors include dietary factors, such as high salt intake, tobacco consumption, and Helicobacter pylori (H. pylori) infection (Smyth et al. 2020). Correa proposed a model for gastric carcinogenesis: chronic atrophic gastritis leads to intestinal metaplasia, dysplasia, and finally GC (Correa 1992). In recent years, multiple bacterial virulence factors have been identified for colonization, persistence, ulcers, and cancer risk, such as CagA, DupA, IceA1, NapA, oipA, and VacA (Malfertheiner et al. 2014). Host factors, e.g., polymorphisms in genes coding for pro- and anti-inflammatory cytokines, environmental factors, and socioeconomic status further modulate the individual cancer risk. Proximal GC and esophagogastric junction carcinoma are associated with gastroesophageal reflux disease and Barrett’s mucosa (Smyth et al. 2020).
Approximately 10% of all GC cases have a familial or hereditary trait (Smyth et al. 2020). In familial GC, the main predisposing factors include H. pylori infection, dietary habits, and gene polymorphisms. The underlying germ line mutations causing hereditary type GC are found in 1–3% of all cases (Moehler et al. 2019). Genes which have been linked with hereditary type GC are CDH1 (coding for E-cadherin), CTNNA1 (α-E-catenin) (Majewski et al. 2013; Petrovchich et al. 2016), FBXO24 (F-Box-protein 24), DOT1L (DOT1-like histone H3K79 methyltransferase), INSR (insulin receptor), MAP3K6 (mitogen-activated protein kinase 6), PRSS1 (protease serine 1) (Donner et al. 2015; Petrovchich et al. 2016), and mutations in the APC promoter 1 B (Worthley et al. 2012; Li et al. 2016). GC can also arise in a setting of other well defined hereditary cancer syndromes, such as familial adenomatous polyposis (APC), Cowden syndrome (PTEN), Lynch syndrome (hMLH1, hMLH2), juvenile polyposis (BMPR1A), MUTYH-associated adenomatous polyposis (MUTYH), Li-Fraumeni syndrome (TP53), Peutz–Jeghers syndrome (STK11), and hereditary breast and ovarian cancer (BRCA1/2) (van der Post et al. 2015).
Histology of gastric cancer
The World Health Organization (WHO) subclassifies adenocarcinomas of the stomach into tubular, parietal cell, mixed type, papillary, micropapillary, mucoepidermoid, mucinous, poorly cohesive (including signet-ring cell carcinoma), medullary carcinoma, hepatoid adenocarcinoma, and Paneth cell carcinoma (Board 2019). In spite of that, the Laurén classification is still used in cancer research and many clinical trials (Fig. 1) (Lauren 1965). Laurén classified GC into intestinal, diffuse, mixed, and unclassified types (Lauren 1965). Most GCs (> 90%) are adenocarcinomas. Molecular characterization studies have been conducted and applied to Laurén’s classification (Wang et al. 2014). The morpho-molecular classification system for GC (see below) corresponds only rudimentarily with the current WHO classification (Cancer Genome Atlas Research 2014; Smyth et al. 2020).

Histology of gastric cancer. Intestinal type (A), diffuse type (B), Epstein–Barr-virus positive (C) and microsatellite instable (D) gastric cancer. Note the tumor-associated inflammatory reaction (C). The microsatellite instable gastric cancer shows expression of PD-L1 (E). An example of immunostaining for Claudin 18.2 is shown in (F). Hematoxylin and eosin-staining (A–D), anti-PD-L1-antibody (E), anti-Claudin 18.2-antibody (F). Original magnifications: 400-fold
Molecular subtypes of gastric cancer
The Cancer Genome Atlas Research Network had performed an integrative genomic analysis and proposed four molecular subtypes, which can be used to stratify patients and design clinical trials for targeted therapy: Epstein–Barr virus-associated (EBV), microsatellite instable (MSI), chromosomal instable (CIN), and genomically stable (GS) GC (Cancer Genome Atlas Research 2014; Wang et al. 2014). These studies linked distinct molecular subtypes with some histological phenotypes. It forms the basis for novel therapeutic strategies and precision medicine (Cancer Genome Atlas Research 2014, Wang et al. 2014).
Microsatellite instability in gastric cancers (MSI-GC)
MSI-GCs are characterized by a very high mutational load and CpG-island methylator phenotype (CIMP). The DNA mismatch repair protein MLH1 is frequently silenced in these tumors. MSI-GCs account for 0–44.5% of all GC cases (Cortes-Ciriano et al. 2017; Mathiak et al. 2017; Guan et al. 2021). They are predominantly observed in elderly patients and located in the distal stomach. MSI-GCs harbor significantly lower numbers of lymph node metastases, and have a better overall and tumor-specific survival (Cortes-Ciriano et al. 2017; Mathiak et al. 2017; Smyth et al. 2017; Guan et al. 2021; Quaas et al. 2021a, b). Different from sporadic microsatellite instable colorectal cancer, MSI-GCs lack BRAF-mutations (Cancer Genome Atlas Research 2014) and BRAF cannot be used as predictive biomarker like in malignant melanomas (Warneke et al. 2013a, b).
Frequently, MSI-GCs exhibit rare and indicative histological phenotypes (Fig. 1) (Mathiak et al. 2017) and an abundant tumor-associated inflammatory cell infiltration comprising either neutrophils and/or lymphocytes is commonly noted, with little or no desmoplastic stromal components (Mathiak et al. 2017; Smyth et al. 2020).
For two reasons, MSI-GC classification is clinically relevant: (1) MSI-GCs predict improved patient outcomes (Mathiak et al. 2017; Pietrantonio et al. 2019; Guan et al. 2021); (2) MSI-GCs frequently express immune checkpoint molecules such as PD-L1 (Fig. 1) making them eligible for treatment with immune checkpoint inhibitors (Cancer Genome Atlas Research 2014; Böger et al. 2016; Pietrantonio et al. 2021). MSI-GCs can be classified and identified using antibodies directed against MLH1, PMS2, MSH2, and MSH6, and mononucleotide markers BAT-25, BAT-26, NR-21, NR-24, and NR-27 (Mathiak et al. 2017). In most cases, histological phenotypes could raise a doubt (see above). However, as one-third of tumors do not show a particular phenotype, a more general approach may be required for MSI-testing, that is, upfront testing irrespective of the histological phenotype. Most MSI-GCs demonstrate loss of MLH1-expression. Thus, detecting loss of MLH1 by immunostaining may be a cost-effective approach to search for MSI-GC (Gonzalez et al. 2016). However, other members of the DNA-repair machinery, such as MSH2, albeit rarely observed in GC, can be lost and may indicate Lynch syndrome (Matsubayashi et al. 2022).
The significantly better outcome of MSI-GC compared with microsatellite stable GC raised concerns regarding the necessity of neoadjuvant/perioperative (radio-)chemotherapy and adjuvant chemotherapy in this patient subgroup (Mathiak et al. 2017). In support of this notion, an explorative analysis of the MAGIC trial showed that patients with non-metastatic MSI-GC had a better prognosis after surgery compared to those with microsatellite stable GCs. However, MSI-GCs had a worse prognosis when treated with perioperative chemotherapy (median overall survival 9.6 vs. 19.5 months; hazard ratio [HR] 2.18), also showing no major pathological responses to chemotherapy (Smyth et al. 2017; Petrillo et al. 2020). These data were further supported by a large meta-analysis performed by Pietrantonio et al. (Pietrantonio et al. 2019) including individual patient data from four multicenter randomized clinical trials. Again, patients with MSI-GC, who were treated with surgery alone, performed well even without adjunctive chemotherapy. In contrast, patients with MSI-GC, who were treated with chemotherapy (perioperative or adjuvant), did not benefit from this treatment (Pietrantonio et al. 2019). However, these studies suffer from their retrospective nature and large prospective trials are urgently needed.
MSI may also serve as a predictive biomarker for the administration of immune checkpoint inhibitors (Pietrantonio et al. 2021). MSI-GC frequently express PD-L1 (Fig. 1) (Böger et al. 2016; Cho et al. 2021), and anti-PD-1 agents with or without chemotherapy significantly and consistently improved overall survival, progression free survival, and objective response rate vs. chemotherapy alone in the subgroup of patients with advanced MSI-GC (Pietrantonio et al. 2021).
Epstein–Bar-virus-associated gastric cancer (EBVaGC)
EBVaGC is another molecular subtype, which accounts for 2–20% of all GC cases (Cancer Genome Atlas Research 2014, Wang et al 2014; Böger et al. 2017a, b; Saito et al. 2021), and also shows a CIMP-phenotype. It frequently harbors mutations in PIK3CA and ARID1A, copy-number amplifications of JAK2 and CD274/PDCD1LG2, and a dysregulation of immune cell signaling molecules. Data show that EBVaGC is more prevalent among Caucasians than Asians, predominantly among males, and younger-aged patients. It occurs primarily in the proximal stomach and postgastrectomy remnant stomach. Multiplicity of EBVaGC is frequently encountered (Fukayama et al. 2011; Saito et al. 2021), and lymph node metastases are less often detected compared with EBV-negative GCs (Tokunaga et al. 1998). In Asian populations, EBVaGC may have a better prognosis than EBV-negative GCs (Liu et al. 2015a, b). EBVaGC and MSI-GC are mutually exclusive (Mathiak et al. 2017). Histologically, EBVaGC may show a tubular and intestinal differentiation. A dense infiltrate of lymphocytes is often noted presenting as an undifferentiated phenotype (lymphoepithelioma-like or medullary) (Fig. 1). Hence, any unusual histological phenotype should be forwarded to EBV-testing. This is best done by EBER-in-situ-hybridization providing a nuclear signal. Immunostaining is less sensitive. GC is commonly of type 1 latency and antibodies directed against EBNA2, LMP1, and ZEBRA may not immunoreact with the tumor cells (Tokunaga et al. 1993; Fukayama et al. 2011). In view of the broad morphological spectrum of EBVaGC, testing should be performed at liberty (Park et al. 2016).
EBVaGCs, like MSI-GC, also express PD-L1 and may respond to immune checkpoint inhibitors (Böger et al. 2016; Wei et al. 2021; Zhang et al. 2021). However, data on the efficacy of immune checkpoint inhibitors in EBVaGC are limited. In a recent review, data from 39 patients were summarized (Wei et al. 2021). Among these 39 patients, 12 had survival information, including progression-free and overall survival. Compared to PD-L1 negative patients, PD-L1 positive patients had superior progression-free survival. Thus, while the identification and diagnosis of EB-VaGC may be clinically relevant, further research is required.
Chromosomal instable gastric cancers (CIN GC)
Chromosomal instability drives intratumoral heterogeneity, which supports microenvironmental selection and evolution of cancer cell populations, leading to cancer resistance (Tannock et al. 2016). Thus, recognition of CIN GC is highly important. Although, it may not constitute a distinct subgroup but may rather be a compilation of a more heterogeneous group of tumors (Maleki et al. 2017). Currently, there is no validated simple diagnostic method for the identification of CIN besides somatic copy number alteration analysis or defined molecular markers. According to Laurén classification, CIN GC often shows an intestinal phenotype. Genetically, CIN GC are characterized by frequent mutations in the TP53-tumor suppressor gene (Cancer Genome Atlas Research 2014). The Cancer Genome Atlas Program identified TP53 mutations and the loss of its protein’s pathway to be one of its key characteristics; 71% of their CIN tumors had a TP53 mutation (Cancer Genome Atlas Research 2014). Detection of TP53 mutations might, therefore, be an accurate method to diagnose CIN in GC. In this context, the immunohistochemical staining of protein p53 was considered a useful diagnostic tool, and some groups tried to classify this type of cancer based on p53 protein status (Cristescu et al. 2015; Gonzalez et al. 2016; Setia et al. 2016). However, our own studies provided evidence that p53-immunostaining is unsuitable to predict TP53-mutational status in individual cases and hence CIN GC (Schoop et al. 2020a, b). Other markers might be needed.
Meanwhile, CIN GC also frequently harbors amplifications of genes coding for RTKs, such as EGFR, FGFR2, HER2, and MET (Deng et al. 2012; Kiyose et al. 2012), which represent either validated or putative therapeutic targets, and several are being explored in ongoing clinical trials.
Human epidermal growth factor receptor 2 (HER2)
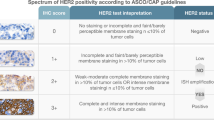
The European Medicines Agency approved in 2010 trastuzumab, a monoclonal anti-body targeting the human epidermal growth factor receptor 2 (HER2; also known as ERBB2), in combination with chemotherapy for first-line treatment of HER2-positive advanced gastric or esophagogastric junction cancer. The addition of trastuzumab to chemotherapy improved survival in these patients’ cancer compared with chemotherapy alone [13.8 months (95% confidence interval (CI) 12–16) vs. 11.1 months (10–13)] (Bang et al. 2010). Following official approval, testing HER2 became the first predictive biomarker for GC (Ruschoff et al. 2012). Many subsequent studies demonstrated that overexpression of HER2 correlates significantly with HER2-gene amplification. Overexpression of HER2 can be heterogeneously distributed in both, the primary tumor as well as in metastases (Fig. 2), and is more commonly found in proximal and intestinal type GCs, respectively (Warneke et al. 2013a, b; Roviello et al. 2022).

Intratumoral heterogeneity of HER2. Ten biopsy specimens with gastric cancer (A) and an intestinal phenotype (B) show overexpression of Her2/neu only in a single biopsy specimen (A, C). Chromogenic in situ hybridization confirms HER2 amplification only in the tumor cells with very strong (3 +) Her2/neu immunostaining (D). Hematoxylin and eosin (B), anti-Her2/neu-antibody (A, C), HER2 chromogenic in situ hybridization (D). Original magnifications 0.5-fold (A), 100-fold (B, C), 400-fold (D)
The assessment of HER2-overexpression is complicated and necessitates training as well as expertise (Haffner et al. 2021). The HER2 scoring system of GC is different from breast cancer scoring and also distinguishes biopsy and resection specimens (Ruschoff et al. 2012). HER2 positive GC resection specimens are defined by ≥ 10% of tumor cells showing basolateral immunostaining intensity of 2 + and a confirmatory in situ hybridization showing HER2 amplification or a strong (3 +) basolateral immunostaining. In biopsy specimens, the cutoff is set at ≥ 5 adjacent cancer cells (Ruschoff et al .2012). Accurate and validated assay methods should be used and participation in external quality assurance schemes is recommended. The difficulties of accurate assessment of the HER2 status and its impact on patient outcome was demonstrated recently by a prospective multicenter study (VARIANZ) (Haffner et al. 2021). The VARIANZ study enrolled prospectively patients at 35 German sites receiving medical treatment for metastatic GC. Follow-up lasted up to 48 months. HER2 status was assessed centrally using immunohistochemistry and chromogenic in situ hybridization. Central HER2 test results were compared with those obtained previously at local sites: central and local test results were available from 374 patients. In 77 cases (20.6%), central testing revealed HER2-positive GC. However, in 22.7% of patients, the locally assessed HER2 status was not confirmed by central testing. In the majority of these cases, a local HER2 + status was not confirmed centrally, whereas, in nine patients, HER2 was tested positive centrally and negative locally. Most interestingly, patients with centrally confirmed HER2-positive GCs had a significantly longer overall survival when treated with trastuzumab plus chemotherapy as compared with patients who tested negative centrally and positive locally [20.5 months (95% CI 15.7–31.5) vs. 10.9 months (95% CI 8.2–14.4; HR 0.42)] (Haffner et al. 2021). The VARIANZ study also refined the test algorithm, and alternative criteria for HER2-directed treatment were proposed. The mean survival in patients with ≥ 40% of HER2-positive tumor cells was 20.5 months (95% CI 15.6–32.6) vs. 11.4 months (95% CI 9.3–15.0; HR 0.46). The optimized HER2/CEP17 ratio for indicating benefit from trastuzumab was 3.0 with a median survival of 22.8 months (95% CI 16.5–81.7) in patients with a HER2/CEP17 ratio ≥ 3.0 vs. 11.7 months (95% CI 9.3–14.8) for patients with a HER2/CEP17 ratio < 3.0 (HR 0.36) (Haffner et al .2021). The VARIANZ study confirms findings made previously by two small Asian studies, in which the effect of homogeneous and heterogeneous expression of HER2 was compared in biopsy and resection specimens (Wakatsuki et al. 2018; Yagi et al. 2019). GCs showing homogeneous overexpression are more likely to respond to HER2-targeted therapy compared with GC overexpressing HER2 heterogeneously (Wakatsuki et al. 2018; Yagi et al. 2019).
Per the HER2 observations and its marked intratumoral heterogeneity (Fig. 2) (Warneke et al. 2013a, b), experts from an interdisciplinary German group accepted GC-specific HER2 testing protocols and recommended a minimum of five tumor-bearing biopsies from the primary site of the tumor growth (Baretton et al. 2016). Similar recommendations for HER2 testing were released by a group of North American organizations (Bartley et al. 2016). The updated recommendation is testing multiple biopsy fragments from either a primary tumor metastasis site or from the resected primary tumor. In the case of biopsy specimens, the present protocol recommends a minimum of five biopsy specimens, optimally, six to eight are required for interpreting intratumoral heterogeneity and providing necessary tumor material for diagnosis and biomarker testing (Bartley et al. 2016).
Hepatocyte growth factor receptor (MET)
MET has pleiotropic effects and induces proliferation, survival, motility, cell scattering, angiogenesis, tubulogenesis, drives epithelial–mesenchymal transition and tumor invasion (Graveel et al. 2013; Hack et al. 2014). Most commonly, the MET pathway is activated in GC by protein overexpression, which can be detected by immunohistochemistry and occurs in 50–65% of cases. MET can be overexpressed in pre-cancerous intestinal metaplasia and dysplastic lesions, underscoring its critical role in gastric carcinogenesis. It is frequently found in well-differentiated tubular adenocarcinoma (67%), intestinal-type (35%), and less commonly in diffuse-type GCs (15–51%). Overexpression of MET has been linked to a more aggressiveness phenotype, i.e., advanced local tumor growth, nodal spread, distant metastasis, advanced tumor stage, recurrence, and poor survival (Metzger et al. 2016). MET can also be activated by gene amplification and, although infrequent, co-amplification with other RTKs can occur in GC. Other activating genetic mutations of MET remain exceedingly rare in GC (for a review see (El Darsa et al. 2020)).
MET was also explored as druggable target in GC (Lennerz et al. 2011; Shah et al. 2013; Teng et al. 2013; Hack et al. 2014; Jardim et al. 2014; Kang et al. 2014), and efficacy may depend on its expression pattern detected by immunohistochemistry and/or in situ hybridization (Hack et al. 2014). Many studies investigated the tumor-biological and clinicopathological characteristics of MET-positive GCs. The prevalence ranged from 3.8 to 85% (Drebber et al 2008; Betts et al. 2014). In our study, any immunostaining of MET, i.e., weak, moderate or strong, was observed in 192 (42.1%) cases (Metzger et al. 2016). The wide spectrum of immunopositivity in the various studies stems from the usage of different types of antibodies and different, non-standardized scoring systems. In fact, until now, no standardized scoring system was established for MET in GC. Furthermore, like HER2, MET shows intratumoral heterogeneity: amplified and unamplified tumor cell clones occur in the same tumor distinguishable on a cell-by-cell level (Catenacci et al. 2011; Nagatsuma et al. 2015; Metzger et al. 2016), further compromising its usage as a predictive biomarker. However, diverse MET inhibitors have been developed and are also currently under investigation in clinical trials (El Darsa et al. 2020). Until now, none has reached formal approval.
Fibroblast growth factor receptor (FGFR2)
The dysregulation of the fibroblast growth factor receptor (FGFR) pathway has been studied as therapeutic target in many different tumor types (Babina et al. 2017). Activation of FGFR signaling is caused by gene amplification, activating mutation, and chromosomal translocations/fusions. In GC, FGFR2 is most frequently mutated among the FGFR family members occurring in approximately 4.0% of the cases with advanced GC. The frequencies of amplifications, mutations, translocations/fusions, and multiple alterations among FGFR2-dysregulated GCs are given as 72, 13, 8.6, and 6.3%, respectively (Ooki et al. 2021) and gene amplification is the common mechanism of FGFR2 overexpression. So far, no association has been established between FGFR2-amplification and gender, anatomical site, histological subtype, or TNM classification (Deng et al. 2012; Silva et al 2018). FGFR2-amplification is not limited to CIN GCs, it was detected in genomically stable GCs (Schrumpf et al. 2022).
The protein can be detected in the cytoplasm and at the cell membrane in both, intestinal and diffuse type GC (Schrumpf et al. 2022). There are two major FGFR2 isoforms, i.e., FGFR2-IIIb and IIIc, which are determined by alternative splicing of a ternary extracellular immunoglobulin (Ig) domain III. In GC, FGFR2-IIIb is the predominantly overexpressed isoform (Ooki et al. 2021). Ueki et al. were the first to report on the potential prognostic significance of FGFR2 in GC (Ueki et al. 1995). FGFR2 gene amplification occurs in 2–9% of the cases (Jung et al. 2012; Matsumoto et al. 2012; Betts et al. 2014; Cancer Genome Atlas Research 2014; Su et al. 2014; Seo et al. 2017; Hur et al. 2020) and has been shown to be an independent prognostic factor for patient survival (Su et al 2014). Overexpression was found in up to 60% of the patients (Tokunaga et al. 2016; Hosoda et al. 2018), the significance of which has been explored in a number of studies. Matsunoba et al. linked high expression with favourable outcome (Matsunobu et al. 2006). To the contrary, four Asian studies linked FGFR2 overexpression to poor overall and tumor-specific survival (Murase et al. 2014; Nagatsuma et al. 2015; Ahn et al. 2016; Hosoda et al. 2018). Inokuchi et al. demonstrated prognostic significance of FGRR2 overexpression only in diffuse-type GC (Inokuchi et al 2017). Recently, a meta-analysis provided evidence that FGFR2 overexpression is associated with greater depth of tumor invasion, higher rates of lymph node metastasis, more advanced disease stage and worse outcome (Kim et al. 2019). However, FGFR2 protein expression in GC was most commonly studied in Asian populations, and data on White patients are scarce (Kim et al. 2019; Schrumpf et al. 2022).
In our own study on FGFR2 in a Western cohort, protein expression detected by immunohistochemistry did not correlate with patient outcome. However, using different cutoff values, a negative correlation between FGFR2-expression and patient survival was found for diffuse type GC. FGFR2 expression was associated with lower tumor grade and intestinal phenotype (p ≤ 0.0001). FGFR2-positive diffuse type GCs classified as a small subset of patients with a poor tumor specific survival (5.29 ± 1.3 vs. 14.67 ± 1.9 months; p = 0.004) (Schrumpf et al. 2022).
FGFR2 is currently explored for the treatment of GC; however, no standardized test algorithm has been developed yet, and no drug has passed formal approval by the European Medicines Agency for GC treatment.
Epidermal growth factor receptor (EGFR)
The epidermal growth factor receptor (EGFR) is frequently mutated in diverse types of carcinomas, including GC. The signaling pathway consists of several overlapping and interconnecting networks including the phosphatidylinositol 3-kinase (PI3K)/Akt (PKB) pathway, the Ras/Raf/MEK/ERK1/2 pathway, and the phospholipase C (PLCγ) pathway. Overexpression and/or gene amplification of EGFR/EGFR are found in 2–35% of GCs and significantly impact patient prognosis and survival rate. The EGFR-amplified GCs show a preponderance of male patients and affect the distal stomach (Park et al. 2016). EGFR has been and is still being explored for the treatment of GC. Again, no standardized test algorithm has been developed, and no drug has reached routine clinical application.
Ramucirumab is a human IgG1 monoclonal antibody that targets VEGF receptor 2. However, no companion diagnostics is required prior to its administration (Fuchs et al. 2014; Nakamura et al. 2021).
Genomically stable gastric cancers
Genomically stable GCs are characterized by a diffuse histological phenotype according to Laurén classification. They frequently harbor mutations in CDH1 and RHOA, and show rearrangements between CLDN18 and ARHGAP26 or ARHGAP6 (Cancer Genome Atlas Research 2014; Kakiuchi et al. 2014). However, apart from CLDN18 mutations, Claudin 18.2 is also currently explored as therapeutic target in GC irrespective of the molecular subtype.
Claudin 18.2 (CLDN18.2)
CLDN18.2, a member of the claudin family, is a component of tight junctions, regulating paracellular barrier functions (Oshima et al. 2013). The expression of the isoform 2 of CLDN18.2 (CLDN18.2) is restricted to differentiated epithelial cells of the gastric mucosa and primary GC (Fig. 1), underscoring its potential as druggable target. Ectopic expression is also commonly detected in other tumor types, such as lung, esophageal, pancreatic, and ovarian cancer (Sahin et al. 2008). A limited number of studies explored CLDN18.2 in GC. In a Japanese study, moderate-to-strong CLDN18.2 expression [≥ 2 + membrane staining intensity in ≥ 40% of tumor cells (FAST eligibility criterion; see below)] was observed in 52% of primary tumors and 45% of lymph node metastases. Expression was significantly higher in GCs of the diffuse type according to Lauren and in high grade (G3) tumors (Rohde et al. 2019). Moentenich et al. detected CLDN18.2 in 18.4% of their cases (Moentenich et al. 2020). No correlations were found between expression and clinicopathological data (sex, age, local tumor growth, nodal spread, and tumor grade). However, a significantly decreased expression was observed in tumor types with upregulated HER2 expression. Neoadjuvant treatment had no impact on the expression (Moentenich et al. 2020). Arnold et al. observed a high expression of CLDN18.2 in 17.1% of their primary tumors, in 26.7% of lymph nodes, and 16.7% of distant metastasis (Arnold et al. 2020). Expression in lymph node metastasis and primary tumors correlated significantly. High expression did not correlate with histological phenotype, tumor stage, or overall survival (Arnold et al. 2020). In our study, the expression of CLDN18.2 correlated with mucin phenotype, EBV-status, the integrin αvβ5, the EpCAM extracellular domain EpEX and lysozyme. CLDN18.2 status did not correlate with Laurén phenotype, survival, or any other clinicopathological patient characteristic (Dottermusch et al. 2019). These conflicting results largely stem from different antibodies, staining, and scoring systems. However, harmonization is to be expected in the near future due to ongoing clinical trials.
The phase II-FAST study investigated CLDN18.2 tumor expression and therapy with the chimeric monoclonal anti-CLDN18.2 antibody zolbetuximab in combination with first line chemotherapy (EOX: epirubicin + oxaliplatin + capecitabine) in patients with advanced cancer of the esophagogastric junction and stomach and a moderate to strong expression of CLDN18.2 in ≥ 40% tumor cells. Both progression free survival (95% CI 0.29–0.67; HR 0.44;) and overall survival (95% CI 0.39–0.77; HR 0.55) were significantly improved with zolbetuximab + EOX compared with EOX alone. This significant progression free survival benefit was retained in patients with moderate to strong CLDN18.2 expression in > 70% of tumor cells (95% CI 0.23–0.62; HR 0.38) (Sahin et al. 2021).
Next generation sequencing
Molecular tumor boards are now increasingly used to search for druggable targets by next generation sequencing (NGS), which may also include GC (Hoefflin et al. 2021). These studies are often done on small biopsy specimens or a limited number of tissue samples. However, a few studies illustrate the substantial intraprimary and intermetastatic genetic heterogeneity of GC (Röcken et al 2021). A substantial variation in the extent of mutational overlap or mutational heterogeneity between primary and lymph node metastasis genomes was found by Lee et al. in 15 pairs of primary GC and their matched lymph node metastases, which were studied by whole-exome sequencing (Lee et al. 2019). Pectasides et al. studied two independent patient cohorts (Pectasides et al 2018). In the first cohort, a single biopsy sample was obtained from the primary tumor of 11 patients and was compared with biopsies from synchronous metastates. In a second cohort, more than 100 samples obtained from the primary tumors and metastatic sites of 26 patients were forwarded to targeted sequencing (Pectasides et al. 2018). Discrepant pathogenic alterations between primary tumors and paired metastatic lesions were found in 45% of the patients. With regard to RTKs, 9 of 12 cases (75%) were discordant across all matched samples (Pectasides et al. 2018). Four MSI GCs were forwarded to multiregional sequencing by Loga et al. An extreme intratumoral heterogeneity as well as evidence of parallel evolution in this special subtype was discovered (von Loga et al. 2020).
We performed multiregional sequencing in nine GCs and harbored 16,537 non-synonymous mutations (Röcken et al. 2021). Intratumoral heterogeneity of somatic mutations and copy number variants were present in all tumors. 53–91% of the non-synonymous mutations were not present in each patient’s sample; 399 genes harbored 2–4 different non-synonymous mutations in the same patient; 175 genes showed copy number variations, the majority being heterogeneous, including CD274 (PD-L1). Multisample tree-based analyses provided evidence for branched evolution being most complex in a MSI GC (Röcken et al. 2021). Collectively, these data illustrate the risk of misinterpreting tumor genetics in GC based on single sample analysis. Thus, when NGS is utilized, caution must be taken regarding the validity and significance of the findings.
Immune checkpoint molecules
In their seminal updated review on the Hallmarks of Cancer, Hanahan and Weinberg added immune evasion as a strategy of malignant tumors to escape destruction by the immune system (Hanahan et al. 2011). Observational, experimental, and clinical data strongly support the importance of the immune system in combating tumor development and progression (Hanahan et al. 2011). As a result, checkpoint inhibitors gained considerable attention and are now widely explored and used as novel treatment options in cancer, including GC (Bolandi et al. 2021).
The B7 family of immune checkpoint molecules encompasses eleven members: B7-1, B7-2, B7–H1 (PD-L1), B7-DC (PD-L2), B7–H2, B7–H3, B7–H4, B7–H5 (VISTA), B7–H6, B7–H7, and Ig-like domain-containing receptor 2 (ILDR2). The interaction of the B7 family of immune-regulatory ligands with their corresponding receptors induces and inhibits T cell responses by sending co-stimulatory and co-inhibitory signals, respectively (Bolandi et al. 2021). Several of these members are explored as druggable targets or have already been successfully implemented as such in patient care. However, efficacy of immune checkpoint inhibitors varies between patients and patient selection is of crucial importance here as well.
PD-L1
PD-L1 (B7-H1) is a 290 amino acid type I transmembrane surface glycoprotein. It is encoded by CD274, which is located on chromosome 9. Several cell types of the immune system express PD-L1, such as lymphocytes and dendritic cells. Aberrant expression is observed in diverse solid tumors. PD-L1 is the ligand of programmed cell death 1 (PD-1), another member of the immunoglobulin superfamily B7. PD-1 is expressed by activated T-cells on the germinal center of lymph follicles, tumor infiltrating lymphocytes and other immune cells (Keir et al. 2008) and involved in immunemodulation (Freeman et al. 2000). Binding of PD-L1 to PD-1 suppresses T-cell receptor signaling. This in turn down regulates the immune response and enables cancer cells to escape the destruction by the immune system (Zou et al. 2008). Administration of PD-1/PD-L1 checkpoint inhibitors target the PD-1/PD-L1 interaction and restore cancer cell-directed immune response (Poole 2014). PD-L1 is expressed by GCs and is significantly more prevalent in men, GCs of the proximal stomach, un-classified, papillary, Her2/neu-positive, EBVaGc, and MSI-GC (Böger et al. 2016).
In diagnostic surgical pathology, immunohistochemistry is used to assess the PD-L1 status by applying the tumor proportion score (TPS; percentage of PD-L1 positive tumors cells) and the combined positivity score [CPS; number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumor cells, multiplied by 100]. Although CPS can exceed 100, the maximum score is defined as CPS 100.
Recently, the CheckMate-649 study showed that nivolumab, an anti-PD-1-antibody, in combination with chemotherapy significantly improved overall survival (98.4% CI 0.59–0.86; HR 0.71) and progression-free survival (98% CI 0.56–0.81; HR 0.68) vs. chemotherapy alone in patients with a PD-L1 CPS ≥ 5 and advanced gastric, esophagogastric junction, and esophageal adenocarcinoma (Janjigian et al. 2021). Nivolumab was also shown to be efficacious in resected esophageal or esophagogastric junction cancer after neoadjuvant chemoradiotherapy (Kelly et al. 2021). The KEYNOTE-590 study explored the efficacy of prembolizumab, another anti-PD-1 antibody, in advanced esophageal and Siewert type 1 esophagogastric junction adenocarcinomas. Overall survival was longer in the pembrolizumab plus chemotherapy group than in the placebo plus chemotherapy group [median 11.6 months (95% CI 9.7–15.2) vs. 9.9 months (95% CI 0.54–1.02); HR 0.74] (Sun et al. 2021).
Based on these data, pembrolizumab in combination with platinum and fluoropyrimidine based chemotherapy, was approved for the first-line treatment of patients with locally advanced unresectable or metastatic carcinoma of the esophagus or HER2 negative esophagogastric junction adenocarcinoma in adults whose tumors express PD-L1 with a CPS ≥ 10 (https://www.ema.europa.eu/en/medicines/human/EPAR/keytruda). Nivolumab in combination with fluoropyrimidine- and platinum-based combination chemotherapy was granted approval by the European Medicines Agency for first-line treatment of adult patients with HER2-negative advanced or metastatic gastric, esophagogastric junction cancer or esophageal adenocarcinoma whose tumors express PD-L1 with CPS ≥ 5 (https://www.ema.europa.eu/en/medicines/human/EPAR/opdivo).
Following HER2, PD-L1 is the second predictive biomarker for GC. It has to be tested before treatment with approved immune checkpoint inhibitors can be administered. In general, PD-L1 scoring is sensitive to antibody selection, staining protocols, and expertise in the assessment of immunostaining (Munari et al. 2018; Ahn et al. 2021; Narita et al. 2021; Noske et al. 2021a, b; Noske et al. 2021a, b). However, the interchangeability of PD-L1 assays in GC has been demonstrated (Ahn et al. 2021; Narita et al. 2021).
VISTA
V-domain immunoglobulin (Ig)-containing suppressor of T-cell activation (VISTA) is a 311 amino acid type I-membrane protein. Various hematopoietic cells, such as myeloid, granulocytic, and T cells, express predominantly VISTA (Wang et al. 2019). P-selectin glycoprotein ligand 1 (PSGL-1) and V-Set and Immunoglobulin domain containing 3 (VSIG3) were proposed as binding partner(s). In addition, VISTA may function both as a ligand (for antigen presenting cells) and a receptor (for T cells). It suppresses T cell activation. In murine tumor models, monoclonal antibodies targeting VISTA boost antitumor immunity by increasing the number and elevating the function of intratumoral T cells (Le Mercier et al. 2014). It is noteworthy that VISTA-induced T cell activation appears to be nonredundant from the PD-1/PD-L1 pathway. Thus, a blockade of both, VISTA and PD-1, might open novel avenues for cancer treatment, as it was shown in murine tumor models (Liu et al. 2015a, b; Kondo et al. 2016). Recently, one phase I study using an anti-VISTA monoclonal antibody (JNJ-61610588; NCT02671955) and a phase I study that targets both VISTA and PD-L1/PD-L2 in solid tumors using a small molecule (CA-170; NCT02812875) have started.
Data on VISTA are limited. Böger et al. (Böger et al. 2017a, b) showed that the VISTA expression was associated with the tumor localization, Laurén phenotype, EBV, KRAS- and PIK3CA-mutational status, and PD-L1 expression. However, no significant correlation was observed with patient outcomes. A change in immune cell expression of VISTA during tumor progression was observed (Böger et al. 2017a, b). Loeser et al. (Loeser et al. 2019) observed strong positive outcomes for VISTA-positive tumors in the pT1/T2 stages, with lower expression levels of VISTA in pT3/T4 tumor samples. However, the expression of both PD-L1 and VISTA is sensitive to neoadjuvant (radio-) chemotherapy and is associated with poor tumor regression. Schoop et al. compared a cohort of therapy naïve GCs with a cohort of neoadjuvantly/perioperatively treated GCs. They found a major increase in overall immune cell density coupled with an over proportional increase in PD-1 and VISTA positive immune cells in neoadjuvantly/perioperatively treated GCs (Schoop et al. 2020a, b). The frequency of VISTA expression in tumor cells also substantially increased. In contrast, PD-L1 expression was decreased in immune cells and tumor cells of neoadjuvantly treated GCs (Schoop et al. 2020a, b). Currently, two phase 1/2 clinical trials are listed at www.clinicaltrials.gov, exploring monoclonal antibodies targeting VISTA in solid tumors.
B7–H3
B7 homolog 3 protein (B7–H3, or CD276), a 534 amino acid protein, is another member of the B7 family of immune checkpoint molecules involved in immune evasion. The exact receptor of B7–H3 is currently unknown (Castellanos et al. 2017). It is overexpressed in various cancers (Castellanos et al. 2017; Ni et al. 2017) and may inhibit CD8 + T cells (Lee et al. 2017). B7–H3 is found both in tumor cells and the tumor immune microenvironment, i.e., endothelial cells, fibroblasts, B-lymphocytes, macrophages, natural killer cells, and dendritic cells (Zhan et al. 2019). B7–H3 seems to be linked to cancer progression, metastatic behavior, and worse prognosis in several cancers including cancers of the lung, breast, prostate, kidney, and colon (Ni et al. 2017). So far, only a few studies examined the expression of B7–H3 in relation to the distribution of CD8 + T cells in GC (Guo et al . 2019). Again, data are scarce. Recently, we used double immunohistochemical staining to study the spatial distribution of CD8 + T cells in relation to B7–H3 positive cells. B7–H3 was expressed mainly in the tumor stroma of GC (76% of all cases). GCs with high expression of B7–H3 showed larger spatial differences of CD8 + T cells (86.4/mm2 in tumor center vs. 414.9/mm2 in invasive front) compared to the B7–H3-low group (157.7/mm2 vs. 218.7/mm2, respectively) (Ulase et al. 2021).
Several trials are listed in www.clinicaltrials.gov targeting B7–H3. None has reached clinical application, yet.
Sexual dimorphism
The susceptibility of GC shows a striking sex-specific difference. According to the European Network of Cancer Registries, the estimated GC incidence in men is almost double that of women. This also applies to mortality, with an estimated 63,600 gastric cancer deaths in men and 43,700 in women (ENCR Factsheet Stomach Cancer; https://ec.europa.eu/jrc/en/science-update/new-factsheet-stomach-cancer-europe-released). These differences are unlikely related to H. pylori infection, the major risk factor for GC (Group 2001; Brusselaers et al. 2017). More importantly, the immune response exhibits sex-specific differences with regard to infectious diseases, vaccination, and autoimmunity. Both, estrogen and androgen exposure influence the effector functions of immune cells (Markle et al. 2014). This sexual dimorphism in immune response capacity is now well recognized. Immune surveillance competence differ between men and women and may contribute to the sex effect observed in malignant tumors (Dorak et al. 2012).
Sex influences the development and progression of cancer (Mauvais-Jarvis et al. 2020), since men and women differ in their immune response (Mirandola et al. 2015; Klein et al. 2016; De Martinis et al. 2020). Therapy response after an immune checkpoint inhibitor therapy is less effective for women (Conforti et al. 2018). One reason is the higher antigenicity in male cancers. To the contrary, the combination of immune checkpoint inhibitors with chemotherapy is less effective for men (Irelli et al. 2020). Response rates most likely depend on the different innate and adaptive immune systems of men and women (Cook et al. 2009; Klein et al. 2016). Despite this evidence, sexual dimorphism in biomedical science is often not specifically addressed and many studies fail to analyze results by sex (Beery et al. 2011). All this possibly also applies to GC. We and others have shown a gender-specific effect for GC (Caruso et al. 2002; Clausen et al. 2020; Quaas et al. 2021a, b). For women with GC, the density of tumor associated neutrophils especially located in the invasion front is an independent predictor of tumor-specific survival. In contrast to men, where no association was found (Caruso et al. 2002; Clausen et al. 2020). Thus, future studies on the application of immune checkpoint inhibitors also need to consider gender as a “tale-telling” biomarker.
Limitations
This review addresses mainly recent advancements in established and putative predictive biomarkers for tailoring GC treatment. It does not cover research on, e.g., the growing field of long non-coding RNA. So far, research on long non-coding RNA has focused on tumor biology and patient prognosis, rather than predictive biomarker for drug administration.
Conclusions
Predictive biomarkers are the mainstay of precision oncology. In recent years, major achievements have been made in GC treatment. While targeting HER2 remains the main therapy for a limited number of patients with advanced GC, novel targets have been developed, specifically those addressing immune checkpoint molecules. However, immune oncology must consider sexual dimorphism in tissue-based diagnostics, drug regimens, and patient outcomes. Currently, CLDN18.2 is being explored among several other targets, and further advancements are expected in the near future. The major obstacle to precision medicine for GC is intratumoral heterogeneity, which affects tissue-based diagnostics due to the risk of sampling errors and patient outcomes, and this may likely cause primary and secondary drug resistance.
References
Ahn S, Lee J, Hong M, Kim ST, Park SH, Choi MG et al (2016) FGFR2 in gastric cancer: protein overexpression predicts gene amplification and high H-index predicts poor survival. Mod Pathol 29(9):1095–1103. https://doi.org/10.1038/modpathol.2016.96
Ahn S, Kim KM (2021) PD-L1 expression in gastric cancer: interchangeability of 22C3 and 28–8 pharmDx assays for responses to immunotherapy. Mod Pathol 34(9):1719–1727. https://doi.org/10.1038/s41379-021-00823-9
Arnold A, Daum S, von Winterfeld M, Berg E, Hummel M, Rau B et al (2020) Prognostic impact of Claudin 18.2 in gastric and esophageal adenocarcinomas. Clin Transl Oncol 22(12):2357–2363. https://doi.org/10.1007/s12094-020-02380-0
Babina IS, Turner NC (2017) Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer 17(5):318–332. https://doi.org/10.1038/nrc.2017.8
Bang YJ, Van CE, Feyereislova A, Chung HC, Shen L, Sawaki A et al (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376(9742):687–697. https://doi.org/10.1016/S0140-6736(10)61121-X
Baretton G, Dietel M, Gaiser T, Kirchner T, Kreipe HH, Quaas A et al (2016) HER2-Testung beim Magenkarzinom—Ergebnisse eines deutschen Expertentreffens. Pathologe 37(4):361–366. https://doi.org/10.1007/s00292-016-0179-3
Bartley AN, Washington MK, Ventura CB, Ismaila N, Colasacco C, Benson AB 3rd et al (2016) HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: guideline from the college of american pathologists, american society for clinical pathology, and american society of clinical oncology. Arch Pathol Lab Med 140(12):1345–1363. https://doi.org/10.5858/arpa.2016-0331-CP
Beery AK, Zucker I (2011) Sex bias in neuroscience and biomedical research. Neurosci Biobehav Rev 35(3):565–572. https://doi.org/10.1016/j.neubiorev.2010.07.002
Betts G, Valentine H, Pritchard S, Swindell R, Williams V, Morgan S et al (2014) FGFR2, HER2 and cMet in gastric adenocarcinoma: detection, prognostic significance and assessment of downstream pathway activation. Virchows Arch 464(2):145–156. https://doi.org/10.1007/s00428-013-1517-y
Board TE (2019) Digestive System Tumours. Lyon, France, International Agency for Research on Cancer
Böger C, Behrens HM, Mathiak M, Krüger S, Kalthoff H, Röcken C (2016) PD-L1 is an independent prognostic predictor in gastric cancer of Western patients. Oncotarget 7(17):24269–24283
Böger C, Behrens HM, Krüger S, Röcken C (2017a) The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology 6(4):e1293215. https://doi.org/10.1080/2162402X.2017.1293215
Böger C, Krüger S, Behrens HM, Bock S, Haag J, Kalthoff H et al (2017b) Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann Oncol 28(5):1005–1014. https://doi.org/10.1093/annonc/mdx047
Bolandi N, Derakhshani A, Hemmat N, Baghbanzadeh A, Asadzadeh Z, Afrashteh Nour M et al (2021) The positive and negative immunoregulatory role of b7 family: promising novel targets in gastric cancer treatment. Int J Mol Sci. https://doi.org/10.3390/ijms221910719
Brusselaers N, Maret-Ouda J, Konings P, El-Serag HB, Lagergren J (2017) Menopausal hormone therapy and the risk of esophageal and gastric cancer. Int J Cancer 140(7):1693–1699. https://doi.org/10.1002/ijc.30588
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513(7517):202–209. https://doi.org/10.1038/nature13480
Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C (2002) Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol 15(8):831–837. https://doi.org/10.1097/01.MP.0000020391.98998.6B
Castellanos JR, Purvis IJ, Labak CM, Guda MR, Tsung AJ, Velpula KK et al (2017) B7–H3 role in the immune landscape of cancer. Am J Clin Exp Immunol 6(4):66–75
Catenacci DV, Cervantes G, Yala S, Nelson EA, El-Hashani E, Kanteti R et al (2011) RON (MST1R) is a novel prognostic marker and therapeutic target for gastroesophageal adenocarcinoma. Cancer Biol Ther 12(1):9–46
Cho YA, Lee H, Kim DG, Kim H, Ha SY, Choi YL et al (2021) PD-L1 expression is significantly associated with tumor mutation burden and microsatellite instability score. Cancers (basel). https://doi.org/10.3390/cancers13184659
Clausen F, Behrens HM, Krüger S, Röcken C (2020) Sexual dimorphism in gastric cancer: tumor-associated neutrophils predict patient outcome only for women. J Cancer Res Clin Oncol 146(1):53–66. https://doi.org/10.1007/s00432-019-03082-z
Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G et al (2018) Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol 19(6):737–746. https://doi.org/10.1016/S1470-2045(18)30261-4
Cook MB, Dawsey SM, Freedman ND, Inskip PD, Wichner SM, Quraishi SM et al (2009) Sex disparities in cancer incidence by period and age. Cancer Epidemiol Biomarkers Prev 18(4):1174–1182. https://doi.org/10.1158/1055-9965.EPI-08-1118
Correa P (1992) Human gastric carcinogenesis: a multistep and multifactorial process–first American cancer society award lecture on cancer epidemiology and prevention. Cancer Res 52(24):6735–6740
Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ (2017) A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8:15180. https://doi.org/10.1038/ncomms15180
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS et al (2015) Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 21(5):449–456. https://doi.org/10.1038/nm.3850
De Martinis M, Sirufo MM, Suppa M, Di Silvestre D, Ginaldi L (2020) Sex and gender aspects for patient stratification in allergy prevention and treatment. Int J Mol Sci. https://doi.org/10.3390/ijms21041535
Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB et al (2012) A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 61(5):673–684. https://doi.org/10.1136/gutjnl-2011-301839
Donner I, Kiviluoto T, Ristimaki A, Aaltonen LA, Vahteristo P (2015) Exome sequencing reveals three novel candidate predisposition genes for diffuse gastric cancer. Fam Cancer 14(2):241–246. https://doi.org/10.1007/s10689-015-9778-z
Dorak MT, Karpuzoglu E (2012) Gender differences in cancer susceptibility: an inadequately addressed issue. Front Genet 3:268. https://doi.org/10.3389/fgene.2012.00268
Dottermusch M, Krüger S, Behrens HM, Halske C, Röcken C (2019) Expression of the potential therapeutic target claudin-182 is frequently decreased in gastric cancer: results from a large Caucasian cohort study Virchows. Arch 475(5):563–571
Drebber U, Baldus SE, Nolden B, Grass G, Bollschweiler E, Dienes HP et al (2008) The overexpression of c-met as a prognostic indicator for gastric carcinoma compared to p53 and p21 nuclear accumulation. Oncol Rep 19(6):1477–1483
El Darsa H, El Sayed R, Abdel-Rahman O (2020) MET inhibitors for the treatment of gastric cancer: what’s their potential? J Exp Pharmacol 12:349–361. https://doi.org/10.2147/jep.S242958
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H et al (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192(7):1027–1034
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C et al (2014) Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 383(9911):31–39. https://doi.org/10.1016/S0140-6736(13)61719-5
Fukayama M, Ushiku T (2011) Epstein-Barr virus-associated gastric carcinoma. Pathol Res Pract 207(9):529–537. https://doi.org/10.1016/j.prp.2011.07.004
Gonzalez RS, Messing S, Tu X, McMahon LA, Whitney-Miller CL (2016) Immunohistochemistry as a surrogate for molecular subtyping of gastric adenocarcinoma. Hum Pathol 56:16–21. https://doi.org/10.1016/j.humpath.2016.06.003
Graveel CR, Tolbert D, Vande Woude GF (2013) MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a009209
Group HaCC (2001) Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut 49(3):347–353
Guan WL, Ma Y, Cui YH, Liu TS, Zhang YQ, Zhou ZW et al (2021) The impact of mismatch repair status on prognosis of patients with gastric cancer: a multicenter analysis. Front Oncol 11:712760. https://doi.org/10.3389/fonc.2021.712760
Guo L, Liu Z, Zhang Y, Quan Q, Huang L, Xu Y et al (2019) Association of increased B7 protein expression by infiltrating immune cells with progression of gastric carcinogenesis. Medicine (baltimore) 98(8):e14663. https://doi.org/10.1097/md.0000000000014663
Hack SP, Bruey JM, Koeppen H (2014) HGF/MET-directed therapeutics in gastroesophageal cancer: a review of clinical and biomarker development. Oncotarget 5(10):2866–2880
Haffner I, Schierle K, Raimúndez E, Geier B, Maier D, Hasenauer J et al (2021) HER2 expression, test deviations, and their impact on survival in metastatic gastric cancer: results from the prospective multicenter VARIANZ study. J Clin Oncol 39(13):1468–1478. https://doi.org/10.1200/jco.20.02761
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hoefflin R, Lazarou A, Hess ME, Reiser M, Wehrle J, Metzger P et al (2021) Transitioning the molecular tumor board from proof of concept to clinical routine: a german single-center analysis. Cancers (Basel). https://doi.org/10.3390/cancers13051151
Hosoda K, Yamashita K, Ushiku H, Ema A, Moriya H, Mieno H et al (2018) Prognostic relevance of FGFR2 expression in stage II/III gastric cancer with curative resection and S-1 chemotherapy. Oncol Lett 15(2):1853–1860. https://doi.org/10.3892/ol.2017.7515
Hur JY, Chao J, Kim K, Kim ST, Kim KM, Klempner SJ et al (2020) High-level FGFR2 amplification is associated with poor prognosis and Lower response to chemotherapy in gastric cancers. Pathol Res Pract 216(4):152878. https://doi.org/10.1016/j.prp.2020.152878
Inokuchi M, Murase H, Otsuki S, Kawano T, Kojima K (2017) Different clinical significance of FGFR1-4 expression between diffuse-type and intestinal-type gastric cancer. World J Surg Oncol 15(1):1–9. https://doi.org/10.1186/s12957-016-1081-4
Irelli A, Sirufo MM, D’Ugo C, Ginaldi L, De Martinis M (2020) Sex and gender influences on cancer immunotherapy response. Biomedicines 8(7):232. https://doi.org/10.3390/biomedicines8070232
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L et al (2021) First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398(10294):27–40. https://doi.org/10.1016/s0140-6736(21)00797-2
Jardim DL, de Melo GD, Falchook GS, Janku F, Zinner R, Wheler JJ et al (2014) MET aberrations and c-MET inhibitors in patients with gastric and esophageal cancers in a phase I unit. Oncotarget 5(7):1837–1845
Jung EJ, Jung EJ, Min SY, Kim MA, Kim WH (2012) Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum Pathol 43(10):1559–1566. https://doi.org/10.1016/j.humpath.2011.12.002
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A et al (2014) Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet 46(6):583–587. https://doi.org/10.1038/ng.2984
Kang YK, Muro K, Ryu MH, Yasui H, Nishina T, Ryoo BY et al (2014) A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Invest New Drugs 32(2):355–361. https://doi.org/10.1007/s10637-013-0057-2
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331
Kelly RJ, Ajani JA, Kuzdzal J, Zander T, Van Cutsem E, Piessen G et al (2021) Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med 384(13):1191–1203. https://doi.org/10.1056/NEJMoa2032125
Kim HS, Kim JH, Jang HJ, Han B, Zang DY (2019) Pathological and prognostic impacts of fgfr2 overexpression in gastric cancer: a meta-analysis. J Cancer 10(1):20–27. https://doi.org/10.7150/jca.28204
Kiyose S, Nagura K, Tao H, Igarashi H, Yamada H, Goto M et al (2012) Detection of kinase amplifications in gastric cancer archives using fluorescence in situ hybridization. Pathol Int 62(7):477–484. https://doi.org/10.1111/j.1440-1827.2012.02832.x
Klein SL, Flanagan KL (2016) Sex differences in immune responses. Nat Rev Immunol 16(10):626–638. https://doi.org/10.1038/nri.2016.90
Kondo Y, Ohno T, Nishii N, Harada K, Yagita H, Azuma M (2016) Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol 57:54–60. https://doi.org/10.1016/j.oraloncology.2016.04.005
Lauren P (1965) The two histologic main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. Acta Pathol Microbiol Scand 64:31–49
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P et al (2014) VISTA regulates the development of protective antitumor immunity. Cancer Res 74(7):1933–1944. https://doi.org/10.1158/0008-5472.CAN-13-1506
Lee HH, Kim SY, Jung ES, Yoo J, Kim TM (2019) Mutation heterogeneity between primary gastric cancers and their matched lymph node metastases. Gastric Cancer 22(2):323–334. https://doi.org/10.1007/s10120-018-0870-6
Lee YH, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H et al (2017) Inhibition of the B7–H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res 27(8):1034–1045. https://doi.org/10.1038/cr.2017.90
Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K et al (2011) MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol 29(36):4803–4810. https://doi.org/10.1200/JCO.2011.35.4928
Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS et al (2016) Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet 98(5):830–842. https://doi.org/10.1016/j.ajhg.2016.03.001
Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD et al (2015a) Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A 112(21):6682–6687. https://doi.org/10.1073/pnas.1420370112
Liu X, Liu J, Qiu H, Kong P, Chen S, Li W et al (2015b) Prognostic significance of Epstein-Barr virus infection in gastric cancer: a meta-analysis. BMC Cancer 15:782. https://doi.org/10.1186/s12885-015-1813-9
Loeser H, Kraemer M, Gebauer F, Bruns C, Schröder W, Zander T et al (2019) The expression of the immune checkpoint regulator VISTA correlates with improved overall survival in pT1/2 tumor stages in esophageal adenocarcinoma. Oncoimmunology 8(5):e1581546. https://doi.org/10.1080/2162402x.2019.1581546
Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ et al (2013) An alpha-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol 229(4):621–629. https://doi.org/10.1002/path.4152
Maleki SS, Röcken C (2017) Chromosomal instability in gastric cancer biology. Neoplasia 19(5):412–420. https://doi.org/10.1016/j.neo.2017.02.012
Malfertheiner P, Link A, Selgrad M (2014) Helicobacter pylori: perspectives and time trends. Nat Rev Gastroenterol Hepatol 11(10):628–638. https://doi.org/10.1038/nrgastro.2014.99
Markle JG, Fish EN (2014) SeXX matters in immunity. Trends Immunol 35(3):97–104. https://doi.org/10.1016/j.it.2013.10.006
Mathiak M, Warneke VS, Behrens HM, Haag J, Böger C, Kruger S et al (2017) Clinicopathologic characteristics of microsatellite instable gastric carcinomas revisited: urgent need for standardization. Appl Immunohistochem Mol Morphol 25:12–24. https://doi.org/10.1097/PAI.0000000000000264
Matsubayashi H, Higashigawa S, Kiyozumi Y, Oishi T, Sasaki K, Ishiwatari H et al (2022) Microsatellite instability is biased in Amsterdam II-defined Lynch-related cancer cases with family history but is rare in other cancers: a summary of 1000 analyses. BMC Cancer 22(1):73. https://doi.org/10.1186/s12885-022-09172-5
Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I et al (2012) FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer 106(4):727–732. https://doi.org/10.1038/bjc.2011.603
Matsunobu T, Ishiwata T, Yoshino M, Watanabe M, Kudo M, Matsumoto K et al (2006) Expression of keratinocyte growth factor receptor correlates with expansive growth and early stage of gastric cancer. Int J Oncol 28(2):307–314
Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero JJ, DeMeo DL et al (2020) Sex and gender: modifiers of health, disease, and medicine. Lancet 396(10250):565–582. https://doi.org/10.1016/S0140-6736(20)31561-0
Metzger ML, Behrens HM, Böger C, Haag J, Kruger S, Röcken C (2016) MET in gastric cancer—discarding a 10% cutoff rule. Histopathology. 68(2): 241 253.
Mirandola L, Wade R, Verma R, Pena C, Hosiriluck N, Figueroa JA et al (2015) Sex-driven differences in immunological responses: challenges and opportunities for the immunotherapies of the third millennium. Int Rev Immunol 34(2):134–142. https://doi.org/10.3109/08830185.2015.1018417
Moehler M, Al-Batran SE, Andus T, Arends J, Arnold D, Baretton G et al (2019) S3-Leitlinie Magenkarzinom—Diagnostik und Therapie der Adenokarzinome des Magens und des ösophagogastralen Übergangs—Langversion 2.0—August 2019. AWMF-Registernummer: 032/009OL. Z Gastroenterol 57(12):1517–1632. https://doi.org/10.1055/a-1018-2516
Moentenich V, Gebauer F, Comut E, Tuchscherer A, Bruns C, Schroeder W et al (2020) Claudin 18.2 expression in esophageal adenocarcinoma and its potential impact on future treatment strategies. Oncol Lett 19(6):3665–3670. https://doi.org/10.3892/ol.2020.11520
Munari E, Rossi G, Zamboni G, Lunardi G, Marconi M, Sommaggio M et al (2018) PD-L1 assays 22C3 and SP263 are not interchangeable in non-small cell lung cancer when considering clinically relevant cutoffs: an interclone evaluation by differently trained pathologists. Am J Surg Pathol 42(10):1384–1389. https://doi.org/10.1097/pas.0000000000001105
Murase H, Inokuchi M, Takagi Y, Kato K, Kojima K, Sugihara K (2014) Prognostic significance of the co-overexpression of fibroblast growth factor receptors 1, 2 and 4 in gastric cancer. Mol Clin Oncol 2(4):509–517. https://doi.org/10.3892/mco.2014.293
Nagatsuma AK, Aizawa M, Kuwata T, Doi T, Ohtsu A, Fujii H et al (2015) Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer 18(2):227–238. https://doi.org/10.1007/s10120-014-0360-4
Nakamura Y, Kawazoe A, Lordick F, Janjigian YY, Shitara K (2021) Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: an emerging paradigm. Nat Rev Clin Oncol 18(8):473–487. https://doi.org/10.1038/s41571-021-00492-2
Narita Y, Sasaki E, Masuishi T, Taniguchi H, Kadowaki S, Ito S et al (2021) PD-L1 immunohistochemistry comparison of 22C3 and 28–8 assays for gastric cancer. J Gastrointest Oncol. 12(6):2696–2705
Ni L, Dong C (2017) New B7 family checkpoints in human cancers. Mol Cancer Ther 16(7):1203–1211. https://doi.org/10.1158/1535-7163.Mct-16-0761
Noske A, Ammann JU, Wagner DC, Denkert C, Lebeau A, Sinn P et al (2021a) A multicentre analytical comparison study of inter-reader and inter-assay agreement of four programmed death-ligand 1 immunohistochemistry assays for scoring in triple-negative breast cancer. Histopathology 78(4):567–577. https://doi.org/10.1111/his.14254
Noske A, Wagner DC, Schwamborn K, Foersch S, Steiger K, Kiechle M et al (2021b) Interassay and interobserver comparability study of four programmed death-ligand 1 (PD-L1) immunohistochemistry assays in triple-negative breast cancer. Breast 60:238–244. https://doi.org/10.1016/j.breast.2021.11.003
Ooki A, Yamaguchi K (2021) The beginning of the era of precision medicine for gastric cancer with fibroblast growth factor receptor 2 aberration. Gastric Cancer 24(6):1169–1183. https://doi.org/10.1007/s10120-021-01235-z
Oshima T, Shan J, Okugawa T, Chen X, Hori K, Tomita T et al (2013) Down-regulation of claudin-18 is associated with the proliferative and invasive potential of gastric cancer at the invasive front. PLoS ONE 8(9):e74757. https://doi.org/10.1371/journal.pone.0074757
Park CK, Park JS, Kim HS, Rha SY, Hyung WJ, Cheong JH et al (2016) Receptor tyrosine kinase amplified gastric cancer: Clinicopathologic characteristics and proposed screening algorithm. Oncotarget 7(44):72099–72112
Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M et al (2018) Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov 8(1):37–48. https://doi.org/10.1158/2159-8290.CD-17-0395
Petrillo A, Smyth EC (2020) Biomarkers for precision treatment in gastric cancer. Visc Med 36(5):364–372. https://doi.org/10.1159/000510489
Petrovchich I, Ford JM (2016) Genetic predisposition to gastric cancer. Semin Oncol 43(5):554–559. https://doi.org/10.1053/j.seminoncol.2016.08.006
Pietrantonio F, Miceli R, Raimondi A, Kim YW, Kang WK, Langley RE et al (2019) Individual patient data meta-analysis of the value of microsatellite instability as a biomarker in gastric cancer. J Clin Oncol 37(35):3392–3400. https://doi.org/10.1200/jco.19.01124
Pietrantonio F, Randon G, Di Bartolomeo M, Luciani A, Chao J, Smyth EC et al (2021) Predictive role of microsatellite instability for PD-1 blockade in patients with advanced gastric cancer: a meta-analysis of randomized clinical trials. ESMO Open 6(1):100036. https://doi.org/10.1016/j.esmoop.2020.100036
Poole RM (2014) Pembrolizumab: first global approval. Drugs 74(16):1973–1981. https://doi.org/10.1007/s40265-014-0314-5
Quaas A, Pamuk A, Klein S, Quantius J, Rehkaemper J, Barutcu AG et al (2021a) Sex-specific prognostic effect of CD66b-positive tumor-infiltrating neutrophils (TANs) in gastric and esophageal adenocarcinoma. Gastric Cancer 24(6):1213–1226. https://doi.org/10.1007/s10120-021-01197-2
Quaas A, Rehkaemper J, Rueschoff J, Pamuk A, Zander T, Hillmer A et al (2021b) Occurrence of high microsatellite-instability/mismatch repair deficiency in nearly 2,000 human adenocarcinomas of the gastrointestinal tract, pancreas, and bile ducts: a study from a large german comprehensive cancer center. Front Oncol 11:569475. https://doi.org/10.3389/fonc.2021.569475
Röcken C, Amallraja A, Halske C, Opasic L, Traulsen A, Behrens HM et al (2021) Multiscale heterogeneity in gastric adenocarcinoma evolution is an obstacle to precision medicine. Genome Med 13(1):177. https://doi.org/10.1186/s13073-021-00975-y
Rohde C, Yamaguchi R, Mukhina S, Sahin U, Itoh K, Türeci Ö (2019) Comparison of Claudin 18.2 expression in primary tumors and lymph node metastases in Japanese patients with gastric adenocarcinoma. Jpn J Clin Oncol 49(9):870–876. https://doi.org/10.1093/jjco/hyz068
Roviello G, Catalano M, Iannone LF, Marano L, Brugia M, Rossi G et al (2022) Current status and future perspectives in HER2 positive advanced gastric cancer. Clin Transl Oncol. https://doi.org/10.1007/s12094-021-02760-0
Ruschoff J, Hanna W, Bilous M, Hofmann M, Osamura RY, Penault-Llorca F et al (2012) HER2 testing in gastric cancer: a practical approach. Mod Pathol 25(5):637–650. https://doi.org/10.1038/modpathol.2011.198
Sahin U, Koslowski M, Dhaene K, Usener D, Brandenburg G, Seitz G et al (2008) Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res 14(23):7624–7634. https://doi.org/10.1158/1078-0432.Ccr-08-1547
Sahin U, Türeci Ö, Manikhas G, Lordick F, Rusyn A, Vynnychenko I et al (2021) FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann Oncol 32(5):609–619. https://doi.org/10.1016/j.annonc.2021.02.005
Saito M, Kono K (2021) Landscape of EBV-positive gastric cancer. Gastric Cancer 24(5):983–989. https://doi.org/10.1007/s10120-021-01215-3
Schoop H, Bregenzer A, Halske C, Behrens HM, Krüger S, Egberts JH et al (2020a) Therapy resistance in neoadjuvantly treated gastric cancer and cancer of the gastroesophageal junction is associated with an increased expression of immune checkpoint inhibitors-comparison against a therapy naive cohort. Transl Oncol 13(2):165–176. https://doi.org/10.1016/j.tranon.2019.11.004
Schoop I, Maleki SS, Behrens HM, Krüger S, Haag J, Röcken C (2020b) p53 immunostaining cannot be used to predict TP53 mutations in gastric cancer: results from a large Central European cohort. Hum Pathol 105:53–66. https://doi.org/10.1016/j.humpath.2020.09.006
Schrumpf T, Behrens HM, Haag J, Kruger S, Rocken C (2022) FGFR2 overexpression and compromised survival in diffuse-type gastric cancer in a large central European cohort. PLoS ONE 17(2):e0264011. https://doi.org/10.1371/journal.pone.0264011
Seo S, Park SJ, Ryu MH, Park SR, Ryoo BY, Park YS et al. (2017) Prognostic impact of fibroblast growth factor receptor 2 gene amplification in patients receiving fluoropyrimidine and platinum chemotherapy for metastatic and locally advanced unresectable gastric cancers. Oncotarget. 8(20): 33844–33854. https://doi.org/10.18632/oncotarget.12953.
Setia N, Agoston AT, Han HS, Mullen JT, Duda DG, Clark JW et al (2016) A protein and mRNA expression-based classification of gastric cancer. Mod Pathol 29(7):772–784. https://doi.org/10.1038/modpathol.2016.55
Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P et al (2013) Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS ONE 8(3):e54014. https://doi.org/10.1371/journal.pone.0054014
Silva AN, Coffa J, Menon V, Hewitt LC, Das K, Miyagi Y et al (2018) Frequent coamplification of receptor tyrosine kinase and downstream signaling genes in japanese primary gastric cancer and conversion in matched lymph node metastasis. Ann Surg 267:114–121. https://doi.org/10.1097/SLA.0000000000002042
Smyth EC, Wotherspoon A, Peckitt C, Gonzalez D, Hulkki-Wilson S, Eltahir Z et al (2017) Mismatch repair deficiency, microsatellite instability, and survival: an exploratory analysis of the medical research council adjuvant gastric infusional chemotherapy (MAGIC) trial. JAMA Oncol 3(9):1197–1203. https://doi.org/10.1001/jamaoncol.2016.6762
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F (2020) Gastric cancer. Lancet 396(10251):635–648. https://doi.org/10.1016/S0140-6736(20)31288-5
Su X, Zhan P, Gavine PR, Morgan S, Womack C, Ni X et al (2014) FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer 110(4):967–975. https://doi.org/10.1038/bjc.2013.802
Sun JM, Shen L, Shah MA, Enzinger P, Adenis A, Doi T et al (2021) Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet 398(10302):759–771. https://doi.org/10.1016/s0140-6736(21)01234-4
Tannock IF, Hickman JA (2016) Limits to personalized cancer medicine. N Engl J Med 375(13):1289–1294. https://doi.org/10.1056/NEJMsb1607705
Teng L, Lu J (2013) cMET as a potential therapeutic target in gastric cancer (Review). Int J Mol Med 32(6):1247–1254. https://doi.org/10.3892/ijmm.2013.1531
Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E (1993) Epstein-Barr virus in gastric carcinoma. Am J Pathol 143(5):1250–1254
Tokunaga M, Land CE (1998) Epstein-Barr virus involvement in gastric cancer: biomarker for lymph node metastasis. Cancer Epidemiol Biomarkers Prev 7(5):449–450
Tokunaga R, Imamura Y, Nakamura K, Ishimoto T, Nakagawa S, Miyake K et al (2016) Fibroblast growth factor receptor 2 expression, but not its genetic amplification, is associated with tumor growth and worse survival in esophagogastric junction adenocarcinoma. Oncotarget 7(15):19748–19761
Ueki T, Koji T, Tamiya S, Nakane PK, Tsuneyoshi M (1995) Expression of basic fibroblast growth factor and fibroblast growth factor receptor in advanced gastric carcinoma. J Pathol 177(4):353–361. https://doi.org/10.1002/path.1711770405
Ulase D, Behrens H-M, Krüger S, Zeissig S, Röcken C (2021) Gastric carcinomas with stromal B7–H3 expression have lower intratumoural CD8+ T cell density. Int J Mol Sci 22(4):2129
van der Post RS, Vogelaar IP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N et al (2015) Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 52(6):361–374. https://doi.org/10.1136/jmedgenet-2015-103094
von Loga K, Woolston A, Punta M, Barber LJ, Griffiths B, Semiannikova M et al (2020) Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat Commun 11(1):139. https://doi.org/10.1038/s41467-019-13915-7
Wakatsuki T, Yamamoto N, Sano T, Chin K, Kawachi H, Takahari D et al (2018) Clinical impact of intratumoral HER2 heterogeneity on trastuzumab efficacy in patients with HER2-positive gastric cancer. J Gastroenterol 53(11):1186–1195. https://doi.org/10.1007/s00535-018-1464-0
Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C et al (2019) VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 156(1):74–85. https://doi.org/10.1111/imm.13001
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST et al (2014) Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 46(6):573–582. https://doi.org/10.1038/ng.2983
Warneke V, Behrens HM, Haag J, Balschun K, Böger C, Becker T et al (2013a) Prognostic and putative predictive biomarkers of gastric cancer for personalized medicine. Diagn Mol Pathol 22(3):127–137. https://doi.org/10.1097/PDM.0b013e318284188e
Warneke VS, Behrens HM, Boger C, Becker T, Lordick F, Ebert MP et al (2013b) Her2/neu testing in gastric cancer: evaluating the risk of sampling errors. Ann Oncol 24(3):725–733. https://doi.org/10.1093/annonc/mds528
Wei XL, Liu QW, Liu FR, Yuan SS, Li XF, Li JN et al (2021) The clinicopathological significance and predictive value for immunotherapy of programmed death ligand-1 expression in Epstein-Barr virus-associated gastric cancer. Oncoimmunology 10(1):1938381. https://doi.org/10.1080/2162402x.2021.1938381
Worthley DL, Phillips KD, Wayte N, Schrader KA, Healey S, Kaurah P et al (2012) Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): a new autosomal dominant syndrome. Gut 61(5):774–779. https://doi.org/10.1136/gutjnl-2011-300348
Yagi S, Wakatsuki T, Yamamoto N, Chin K, Takahari D, Ogura M et al (2019) Clinical significance of intratumoral HER2 heterogeneity on trastuzumab efficacy using endoscopic biopsy specimens in patients with advanced HER2 positive gastric cancer. Gastric Cancer 22(3):518–525. https://doi.org/10.1007/s10120-018-0887-x
Zhan S, Liu Z, Zhang M, Guo T, Quan Q, Huang L et al (2019) Overexpression of B7–H3 in α-SMA-positive fibroblasts is associated with cancer progression and survival in gastric adenocarcinomas. Front Oncol 9:1466. https://doi.org/10.3389/fonc.2019.01466
Zhang L, Wang Y, Li Z, Lin D, Liu Y, Zhou L et al (2021) Clinicopathological features of tumor mutation burden, Epstein-Barr virus infection, microsatellite instability and PD-L1 status in Chinese patients with gastric cancer. Diagn Pathol 16(1):38. https://doi.org/10.1186/s13000-021-01099-y
Zou W, Chen L (2008) Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 8(6):467–477. https://doi.org/10.1038/nri2326
Funding
Open Access funding enabled and organized by Projekt DEAL. The author have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
C.R. wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Röcken, C. Predictive biomarkers in gastric cancer. J Cancer Res Clin Oncol 149, 467–481 (2023). https://doi.org/10.1007/s00432-022-04408-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04408-0