
Abstract
The MDM2 proto-oncogene (MDM2) is a primary negative regulator of p53. The latter is frequently mutated in gastric cancer (GC). In the present study, we aimed to validate gene amplification, protein expression, and the putative tumor biological function of MDM2 in a well-characterized Western GC cohort. MDM2 amplification and protein expression were studied in a cohort of 327 GCs by fluorescence in situ hybridization (FISH) and immunohistochemistry. Gene amplification and protein expression were correlated with diverse clinicopathological patient characteristics including patient outcome. Immunohistochemically, 97 GCs (29.7%) were categorized as MDM2 positive and 230 GCs (70.3%) as negative. An amplification of MDM2 was found in 11 (3.4%) cases without evidence of intratumoral heterogeneity. Nine of these eleven (81.8%) cases showed MDM2 protein expression. MDM2 amplification correlated significantly with MDM2 protein expression (p < 0.001). On a case-by-case analysis, MDM2-amplified cases showed varied histological phenotypes and were most commonly microsatellite stable; EBV, HER2, and MET negative; and FGFR2 positive. A single case harbored both, MDM2 amplification and TP53 mutation. MDM2 amplification and MDM2 expression, respectively, did not correlate with overall or tumor-specific survival. Our targeted analysis of MDM2 in a well-characterized cohort of GC patients showed that MDM2 amplification is rare, of no specific histological phenotype, and may not be always mutually exclusive with TP53 mutations. Given the low number of cases, currently, no diagnostic or therapeutic recommendation related to MDM2 amplification can be given for GC of Western origin.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is the fifth most common cancer worldwide [1]. Its 5-year survival rate is still poor, ranging between 34 and 37% for men and women, respectively [2]. The vast majority are adenocarcinomas, which can be hereditary, familial, or sporadic. Common risk factors are chronic gastritis due to infection with Helicobacter pylori, a diet rich in salt, and lack of fresh fruits and vegetables. Less commonly, GC may be related to infection with Epstein-Barr virus (EBV). Gene polymorphisms and germline mutations modulate the individual susceptibility for GC [3].
Gastric cancer is a genetic disease affecting numerous oncogenes and tumor suppressor genes and was categorized into four molecular subtypes, i.e., chromosomal instable (CIN), genomically stable (GS), EBV-positive, and microsatellite-instable (MSI) GCs [4]. CIN-GC often has an intestinal histological phenotype according to Lauren and is associated with TP53 mutations and activation of receptor tyrosine kinases (RTK). GS-GC often exhibits a diffuse phenotype and mutations in CDH1 and RHOA, as well as CLDN18-ARHGAP fusion.
Using multiregional whole exome sequencing, we recently investigated the effect of somatic evolution on intratumoral heterogeneity aiming to shed light on the evolutionary biology of GC and noted that two cases harbored MDM2 amplifications [5]. The MDM2 proto-oncogene (MDM2) is located on chromosome 12 (12q15) and encodes a protein that inhibits p53’s tumor suppressor function by blocking the transcriptional activation domain, targeting ubiquitination, and exporting to the cytoplasm [6]. TP53 is among the most commonly mutated genes in GC accounting for 50–73% of all cases. MDM2, in turn, can be “activated” by gene amplification or promoter single nucleotide polymorphisms [7]. MDM2 amplification and TP53 mutations are mutually exclusive [8]. MDM2 amplification is a common finding in malignant soft tissue tumors and infrequent in other tumor types [7,8,9,10]. Promoter polymorphism and MDM2 amplification represent the two most extensively studied alterations. A germline single nucleotide polymorphism of the MDM2 promoter increased MDM2 expression, increased cancer risk, and accelerated tumor progression [11]. Amplification can be assessed by comparative genomic hybridization; quantitative polymerase chain reaction; fluorescence (FISH), automated dual-color (DISH), or chromogenic in situ hybridization (CISH); and immunohistochemistry with intense diffuse nuclear staining [7, 12,13,14,15]. Few studies addressed the role of MDM2 in GC [16,17,18].
Resistance to radiation and chemotherapy, to immune checkpoint inhibitor therapy, and to a RTK inhibition may be linked to MDM2 amplification [19]. A combination of radiotherapy and MDM2-p53 inhibitor (APG-115) could boost the effect of antitumor activity in vitro and in vivo [20]. MDM2 may also predict efficacy of adjuvant fluorouracil-leucovorin-oxaliplatin (FLO) chemotherapy in resectable GC [21].
In this study, we aimed to shed further light on the prevalence and putative tumor biological function of MDM2 in GC and studied protein expression and gene amplification in a large and well-characterized cohort of Western patients with GC. We finally demonstrate that MDM2 amplification is rare in GC, more commonly associated with microsatellite stability, FGR2 positivity, and HER2 negativity.
Material and methods
Statement on ethics
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent for the therapeutic surgeries had been obtained from all patients. Ethical approval was obtained from the local ethical review board (D 453/10 and D 525/15) of the University Hospital Schleswig-Holstein, Kiel, Germany.
Patients and tumor samples
From the archive of the Department of Pathology, University Hospital Schleswig-Holstein, Campus Kiel, we retrieved all patients who have undergone partial or complete gastrectomy for adenocarcinoma of the stomach or gastroesophageal junction between 1997 and 2009. Inclusion criteria were histologically confirmed primary adenocarcinoma. Criteria of exclusion were perioperative radiotherapy or chemotherapy, and histology identified a tumor type other than adenocarcinoma. The study was based on the assumption that adenocarcinomas of the stomach and gastroesophageal junction are very alike since both show the same four molecular subtypes proposed by The Cancer Genome Atlas (TCGA) [4, 22]. The following data were retrieved from the electronic database: patient age and gender, anatomical tumor location, tumor type according to Lauren [23], tumor grade (intestinal type only), depth of local tumor invasion (pT category), number of resected lymph nodes, number of lymph nodes with metastases (pN category), lymph node ratio, presence or absence of distant metastases (pM category), tumor stage according to UICC [24], lymphatic (pL category) or vascular (pV category) invasion, and residual tumor status (pR category) [25]. Date of patient death was obtained from Epidemiological Cancer Registry of the state of Schleswig-Holstein, Germany. Hospital records and general practitioners provided the follow-up data of patients still alive. All patient-related data were pseudonymized after inclusion in the study.
Assessing further clinicopathological characteristics
Assessments of mucin type, insulin receptor (IR) expression, FGFR2, human epidermal growth factor receptor 2 (HER2), MET, and p53 status, as well as the RHOA, PIK3CA, and KRAS genotype, were performed as previously described [26,27,28,29,30,31,32,33]. Modified Giemsa staining was used to evaluate infection with H. pylori and confirmed by polymerase chain reaction as described in detail previously [26]. BondMax detection system and EBER probe (Novocaster, Leica Microsystems GmbH, Wetzlar, Germany) were used for detecting EBV-encoded RNA based on the manufacturer’s instructions (Leica microsystems GmbH, Wetzlar, Germany) [27]. The study applied immunohistochemistry to assess microsatellite instability status using antibodies directed against MSH2, MSH6, MLH1, and PMS2 and performed subsequent molecular comparisons of the mononucleotide repeat markers NR-21, NR-24, NR-27, BAT-25, and BAT-26’s allelic profiles in the tumor for every case with absent or minimal nuclear staining [34].
Tissue microarray construction
Formalin-fixed and paraffin-embedded tissue samples were used to generate tissue microarrays (TMA) as described previously [35]. Briefly, five separated, morphologically representative regions of the paraffin “donor” block were chosen. Tissue cylinders of 1 mm diameter were punched from these areas and precisely arrayed into a new “recipient” paraffin block using a customer-built instrument (Beecher Instruments, Silver Spring, MD, USA). After completing the block construction, 4-μm sections of the resulting tumor TMA block were cut for further analysis. Hematoxylin and eosin staining was performed to control for successful transfer of tumor tissue.
Immunohistochemistry
Immunostaining was performed with a monoclonal mouse antibody directed against MDM2 (clone: 2A10, dilution 1:5000, Abcam, Berlin, Germany) using the BondMax Autostainer (Leica Microsystems GmbH, Wetzlar, Germany). ER2 antigen retrieval solution (20 min, Leica Microsystems GmbH, Wetzlar, Germany) was used for antigen retrieval. Staining was visualized with the Bond Polymer Refine Detection Kit (Leica Microsystems GmbH, Wetzlar, Germany). Hematoxylin served as counterstain. Immunostaining was assessed using a Zeiss microscope (Axioskop 40; Carl Zeiss AG, Oberkochen, Germany).
Assessing immunostaining
During evaluation of the immunostaining results, the pathologist was blinded to the clinical data. A scoring system was applied as outlined elsewhere [21]. In brief, only nuclear staining was considered and categorized as negative (0; no staining or staining of <5% of tumor cells), weak (1+), moderate (2+), or strong (3+) (Fig. 1). In addition, the percentage of stained tumor cells was recorded as 0 (<5% of tumor cells), 1+ (5–25%), 2+ (26–50%), 3+ (51–75%), and 4+ (76–100%), as previously described. Finally, a sum score was calculated combining intensity of nuclear staining and percentage of positive tumor cells. The minimum sum score was 0 and the maximum score was 7, lacking a sum score of 1.

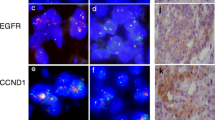
MDM2 fluorescence in situ hybridization and MDM2 immunostaining. MDM2 immunohistochemistry: case immunonegative for MDM2 (a) and case immunopositive for MDM2 (b). Anti-MDM2 antibody, hematoxylin counterstain, original magnification × 400. Case #7 with MDM2 amplification (c). One case without MDM2 amplification (d). Fluorescence in situ hybridization, original magnification × 1000
Fluorescence in situ hybridization
Three- to 5-μm-thick paraffin sections obtained from TMAs were used for FISH. Following dewaxing, tissue sections were re-hydrated in a descending alcohol series, incubated in 0.1 N citrate buffer 2 × 10 min, washed 2 × 2 min in dH2O, and incubated in 0.1 N HCl with 0.01% proteinase K for 10 min. The slides were then washed in dH2O for 5 min, dehydrated in descending alcohol series, dried, and incubated with 100 μl of fluorochrome-labeled probe mixture (Vysis MDM2/CEP 12 FISH Probe Kit, Abbott Molecular Inc., Des Plaines, IL 60018, USA) in a ThermoBrite hybridizer (Abbott) at 95 °C for 10 min, cooling to 37 °C and subsequently overnight. Following strict washing, dehydration, and air drying, tissue sections were finally counterstained with fluorescence mounting media containing 4′,6-diamidino-2-phenilindole (DAPI).
Assessment of fluorescence in situ hybridization
Fluorescence in situ hybridization was evaluated with a fluorescence microscope (Axio Imager.Z2, Carl Zeiss Microscopy GmbH, Göttingen, Germany) using suitable filter sets (AHF-filter set F56-700 dual-band filter green and orange red) and the following criteria: cell nuclei had to be generally intact and distinctively visible with clear borders. The background had to be black or dark and relatively free of haziness or fluorescence particles. The signals had to be easily evaluable, distinct, oval or round shaped, compact, and bright. If these criteria were not met, the specimen was not evaluated. The number of red (MDM2) and green (CEP12) fluorescence signals was counted in a minimum of 30 tumor cells (Fig. 1). Subsequently, the MDM2/CEP12 ratio was calculated for each case. MDM2 amplification was defined as MDM2/CEP12 ratio ≥ 2. A ratio <2 was classified as unamplified. MDM2 clusters precluding exact counting of MDM2 fluorescence signals were classified as “amplified, cannot be quantitated (CBQ)”. Immunostaining of whole mount tissue sections (“full slide section”) were then additionally performed on all MDM2-amplified cases.
Next-generation sequencing
Genomic DNA was extracted from formalin-fixed and paraffin-embedded tissue using the QIAamp DNA mini kit (Qiagen, Hilden, Germany) for all cases except case #4, in which no tumor DNA was available. Tissue sections were manually microdissected prior to DNA isolation to ensure a tumor cell content of higher than 80%. Library preparation was performed with AmpliSeq™ Library PLUS for Illumina using the AmpliSeq Focus Panel for Illumina and the AmpliSeq™ CD Indexes Set for Illumina® (Illumina, San Diego, USA). The quality of the libraries was assessed with a TapeStation 4150 and D1000 ScreenTapes (Agilent, Santa Clara, USA). Sequencing was done on the MiSeq System (Illumina) and sequencing data were analyzed with the Illumina VariantStudio 3.0 and the Onco CNV Caller (both Illumina). Mutations were retained if variant allele frequency (VAF) was ≥ 5%. Copy number variations were considered amplifications when total copy number was ≥6.0.
Statistical analysis
SPSS version 25.0.0.2 (IBM Corp., Armonk, NY) was used for statistical analyses. Fisher’s exact test was used to test association between nonordinal variables. Kendall’s tau rank correlation coefficient was used to test correlation of ordinal variables. We accepted a significant level of 0.05. The Simes (Benjamini-Hochberg) procedure was used to correct for false discovery rates (FDR) [36]. All p-values are uncorrected. Survival curves were estimated using the Kaplan-Meier method. Differences between median survival rates were tested using the log-rank (Mantel-Cox) test.
Results
Three hundred twenty-seven patients met all study criteria. One hundred twenty-three were female (37.6%) and 204 were male (62.4%). Median age was 68 years (range 31–92). One hundred nine (33.6%) GCs were localized in the proximal stomach (33.6%), and 215 (66.4%) in the distal stomach. According to Lauren’s classification [23], 163 (49.8%) GCs had an intestinal phenotype, 100 (30.6%) had a diffuse, and 74 (19.5%) cases were unclassifiable or mixed. Table 1 summarizes the clinicopathological patient characteristics.
Expression of MDM2 in gastric cancer
First, we examined the expression of MDM2 in GC by immunohistochemistry (Fig. 1a, b). Nuclear staining of MDM2 was found in 97 (29.7%) cases. Weak immunostaining (MDM2-1+) was observed in 67 (20.5%) cases, moderate (MDM2-2+) in 29 (8.9%), and strong (MDM2-3+) in 1 (0.3%). No immunostaining was found in 230 (70.3%) GCs. The percentage of the immunostained tumor area varied for all three staining intensities (weak to strong), ranging from 5 to 100%, with scores ranging from 1 to 4. The sum score was 2 in 58 (17.7%) cases, 3 in 20 (6.1%), 4 in 9 (2.8%), 5 in 8 (2.4%), and 6 in 2 (0.6%). There was no case with a sum score 7.
MDM2 amplification in gastric cancer
Next, we explored the amplification of MDM2 in GC by FISH (Fig. 1c, d). Eleven cases (3.4%) showed amplification of the MDM2. The mean MDM2/chromosome 12 ratio was 4.6 (range 3–8). In order to assess intratumoral heterogeneity and to avoid sampling error, we repeated FISH analysis using whole-mount tissue sections obtained from the eleven MDM2-amplified cases. MDM2 amplification was homogeneous in all these cases without evidence of intratumoral heterogeneity.
We then correlated MDM2 amplification with MDM2 expression. The MDM2 protein expression (sum score 2–6) was absent in 228 (72.2%) GCs without gene amplification, while MDM2-amplified cases expressed MDM2 protein in 9 of 11 cases (81.8%). In these cases, weak to strong nuclear immunopositivity was found and the sum score ranged from 2 to 5. Two cases with MDM2 amplification had no MDM2 protein expression (18.2%) (Fig. 2). Thus, MDM2 expression was significantly more common in MDM2-amplified cases (p < 0.001).

Case-wise summary of the phenotypic/genotypic co-alterations associated with MDM2 amplification. Green: cases with MDM2 amplification, intestinal phenotype, immunohistochemical reactivity, or mutated; red: cases without immunohistochemical reactivity, wildtype, or diffuse phenotype; gray: not available; yellow: unclassified; and orange: cases with mixed phenotype. Numbers in brackets document the sum score of MDM2 immunostaining
Phenotype of MDM2-amplified gastric cancers
Next, we explored the correlation of genotype with phenotype and reviewed the histology all MDM2-amplified cases. Seven were of an intestinal, two of a diffuse, and one of each mixed or unclassifiable according to Lauren (Fig. 2).
Correlation with clinicopathological patient characteristics
In order to explore the putative tumor biological significance, we correlated both, MDM2 expression and MDM2 amplification, with diverse clinicopathological patient characteristics. Following dichotomization of the MDM2 expression at the median immunoscore (0 vs. >0), MDM2-positive tumors were more commonly MET positive (p=0.033) and showed more commonly a membranous expression of the insulin receptor (p=0.044). No other clinicopathological patient characteristic correlated with the MDM2 expression. Following a separate analysis of intestinal type GCs, no correlation was found between MDM2 expression and any clinicopathological patient characteristic (data not shown).
With regard to copy number variation, it was interesting to note that MDM2-amplified GCs were commonly localized in the proximal stomach, were all negative for H. pylori and HER2, and were a KRAS, PIK3CA, and RHOA wildtype (Fig. 2). Interestingly, 9 (82%) cases were negative for MET and 10 (91%) for EBV. Microsatellite instability was noted in a single, MDM2-amplified case, while FGFR2 positivity was found in 7 (64%) MDM2-amplified cases (Fig. 2). Due to low case numbers, none of these findings was statistically significant.
Next-generation sequencing
To further explore the genotype of MDM2-amplified cases and search for putative druggable targets, tumor DNA of ten cases (case #1–3 and case #5–11) was forwarded to next-generation sequencing using the Oncomine™ Focus Assay, which covers hot spot mutations, copy number variations, and fusions of 52 different genes. As shown in Table 2, an amplification of CDK4 was found in two cases, among which one also showed an amplification of KRAS and ERBB3. No other alteration matched with MDM2 amplification. Two cases (# 6 and #10) were not assessable due to low DNA quality. Noteworthy, NGS did not detect the MET amplification in case #8 and #9 due to intratumoral heterogeneity of MET amplification [29].
Prognostic significance
No significant difference was found in the overall and tumor-specific survival between MDM2-amplified and nonamplified cases nor between cases with or without MDM2 expression (Fig. 3).

Kaplan-Meier curves of patient overall survival and tumor-specific survival using expression of MDM2 in the immunohistochemical examination (IHC) and MDM2 amplification using fluorescence in situ hybridization (FISH). a, b Patients’ overall survival and tumor-specific survival according to MDM2 expression; negative, no expression; positive, cases with a sum score 2–6. c, d Patients’ overall survival and tumor-specific survival according to FISH, amplified, not amplified
Discussion
To the best of our knowledge, our study is the single most extended analysis of MDM2 amplification in GC of White patients. Overall, amplification of MDM2 was rare accounting only for 3.4% of our cases, a prevalence supported by previous findings. MDM2 amplification was present in 1.6% of TCGA cohort and 5.5% of the cohort studied by Kato et al. [7, 10]. Among the 1114 cases with an adenocarcinoma of the esophagus, gastroesophageal junction, or stomach documented in the cBioPortal database (search date 17 September 2023), 72 (6.5%) cases harbored an MDM2 amplification and only 8 (0.7%) a mutation. Structural variants have not been reported [37]. Amplification rates may not be uniform across tumor types. Günther et al. demonstrated MDM2 amplification by Southern blot analysis in 18 of 43 (41.8%) advanced-stage GCs, commonly presenting with a diffuse growth pattern [17]. However, in our series, no evidence of intratumoral heterogeneity for MDM2 amplification was found and we studied a much larger patient cohort.
MDM2 amplification may lead to (over)expression of MDM2, although other mechanisms have been reported, e.g., germline single nucleotide polymorphisms. We were able to demonstrate a significant correlation between MDM2 amplification and protein expression. MDM2-amplified cases expressed MDM2 in 9 cases (81.8%) with gene amplification and MDM2 expression was significantly more common in MDM2-amplified cases. However, eighty-eight GCs were classified as MDM2 positive by immunohistochemistry without MDM2 amplification. Furthermore, the MDM2 protein expression was absent in two tumors with MDM2 amplification. Similar findings were reported from Cordon-Cardo et al. in soft tissue tumors. Twenty-seven percent of the sarcomas with MDM2 expression lacked MDM2 amplification, and vice versa 45% of the tumors with gene amplification lacked MDM2 expression [38]. A putative explanation might be mRNA splicing, which leads to different forms of the MDM2 protein, thereby prohibiting immunohistochemical detection. Marchetti et al. in the same sense indicated that the utilized antibodies might identify varied epitopes only, making immunohistological confirmation sometimes impossible [39]. They observed 21 cases without MDM2 amplification but MDM2 protein immunohistochemical reactivity, which in few cases was quite widespread [39]. Thus, gene amplification does not always correlate with protein expression and lack of immunostaining does not exclude MDM2 amplification. Molecular pathological analysis of MDM2 may give higher specificity and sensitivity compared with immunohistochemistry [40, 41].
Next, we tested the hypothesis that MDM2-amplified and/or MDM2-positive GCs harbor a specific molecular or clinical phenotype. While the overall number of MDM2-amplified GCs was low, it was interesting to note that these tumors frequently were of KRAS, PIK3CA, and RHOA wildtype; microsatellite stable; H. pylori and EBV negative; HER2 and MET negative; and positive for FGFR2 (Fig. 3). In addition, a single case showed both MDM2 amplification and TP53 mutation. Thus, it seems that MDM2 amplification and TP53 mutation are not mutually exclusive in all cases with GC. Supporting our findings, no significant difference in TP53 mutation frequency and MDM2 amplification status was found in esophageal carcinomas [42]. On the contrary, MDM2 alterations are mutually exclusive with TP53 mutations in sarcomas. However, further studies on larger case series are warranted to substantiate these observations. At least in our series, no correlation was found between p53 expression and MDM2 amplification.
MDM2 amplification co-altered with TP53 indicates a noncanonical, p53-independent role of MDM2 in tumor biology, which was apparent in breast cancer [39]. MDM2 facilitates angiogenesis as one of its suggested noncanonical effects [43]. The distribution of p53 alterations in identifying tumor type contrasted with the distribution of MDM2 amplification. According to Zhou et al., MDM2 regulates vascular endothelial growth factor translation, and it is active contributor to increased cancer cell growth and angiogenesis [44].
In summary, our study on a large cohort of GCs of Western origin shows that MDM2 amplification is rare in GC and more common in intestinal type GC. However, on a case-by-case analysis, intriguing findings were made, such as that MDM2-amplified cases were most commonly microsatellite stable; EBV, HER2, and MET negative, and FGFR2 positive.
Conclusion
Whole exome sequencing is becoming a mainstay of precision medicine and provides massive data on tumor genetics. These data are increasingly used to tailor patient treatment, particularly in the palliative setting: molecular tumor boards heavily rely on published data about the putative tumor biological significance of mutated target genes. However, there is a growing gap between the availability of genetic data and validation studies exploring the putative tumor biological significance of the respective gene product. To fill this gap of information, we extended our previous genetic research on nine GCs, two of which harbored an MDM2 amplification [5]. The targeted analysis of MDM2 in a well-characterized cohort of GC patients showed that MDM2 amplification is rare, more commonly of intestinal phenotype, although not exclusively, and may not be mutually exclusive with TP53 mutations. Given the low number of cases, currently, no diagnostic or therapeutic recommendation related to MDM2 amplification can be given for GC of Western origin.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Robert-Koch-Institut (2021) Krebs in Deutschland für 2017/2018. Zentrum für Krebsregisterdaten https://www.krebsdaten.de/Krebs/DE/Content/Publikationen/Krebs_in_Deutschland/krebs_in_deutschland_inhalt.html
Röcken C, Warneke V (2012) Molekulare Pathologie des Magenkarzinoms. Pathologe 33(Suppl 2):235–240. https://doi.org/10.1007/s00292-012-1634-4
The Cancer Genome Atlas Research Network (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513:202–209. https://doi.org/10.1038/nature13480
Röcken C, Amallraja A, Halske C et al (2021) Multiscale heterogeneity in gastric adenocarcinoma evolution is an obstacle to precision medicine. Genome Med 13:177. https://doi.org/10.1186/s13073-021-00975-y
Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ (1998) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J 17:554–564. https://doi.org/10.1093/emboj/17.2.554
Oliner JD, Saiki AY, Caenepeel S (2016) The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb Perpect Med 6:a026336. https://doi.org/10.1101/cshperspect.a026336
Momand J, Jung D, Wilczynski S, Niland J (1998) The MDM2 gene amplification database. Nucleic Acids Res 26(15):3453–3459. https://doi.org/10.1093/nar/26.15.3453
Oliner J, Kinzler K, Meltzer P, George DL, Vogelstein B (1992) Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358:80–83. https://doi.org/10.1038/358080a0
Kato S, Ross JS, Gay L, Dayyani F, Roszik J, Subbiah V, Kuryrock R (2018) Analysis of MDM2 amplification: next-generation sequencing of patients with diverse malignancies. JCO Precis Oncol 2:1–14. https://doi.org/10.1200/PO.17.00235
Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G, Levien AJ (2004) A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119:591–602
Coindre JM, Pedeutour F, Aurias A (2010) Well-differentiated and dedifferentiated liposarcomas. Virchows Arch 456(2):167–179. https://doi.org/10.1007/s00428-009-0815-x
Kobayashi A, Sakuma T, Fujimoto M, Jimbo N, Hirose T (2019) Diagnostic utility and limitations of immunohistochemistry of p16, CDK4, and MDM2 and automated dual-color in situ hybridization of MDM2 for the diagnosis of challenging cases of dedifferentiated liposarcoma. Appl Immunohistochem Mol Morphol 27(10):758–763. https://doi.org/10.1097/PAI.0000000000000677
Jimbo N, Komatsu M, Itoh T, Hirose T (2019) MDM2 dual-color in situ hybridization (DISH) aids the diagnosis of intimal sarcomas. Cardiovasc Pathol 43:107142. https://doi.org/10.1016/j.carpath.2019.07.001
Mardekian SK, Solomides CC, Gong JZ, Peiper SC, Wang Z, Bajaj R (2015) Comparison of chromogenic in situ hybridization and fluorescence in situ hybridization for the evaluation of MDM2 Amplification in adipocytic tumors. J Clin Lab Anal 29:462–468
Blok P, Craanen ME, Dekker W, Offerhaus GJA, Tytgat GNJ (1998) No evidence for functional inactivation of wild type p53 protein by mdm2 overexpression in gastric carcinogenesis. J Pathol 186(1):36–40. https://doi.org/10.1002/(SICI)1096-9896(199809)186:1<36::AID-PATH150>3.0.CO;2-R
Günther T, Schneider-Stock R, Häckel C, Kasper HU, Pross M, Hackelsberger A, Lippert H, Roessner A (2000) Mdm2 gene amplification in gastric cancer correlation with expression of Mdm2 protein and p53 alterations. Mod Pathol 13(6):621–626
Bartpho TS, Wattanawongdon W, Tongtawee T (2021) The mouse double minute 2 polymorphism is associated with both decreased p53 expression and poor clinicopathological outcomes of gastric cancer. J Can Res Ther 17:1438–1444
Hou H, Sun D, Zhang X (2019) The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int 19:216. https://doi.org/10.1186/s12935-019-0937-4
Yi H, Yan X, Luo Q, Yuan L, Li B, Pan W, Zhang Y, Chen H, Wang J, Zhang Y, Zhai Y, Qiu M, Yang D (2018) A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J Exp Clin Can Res 37:97. https://doi.org/10.1186/s13046-018-0765-8
Ye Y, Li X, Yang J, Miao S, Chen Y, Xia X, Wu X, Zhang J, Zhou Y, He S, Tan Y, Qiang F, Li G, Roe OD, Zhou J (2013) MDM2 is a useful prognostic biomarker for resectable gastric cancer. Cancer Sci 104(5):590–598
The Cancer Genome Atlas Research Network (2017) Integrated genomic characterization of esophageal carcinoma. Nature 541:169–175. https://doi.org/10.1038/nature20805
Lauren P (1965) The two histologic main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. Acta Pathol Microbiol Scand 64:31–49
Brierley J, Gospodarowicz MK, Wittekind C (2017) TNM classification of malignant tumours, 8rd edn. Wiley-Blackwell, USA
Hermanek P, Wittekind C (1994) Residual tumor (R) classification and prognosis. Semin Surg Oncol 10(1):12–20. https://doi.org/10.1002/ssu.2980100105
Warneke VS, Behrens HM, Haag J, Balschun K, Böger C, Becker T, Ebert MPA, Lordick F, Röcken C (2013) Prognostic and putative biomarkers of gastric cancer for personalized medicine. Diagn Mol Pathol 22(3):127–137. https://doi.org/10.1097/PDM.0b013e318284188e
Böger C, Krüger S, Behrens HM, Bock S, Haag J, Kalthoff H, Röcken C (2017) Epstein-Barr virus associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann Oncol 28:1005–1014. https://doi.org/10.1093/annonc/mdx047
Warneke VS, Behrens HM, Böger C, Becker T, Lordick F, Ebert MPA, Röcken C (2013) Her2/neu testing in gastric cancer: evaluating the risk of sampling errors. Ann Oncol 24:725–733. https://doi.org/10.1093/annonc/mds528
Metzger ML, Behrens HM, Böger C, Haag J, Krüger S, Röcken C (2016) MET in gastric cancer – discarding a 10% cutoff rule. Histopathology 68:241–253. https://doi.org/10.1111/his.12745
Schoop I, Maleki SS, Behrens HM, Krüger S, Haag J, Röcken C (2020) p53 immunostaining cannot be used to predict TP53 mutations in gastric cancer: results from a large Central European cohort. Human Pathol 105:53–66. https://doi.org/10.1016/j.humpath.2020.09.006
Schrumpf T, Behrens HM, Haag J, Krüger S, Röcken C (2022) FGFR2 overexpression and compromised survival in diffuse-type gastric cancer in a large central European cohort. PLoS ONE 17(2):e0264011. https://doi.org/10.1371/journal.pone.0264011
Röcken C, Behrens HM, Böger C, Krüger S (2016) Clinicopathological characteristics of RHOA mutations in a Central European gastric cancer cohort. J Clin Pathol 69:70–75. https://doi.org/10.1136/jclinpath-2015-202980
Heckl SM, Wiesener V, Behrens HM, Ulase D, Krüger S, Röcken C (2019) The expression of the insulin receptor in gastric cancer correlates with the HER2 status and may have putative therapeutic implications. Gastric Cancer 22(6):1130–1142. https://doi.org/10.1007/s10120-019-00964-6
Mathiak M, Warneke VS, Behrens HM, Haag J, Böger C, Krüger S, Röcken C (2017) Clinicopathologic characteristics of microsatellite instable gastric carcinomas revisited: urgent need for standardization. Appl Immunhistochem Mol Morphol 25:12–24. https://doi.org/10.1097/PAI.0000000000000264
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP (1998) Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 4:844–847
Simes RJ (1986) An improved Bonferroni procedure for multiple tests if significance. Biometrika 73:751–754
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):l1. https://doi.org/10.1126/scisignal.2004088
Cordon-Cardo C, Latres E, Drobnjak M, Oliva MR, Pollack D, Woodruff VM, Marechal V, Chen J, Brennan MF, Levine AJ (1994) Molecular abnormalities of MDM2 and p53 genes in adult soft tissue sarcomas. Cancer Res 54:794–799
Marchetti A, Buttitta F, Girlando S, Dalla Palma P, Pellegrini S, Fina P, Doglioni C, Bevilacqua G, Barbareschi M (1995) MDM2 gene alterations and MDM2 protein expression in breast carcinomas. J Pathol 175:31–38. https://doi.org/10.1002/path.1711750106
Machado I, Vargas AC, Maclean F, Llombart-Bosch A (2022) Negative MDM2/CDK4 immunoreactivity does not fully exclude MDM2/CDK4 amplification in a subset of atypical lipomatous tumor/well differentiated liposarcoma. Pathol Res Pract 232:153839. https://doi.org/10.1016/j.prp.2022.153839
Weaver J, Sowns-Kelly E, Goldblum JR, Turner S, Kulkarni S, Tubbs RR, Rubin BP, Skacel M (2008) Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol 21:943–949. https://doi.org/10.1038/modpathol.2008.84
Michalk M, Meinrath J, Künstlinger H, Koitzsch U, Drebber U, Merkelbach-Bouse S, Bollschweller E, Kloth M, Hartmann W, Hölschner A, Quaas A, Grimminger PP, Odenthal M (2016) MDM2 gene amplification in esophageal carcinoma. Oncology Rep 35:2223–2227. https://doi.org/10.3892/or.2016.4578
Kim ES, Shohet JM (2015) Reactivation of p53 via MDM2 inhibition. Cell Death Dis 6:e1936. https://doi.org/10.1038/cddis.2015.302
Zhou S, Gu L, He J, Zhang H, Zhou M (2011) MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Mol Cell Biol 31:4928–4937. https://doi.org/10.1128/MCB.06085-11
Acknowledgements
We thank Silke Lüschen, Christin Dewitz, and Franziska Wilhelm for their technical assistance with next-generation sequencing.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Study concept and design were contributed by S.A. and C.R. Surgical pathological data were acquired by S.A., S.K., and C.R. Molecular pathological analysis was conducted by S.A., S.K., and J.H. The data were analyzed and interpreted by S.A., H.-M.M., and C.R. Drafting of the manuscript and critical revision of the manuscript were carried out by all authors. Administrative, technical, and material support was provided by S.K. and C.R.
Corresponding author
Ethics declarations
Ethical statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent for the therapeutic surgeries had been obtained from all patients. Ethical approval was obtained from the local ethical review board (D 453/10 and D 525/15) of the University Hospital Schleswig-Holstein, Kiel, Germany.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdullazade, S., Behrens, HM., Krüger, S. et al. MDM2 amplification is rare in gastric cancer. Virchows Arch 483, 795–807 (2023). https://doi.org/10.1007/s00428-023-03674-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-023-03674-8