
Abstract
A repeat expansion in C9orf72 is responsible for the characteristic neurodegeneration in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) in a still unresolved manner. Proposed mechanisms involve gain-of-functions, comprising RNA and protein toxicity, and loss-of-function of the C9orf72 gene. Their exact contribution is still inconclusive and reports regarding loss-of-function are rather inconsistent. Here, we review the function of the C9orf72 protein and its relevance in disease. We explore the potential link between reduced C9orf72 levels and disease phenotypes in postmortem, in vitro, and in vivo models. Moreover, the significance of loss-of-function in other non-coding repeat expansion diseases is used to clarify its contribution in C9orf72 ALS/FTD. In conclusion, with evidence pointing to a multiple-hit model, loss-of-function on itself seems to be insufficient to cause neurodegeneration in C9orf72 ALS/FTD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A repeat expansion in the C9orf72 gene is associated with two neurodegenerative disorders, which represent the extremes of a disease spectrum: amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [108]. In ALS, loss of motor neurons in the spinal cord, brainstem, and primary motor cortex causes muscle weakness. FTD is characterized by degeneration of cortical neurons in the frontal and temporal lobe resulting mainly in behavioral aberrations. Characteristic for both diseases is the large phenotypic variability, including the heterogeneous involvement of upper and/or lower motor neurons [90, 108]. In contrast, a common pathological hallmark in 97% of ALS and 45% of FTD patients is the occurrence of cytoplasmic inclusions containing the hyperphosphorylated and mislocalized RNA-binding protein TDP-43 [15, 69].
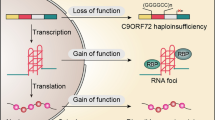
The ‘GGGGCC’ hexanucleotide repeat expansion in the C9orf72 gene is the most common mutation in both familial (± 30%) and sporadic (± 5–10%) disease forms [28, 92, 114]. Despite extensive mechanistic research, the exact disease mechanism remains elusive. Three not mutually exclusive mechanisms have been proposed (Fig. 1) [43]. First, a gain-of-function (GOF) might result from repeat RNA sequestering functionally important RNA binding proteins. A second GOF mechanism is based on the observation of dipeptide repeat protein (DPR) species generated by repeat-associated non-ATG (RAN) translation of the GGGGCC repeat sequence. Third, a loss-of-function (LOF) mechanism might result from reduced C9orf72 protein levels.

Three disease mechanisms proposed to underlie C9orf72 ALS/FTD. First, the repeat expansion (GGGGCC)n could impede transcription processes leading to a reduction in C9orf72 protein levels and hence a loss-of-function of the gene. Second, through bidirectional transcription, the repeat expansion might form secondary sense (GGGGCC) and antisense (CCCCGG) RNA structures sequestering regulatory RNA binding proteins (RBPs). This process, called RNA toxicity, could induce a loss-of-function of these proteins. Third, the non-ATG repeat-associated (RAN) translation process forms potentially toxic dipeptide repeat proteins from the sense strand (GP, GA, GR) and the antisense strand (GP, PA, PR)
Elucidating which (combination) of these mechanisms is the culprit for C9orf72 ALS/FTD (C9 ALS/FTD) is crucial for two main reasons. On one hand, it might shed light on the disease mechanism of other monogenetic as well as sporadic forms of ALS/FTD. On the other hand, it is pivotal to tailor the appropriate treatment of C9 ALS/FTD. Several therapeutic approaches targeting one or more possible mechanisms have been proposed. However, their efficacy and safety still need to be demonstrated. So far, it is unclear whether targeting only one mechanism (e.g. DPR toxicity through DPR antibodies or LOF through C9orf72 gene therapy) will be sufficient to have a disease-modifying effect in patients. Moreover, it is still debated whether targeting the GOF mechanisms using a knockdown approach (e.g. with antisense oligonucleotides (ASOs)) might have detrimental effects by exacerbating C9orf72 LOF.
Whereas the contribution of the GOF mechanisms has already extensively been investigated and reviewed [37, 109] a critical appraisal of C9orf72 LOF is currently lacking. In this review, we will provide an overview of the current knowledge on the physiological function of the C9orf72 protein, as well as the contribution of C9orf72 LOF in C9orf72 ALS/FTD pathogenesis.
The physiological role of C9orf72
The transcription and translation of C9orf72
The C9orf72 gene (Fig. 2) is located at the short arm of chromosome 9. Its sequence has a high degree of homology compared to different species commonly used as model systems (98.1% with Mus musculus, 97.7% with Rattus norvegicus and 76.0% with Danio rerio [26]). This suggests that the protein is involved in essential cellular processes.

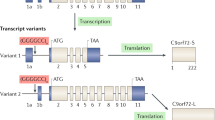
C9orf72 gene structure, transcripts, and isoforms. The C9orf72 gene contains 11 exons. Transcription of this gene results in three main transcripts (V1, V2, V3). The repeat region is located in the first intron of transcripts V1 and V3, whereas the V2 transcript contains the repeat in its promoter region. V4 and V5 are non-coding transcript variants with V5 being a truncated form of V4. Upon translation, two C9orf72 isoforms are formed. The short isoform (24 kDa) arises from the V1 transcript and the long isoform (54 kDa) is translated from V2 or V3
At the mRNA level, diverse transcription start sites trigger the formation of three transcript variants (Fig. 2). Variant 1 (V1; NM_145005.7) and variant 3 (V3; NM_001256054.3), starting from exon 1a, contain the repeat expansion in the first intron, while variant 2 (V2; NM_018325.5) initiates its coding region from exon 1b and harbors the expansion in the promoter region [10, 30, 42]. In addition to these three conventional transcripts, at least two non-coding transcript variants (lncRNAs) have been described, including their natural antisense transcripts [30, 41, 93]. Due to the alternative transcription start sites, exon 1c is the first of transcript variant 4 (V4) and 5 (V5) with V5 representing a truncated form of V4 (Fig. 2). The contribution of these lncRNAs to the physiological function of C9orf72 and the C9 ALS/FTD disease mechanism has not yet been identified.
At the protein level, translation results in two isoforms: transcript variant 1 encodes a 222 amino acid isoform (called ‘C9-short’ or ‘C9-S’; 24 kDa), while transcript variants 2 and 3 encode a 481 amino acid isoform (called ‘C9-long’ or ‘C9-L’; 54 kDa) (Fig. 2).
The cellular and subcellular distribution of C9orf72
At the cellular level, the highest C9orf72 transcript expression is detected in a subset of myeloid cells [93], whereas protein levels (i.e. the long isoform) seem to be most abundant in the brain and spinal cord [38]. Because of the differential use of transcription start sites for C9orf72 transcripts between myeloid and CNS tissues, C9orf72 function might differ between cell types [93]. In the central nervous system (CNS), C9orf72 promoter activity is most abundant in neuronal and glial cell types recognized to degenerate in ALS/FTD which might render them more susceptible to the effects of C9orf72 LOF as well as GOF toxicity [65]. Notably, quantitative mass-spectrometry-based proteomics revealed that C9orf72 has a 200-fold lower baseline expression compared to proteins like TDP-43 and FUS [31, 51], which also complicates the detection of endogenous C9orf72.
At the subcellular level, the highly soluble C9orf72 is detected in both the nucleus and the cytoplasm [38]. The long isoform is the most abundant one and mainly exhibits a diffuse nuclear distribution [123]. Both isoforms also exhibit a cytoplasmic vesicular distribution [33, 74], representing endosomes, lysosomes, and other autophagy-related vesicles, such as autophagosomes [33, 104]. In neurons, C9orf72 has also been localized to the presynaptic region [38].
The protein function of C9orf72
Involvement of C9orf72 in membrane trafficking
To fully understand the underlying disease mechanism of C9orf72 ALS/FTD, it is essential to elucidate the physiological function of C9orf72. Computational analyses revealed a structural homology between C9orf72 and DENN (differentially expressed in normal and neoplastic cells) proteins, implying C9orf72 to act as a RabGEF (Guanine Exchange Factor of Rab proteins) [68, 75]. These factors are well known to activate Rab GTPases (‘Rabs’), hence controlling membrane trafficking events (e.g. endocytosis and autophagy) in the cell. As C9orf72 also colocalizes with several of these Rabs, it is likely to have a function in membrane trafficking [33]. Association of C9orf72 with these Rab proteins indicates that C9orf72 may be specifically involved in endocytic and phagocytic trafficking [6, 33].
Disposing of cellular garbage (e.g. damaged organelles and other dysfunctional cellular components) is essential for cellular homeostasis and for recycling various factors and proteins. This process, called autophagy, can be mediated by several routes. One of these routes, macroautophagy, plays a role in neurodegeneration [39, 120]. One example is that depletion of essential autophagic factors in mice results in protein aggregation, motor neuron loss, and motor dysfunction [48, 60, 84]. C9orf72 forms a stable tripartite complex with WD repeat-containing protein 41 (WDR41) and Smith-Magenis chromosome region 8 (SMCR8) proteins [107, 117, 124]. This complex recruits Rab proteins and subsequently controls early stages of autophagy at four possible levels (Fig. 3a) [33, 101, 107]. A first level is the p62-mediated recruitment of ubiquitinated substrates to the phagophore through the interaction of Rab39b with the complex [84, 101, 110, 124]. Second, C9orf72 regulates the Rab1 and Rab5-dependent trafficking of the ULK1 autophagy initiation complex to induce autophagosome formation [4, 107, 117]. Third, the maturation and closure of the autophagosome by Rab7 and Rab11 might be regulated by C9orf72 [33]. Fourth, the interaction with Rab8a could trigger autophagosome-lysosome fusion [4]. Interestingly, C9orf72 and p62 colocalize with stress granules and Rab1a was identified to be part of these structures (Fig. 3a) [21, 54, 118]. Moreover, mass spectrometry revealed C9orf72 to interact with the heat-shock chaperone, HSC70, essential for the initiation of aggrephagy, a mechanism responsible for clearing aggregating proteins. Hence, it appears that C9orf72 might also be relevant in selective protein aggregation macroautophagy [84, 101]. Despite these rather descriptive associations with key autophagy players, the net impact of C9orf72 on autophagy is still unclear.

The function of C9orf72 in the central nervous system. a In neurons, C9orf72 (or tripartite complex C9orf72/SMCR8/WDR41) directly or indirectly regulates autophagy at four levels; recruitment of substrates to the phagophore (1), phagophore formation (2), maturation and closure of the autophagosome (3) and fusion of the autophagosome with lysosomes (4). Vesicle trafficking in the Golgi apparatus could also be controlled by C9orf72 (5). Interaction with stress granules (6), RNA binding proteins (7) and nucleocytoplasmic transport factors (8) points towards a possible regulating role of C9orf72 in phase separation (6), RNA metabolism (7) and nucleocytoplasmic transport (8). b C9orf72 also localizes presynaptically where it might interact with extracellular vesicle secretion (9) and the cytoskeleton (i.e. cofilin) (10). c In microglia, phagocytosis of dying neurons and other cells has been associated with C9orf72/SMCR8/WDR41 (11). C9orf72 is involved in the clearing of aggregated proteins (12) and the release of cytokines (13)
Next to its role in the initiation phase, C9orf72 seems to be involved in later phases of autophagy as well. Illustrating this, various C9orf72 knockout models exhibit a general dysregulation of lysosomal homeostasis, including defects in lysosome morphology, lysosomal accumulation, and defective fusion of lysosomes to phagosomes [4, 5, 25]. Mouse studies indicate that C9orf72 regulates lysosomal exocytosis in macrophages and microglia [87, 127].
Based on work in C. elegans and neuroblastoma cell lines, C9orf72 was also associated with endocytic pathways [25]. C9orf72 interacts with Rab5 (part of early endosomes), Rab7 (part of late endosomes) and Rab11 (regulates recycling of endocytosed proteins), each associated with a critical phase in the formation of endosomes [50, 73, 129]. Next to autophagy initiation, lysosomes, and endosomes, C9orf72 could also be involved in other membrane trafficking events like extracellular vesicle secretion (Fig. 3b) and trans-Golgi vesicle trafficking (Fig. 3a) [6].
C9orf72 and the immune system
C9orf72 might also play a role in the immune system (Fig. 3c). This is mainly based on the observation of C9orf72 knockout mice (as well as SMCR8 knockout mice) developing inflammatory and autoimmune phenotypes, such as enlarged lymph nodes, splenomegaly, immune-mediated glomerulonephropathy and excessive release of inflammatory cytokines and autoantibodies [7, 16, 64, 79, 106]. These findings suggest that C9orf72 is involved in the immune homeostasis in macrophages and microglia [7, 87]. The exact underlying mechanism of this immune dysfunction is still unclear but might be related to the high expression level of C9orf72 in myeloid cells. In addition, a link with the role of C9orf72 in membrane trafficking has been suggested. In the absence of C9orf72, microglia display a diminished ability to clear aggregated proteins [87, 93]. Moreover, defects in autophagy and lysosomal exocytosis have been observed in C9orf72 and SMCR8 double knockout mice [7, 64, 87, 107, 127].
Other functions of C9orf72
As mentioned before, C9orf72 has been reported to be involved in stress granules (Fig. 3a). In the event of stress signals, C9orf72 is recruited to stress granules [74]. The C9orf72 long isoform has been shown to be essential for the assembly of these granules [74]. More specifically, in the absence of C9orf72, cells become hypersensitive to stress signals, such as endoplasmic reticulum (ER) stress [74]. This might render the cells more vulnerable to toxicity induced by the products of gain-of-function mechanisms, including RNA foci and DPRs [66]. C9orf72 also localizes at the nuclear pore and interacts with components of the nuclear pore complex (Fig. 3a) [122]. Hence, C9orf72 might have a role in nucleocytoplasmic transport. Notwithstanding that nucleocytoplasmic transport defects are especially well studied in the context of gain-of-function mechanisms [11, 36, 59, 66, 115, 126], it remains ambiguous whether these defects could be related to or enhanced by C9orf72 loss-of-function. Moreover, the interaction of C9orf72 with cofilin and other actin binding proteins [105] indicates that C9orf72 could be involved in the organization of actin filaments in neuronal axons (Fig. 3b). Last but not least, RNA binding proteins (e.g. hnRNPK, hnRNPA3…) are associated with C9orf72, suggesting that, next to their role in RNA toxicity, these proteins might also be involved in the LOF mechanism (Fig. 3a) [24, 47, 83].
Assessment of the loss-of-function mechanism in C9orf72 ALS/FTD
To assess the contribution of C9orf72 LOF in C9 ALS/FTD, we will address two main aspects. On one hand, the impact of the repeat expansion on C9orf72 physiology (i.e. epigenetics, transcription, protein levels et cetera) and functionality (i.e. downstream targets) in C9 ALS/FTD patients needs to be assessed. On the other hand, determining whether this degree of impact is sufficient to induce (or contribute to) neurodegeneration is crucial as well.
Postmortem studies
Homozygous C9orf72 patients and coding mutations in C9 ALS/FTD
Classical genetic indications of a LOF mutation include increased clinical severity in the case of homozygosity of the mutation, as well as mutations occurring in the coding region (e.g. nonsense mutations or missense mutations). Although very rare, two independent studies each reported one patient homozygous for the C9orf72 repeat expansion. These patients did not exhibit a more severe clinical phenotype compared to heterozygous cases [23, 35]. Moreover, no coding mutations in the C9orf72 gene were found in C9 ALS/FTD [49], except for one solitary patient harboring a splice site mutation in exon 5 [71]. Nonetheless, a neurological clinical examination, autopsy, and assessment of a C9orf72 repeat expansion have not been performed in this patient.
Epigenetics
Epigenetic modifications are known to alter gene expression through e.g. DNA methylation and histone modifications [46]. Several studies investigated methylation levels of the C9orf72 promoter and the repeat expansion itself in genomic DNA from the patient brain and/or blood samples [8, 45, 53, 70, 80, 94, 121]. Hypermethylation of CpG sites in the promoter region of the C9orf72 gene, leading to decreased promoter activity, is associated with shorter disease duration (i.e. a more aggressive disease course) [45, 53, 70], supporting LOF. However, other studies contradict this finding and suggest an association of C9orf72 promoter hypermethylation with increased survival [80, 94]. As the majority of these studies only investigated methylation patterns of the promoter region, uncertainty about a protective or detrimental effect might be explained by additional hypermethylated regions elsewhere in the gene. Next to the promoter, the C9orf72 repeat expansion itself can be hypermethylated [121], which is the case in almost all (97%) repeat expansion carriers, whereas only one third has a hypermethylated C9orf72 promoter region [70]. As hypermethylated patterns can spread along a gene [58], this suggests that the epigenetic alteration originated in the repeat region and subsequently spread towards the promoter region. Moreover, methylation can be seen as a neuroprotective response by preventing expression of the C9orf72 repeat expansion and hence suppressing the formation of potentially toxic RNA foci and/or DPRs [80, 121].
Transcript levels
Several studies evaluated C9orf72 transcript levels in postmortem tissue, examining the total C9orf72 transcript level and/or individual transcript levels of V1 and V3, containing the repeat in the first intron, and V2, harboring the repeat in the promoter region (Table 1) [9, 10, 17, 22, 28, 34, 44, 53, 82, 116]. The frontal cortex, cerebellum, and lumbar spinal cord are the most frequently analyzed CNS regions in these studies. Considering all reports, an overall decrease (~ 50%) in total sense C9orf72 transcript levels in frontal cortex and cerebellum is evident and, interestingly, independent of age-at-onset or age-at-death [9, 10, 22, 34, 44, 82, 116]. However, measuring individual transcripts reveals that V3 remains unaltered, while V1 and especially V2 levels are significantly decreased [10, 28, 116]. Moreover, similar reductions in V1 and V2 were observed in blood samples [53]. The obvious decrease of V2 levels in both blood and CNS samples is probably due to the repeat being located in its promoter region. The repeat expansion might inhibit the binding and/or function of polymerases and transcription initiation factors. V1 and V3 comprise the expansion in the first intron and might, therefore, be less susceptible to transcriptional defects.
Protein levels
Decreased transcript levels do not necessarily implicate decreased protein levels (Table 1). In the frontal cortex of C9 ALS/FTD patients, a 25% decrease (compared to healthy controls, sALS cases and neurodegenerative disease controls) of the long protein isoform C9orf72 was consistently observed [95, 116, 122]. Measurements of cerebellar protein levels are rather inconsistent, ranging from 80% reduction [38, 105] to unaltered protein levels [95, 116, 122]. Moreover, the reduced cerebellar levels do not associate with clinical findings, questioning the disease relevance of cerebellar protein levels [38]. Altogether, a rather modest (25%) decrease of C9orf72 protein levels seems to be rather small to support a LOF mechanism. However, one needs to keep in mind that protein levels might be lower in disease-relevant cell types (e.g. affected neurons) but might escape detection due to a dilution effect. Also, disease-relevance of the examined tissue in most studies is not so straightforward as the denominator ‘frontal cortex’ encompasses both the prefrontal cortex as well as the primary motor cortex, the former being more relevant in FTD and the latter in ALS. Additionally, issues regarding C9orf72 antibody specificity and small sample size further complicate this matter. C9orf72 protein levels do not seem to correlate with age [38]. On the other hand, patients with large repeat expansions show an increased expression of the C9orf72 short isoform, whereas intermediate repeat carriers contain a higher expression of the long isoform [17, 122]. This suggests a possible repeat length-dependent effect on C9orf72 expression level and isoform distribution. Hence, the presence of repeat mosaicism (i.e. different C9orf72 repeat sizes in different tissues) between and within patients might obscure the effect on the expression level of the disease-relevant protein products.
Conclusions
Altogether, genetic and epigenetic data are difficult to reconcile with a predominant LOF mechanism. Most importantly, while C9orf72 transcript levels seem to be reduced by approximately 50%, protein levels are probably reduced by only 25% and there is no indication that C9orf72 functionality is impaired in C9 ALS/FTD patients. As a consequence, further research is required. Specifically, assessment of all individual transcript levels, the contribution of long versus short protein isoform, development of specific antibodies, increased sample sizes, and assessment of disease-relevant brain regions and cell types will be indispensable.
In vitro studies
Whereas postmortem material can be regarded as the end-stage of the disease mechanism, in vitro models with a disease-relevant phenotype might be able to provide earlier mechanistic insights. Additionally, various knockdown approaches (i.e. siRNA, antisense oligonucleotides, CRISPR/Cas9) can be harnessed to assess whether C9orf72 LOF could be harmful.
C9 patient-derived models
Epigenetics
An asset for the use of iPSC-derived neurons as a model system is that the C9orf72 promoter hypermethylation observed in patients is reacquired upon neuronal differentiation [32]. Therefore, this model might be used to study the role of epigenetics in C9 ALS/FTD. In patient lymphoblastoid cell lines, the demethylation of C9orf72 promoter (associated with increased gene expression) increases the cell vulnerability to oxidative and autophagic stress [70]. This implies that the observed hypermethylation probably represents an epigenetic silencing as a protective regulatory reaction to transcriptional instability due to the hexanucleotide repeat expansion. As a consequence, epigenetic in vitro data are difficult to reconcile with a predominant LOF mechanism.
Transcript and protein levels
In keeping with postmortem findings (cf. supra), most C9 patient iPSC-derived neurons exhibit an overall reduction (50%) in C9orf72 transcripts (Table 2) [3, 29, 78, 104, 117]. On the contrary, a recent study did not observe these changes in individual and total transcript levels [40]. Also, fibroblasts and iPSCs do not show these consistent transcript alterations (Table 2) [3, 29, 40, 98]. Moreover, protein levels seem to be reduced in fibroblasts, but not in C9 iPSC-derived motor neurons (MNs) (Table 2) [1, 92, 98, 102]. As a consequence, there are discrepancies between transcript and protein alterations between models and decreased C9orf72 transcript levels do not always correlate with decreased C9orf72 protein levels. Issues regarding C9orf72 antibody specificity could contribute to this discrepancy [28, 92]. Numerous C9orf72 antibodies (including polyclonal, monoclonal and recombinant) have been developed, but remain largely unverified. Interestingly, a novel powerful CRISPR genome-editing approach validated multiple commercially available C9orf72 antibodies, which could be of use and may be considered for future C9orf72 loss-of-function studies [62].
Impaired autophagy
In C9 patient iPSC-derived MNs, several hallmarks of impaired autophagy were observed, including lysosomal alterations, p62 aggregation, proteasomal degradation, and reduced autophagic flux (Table 2) [1, 3, 6, 27, 104, 117]. However, it is unclear so far whether this autophagic disturbance is caused by the GOF or LOF mechanism as these cells contain the ‘GGGGCC’ repeat expansion. Furthermore, a synergistic mechanism is not excluded as autophagy is needed to clear toxic GOF products (i.e. DPRs) [1, 12, 88].
Modelling C9orf72 knockdown and knockout
Wild type models (i.e. in the absence of a GGGGCC repeat expansion) have the advantage of evaluating the effect of C9orf72 knockdown without confounding GOF mechanisms. This allows a clearer evaluation of the effect of C9orf72 knockdown. However, one needs to keep in mind that these models are artificial, hence, disease relevance might be lacking. Therefore, efforts should be done to make these models as reminiscent as possible to C9 patient-derived models. Methods including siRNA, shRNA, and ASOs to knock down or CRISPR/Cas9 to knock out C9orf72 were used to model and investigate the C9orf72 LOF hypothesis in vitro (Table 3). Whereas most knockdown approaches do not or only partially (30%) affect C9orf72 protein levels, knockout models represent a full ablation of the protein. Importantly, reduction in transcript levels do not correlate with a similar reduction in protein levels. To illustrate this, a siRNA-mediated reduction of 50% in total transcript levels does not affect protein levels in primary neuronal cultures [101], whereas an ASO-induced 90% transcript reduction only corresponds to a 30% decrease in protein level in iPSC-derived neurons (iPSNs) [98]. A number of reasons could explain the discrepancy between transcript and protein levels [72], including technical biases that account for different quantitation methods. Additionally, post-translational modifications, half-life of C9orf72 proteins, and the fact that levels often represent a snapshot in disease progression are reasons to consider the lack of transcript-protein level correlation even more critically.
Effect of C9orf72 depletion on neuronal survival
In most studies, survival of iPSNs and primary cortical neurons is largely unaffected upon reduction of C9orf72 (Table 3) [98, 101, 119]. However, heterozygous and homozygous C9orf72 deletion (as well as ASO-mediated knockdown) induces decreased survival of iPSNs in one study [104]. Exogenously adding C9orf72 rescues the survival of these neurons, indicating that C9orf72 depletion might play a role in the observed neurodegeneration in patients [104]. Interestingly, mutations in the ALS-related Ataxin-2 gene synergizes with the effect of C9orf72 knockdown on motor neuron dysfunction and cell death [101]. This is in favor of a multiple-hit mechanism, since lowering C9orf72 transcript levels on itself is not deleterious. TDP-43 pathology (i.e. a hallmark of C9orf72 ALS/FTD) was assessed in only one study, in which increased TDP-43 accumulation was reported upon knockdown of C9orf72 [101].
Effect of C9orf72 depletion on gene expression
An overall depletion of C9orf72 (i.e. 90–100% protein reduction) results in transcriptome alterations in different cell types (Table 3) [34, 104]. Amongst others, alterations in immune system pathways, endothelin signaling, and multiple glutamate cycling genes were observed [34]. Interestingly, gene expression changes observed in both heterozygous and homozygous knockout models are similar to those observed in postmortem tissue from C9 ALS/FTD patients [104]. In contrast to these findings, gene expression profiles improve upon partial knockdown [98] or remain unaltered upon knockout of C9orf72 [1]. Improved sequencing techniques (e.g. single-cell analyses) might be needed to clarify these discrepancies.
Effect of C9orf72 depletion on autophagy and other C9orf72-related processes
As expected from the supposed physiological function of C9orf72 (cf. supra), autophagic [6, 34, 67, 101, 117, 124], lysosomal [5, 6, 104] and endosomal [33] dysfunction was reported in in vitro C9orf72 knockdown and knockout models (Table 3). However, lysosomal transport in axons of MNs lacking C9orf72 is similar to control lines, whereas this trafficking is clearly impeded in C9 patient iPSC-derived MNs [1, 88]. Interestingly, C9 depletion in the latter exacerbates the phenotype, suggesting a synergistic mechanism with GOF.
Conclusions
In patient-derived models, the most important observation is the presence of autophagic alterations. Given the physiological role of C9orf72, these disturbances might be caused by C9orf72 LOF. However, this autophagic dysfunction has not been demonstrated to be disease-relevant and might as well be caused by the GOF mechanisms. Findings in C9orf72 knockdown models further corroborate the role of C9orf72 in autophagy. However, apart from one report, C9orf72 knockdown does not seem to induce in vitro neurotoxicity. Other ALS hallmarks (like TDP-43 pathology) were mostly not assessed. Interestingly, both types of in vitro models provide a hint towards a synergistic mechanism between LOF and GOF, which needs to be elaborated further.
In vivo studies
As the interplay between different cell types clearly contributes to the ALS/FTD disease mechanism and progression, in vivo studies are of uttermost importance to elucidate pathways. Lower order organisms, such as C. elegans and Danio rerio, expanded our knowledge on the C9orf72 ALS/FTD mechanism (Table 4). However, data validation and more sophisticated studies are needed in higher-order organisms, such as mice and rats.
Modelling C9orf72 loss-of-function in lower-order organisms
The first in vivo C9orf72 null mutation was generated in the invertebrate C. elegans [113]. Age-dependent motor neuron degeneration and stress sensitivity was observed upon deleting the C9 orthologue [113]. Moreover, these defects synergized with toxicity caused by mutant TDP-43 proteins, being in favor of a combined LOF and GOF mechanism [113].
To evaluate the effect of C9orf72 on the developing nervous system in a developmental model and addressing the LOF hypothesis in a vertebrate model, a morpholino-mediated knockdown (50% reduction in transcript level) of C9orf72 was performed in zebrafish embryos [22, 125]. This downregulation induces both cellular (i.e. axonal length and embryo morphology) and behavioral deficits (i.e. swimming distance and velocity) associated with locomotion [22, 125]. Although these findings indicate that C9orf72 is crucial for neuronal development, transgenic zebrafish lines with a stable C9orf72 knockdown are essential to validate these results.
Modelling loss-of-function in higher-order organisms
More mechanistic studies were performed in C9orf72 knockdown and knockout mouse models (Table 4). First, knockdown of C9orf72 induces a 50–70% reduction of C9orf72 RNA, without a decrease in protein level [7, 16, 63]. No behavioral or motor phenotypes are observed. Furthermore, C9orf72 hallmarks including TDP-43 pathology and p62 or ubiquitin aggregates are absent. This is in line with findings in cellular models indicating that reduced RNA levels are insufficient to cause motor neuron disease. Second, knockout mouse models allow long-term observation and analysis of acute and chronic effects of C9orf72 depletion. Remarkably, these models are mainly characterized by the lack of motor neuron degeneration and the presence of immune phenotypes [16, 55, 61, 87, 104, 106, 107].
Immune dysfunction, but no motor phenotype in C9orf72 KO mice
Neuronal-specific depletion of C9orf72 induces a decrease in body weight, without motor impairment or decreased survival [61]. Similarly, mice lacking C9orf72 in all tissues exhibit decreased body weight and immune dysfunction without motor neuron impairments or degeneration [7, 16, 55, 87, 96, 106, 128]. Moreover, in contrast to the C9orf72 KD in primary cortical neurons (cf. supra) [101], no TDP-43 pathology is present in these mouse models [61, 106]. Notably, a possible mild motor phenotype (i.e. weakness of hindlimbs, but no differences on rotarod) has been detected in one study [7]. However, this finding might constitute a secondary effect as the immune phenotype already appeared before the motor symptoms. The immune phenotype comprises splenomegaly, enlarged lymph nodes, infiltration of immune cells, increased expression of inflammatory cytokines, presence of autoantibodies and immune-mediated glomerulonephropathy [7, 16, 55, 106]. In line with these observations, RNA sequencing reveals gene dysregulation in inflammatory pathways [7, 87].
Lysosomal and autophagy-related alterations in C9orf72 KO mice
C9orf72-deficient mice exhibit autophagy defects in the spleen and liver, which are accompanied by increased lysosomal proteins and increased autophagy initiation (Table 4) [87, 107]. Mice deficient in SMCR8, a factor in the tripartite complex with C9orf72, display similar autophagy defects as well as inflammatory and autoimmune phenotypes (Table 4) [64, 79, 103, 127]. Moreover, a combined knockout of C9orf72 and SMCR8 induces even more severe immune disturbances (Table 4) [103]. This confirms that both factors act in the same pathway and are partially redundant upon individual depletion. Strikingly, knocking out one or both C9orf72 alleles in a mouse model characterized by the C9orf72 ALS hallmarks (i.e. RNA foci, DPRs), prevents the likely protective activation of autophagy, which is driven by the presence of DPRs [128]. Moreover, it worsens the neuronal loss and cognitive phenotype [128]. Once more, this implies that a combination of loss- and gain-of-function is at play.
Conclusions
The motor phenotype in zebrafish and C. elegans is in contrast to the findings in C9 LOF mouse models, which display an immunological phenotype but no motor neuron degeneration. The phenotypic discrepancy might be explained by a difference in the homology of the human C9orf72 orthologues in the various disease models (i.e. 21% in C. elegans, 75% in zebrafish and 98% in mice) and differences in RNA and protein processing events (i.e. splicing, post-translational modifications). Moreover, findings in the C9 LOF mice could imply that in case of a primary LOF mechanism, patients should present with some immune dysfunction and/or autoimmunity. Apart from one study showing a 12% prevalence of nonthyroid autoimmune disease in a small cohort of C9 ALS/FTD patients [81], there are currently no studies reporting immunological issues in C9 ALS/FTD. Most importantly, whereas the immune phenotypes occur in mice deficient of C9orf72 (i.e. knockout), this is not the case when C9orf72 is only partially reduced (i.e. knockdown). As C9 ALS/FTD patients also exhibit a partial (at best 25% in protein level) reduction, this does not seem to be in line with a prevailing LOF mechanism.
Loss-of-function in other non-coding repeat expansion disorders
In non-coding repeat expansion disorders, a specific short DNA sequence is repeated multiple times. These disorders exhibit considerable phenotypic and pathogenic differences, probably depending on the genomic location (i.e. promoter region, intron, 5′ and 3′ untranslated regions (UTR)) as well as the sequence of the repeat expansion. Understanding the contribution of LOF mechanisms in the neurodegenerative and neurodevelopmental disorders caused by non-coding repeat expansions might elucidate the likelihood of C9orf72 LOF playing a role in C9 ALS/FTD (Table 5). Here, we will discuss the non-coding repeat expansion disorders in which LOF can be considered.
Friedreich’s ataxia (FRDA)
The autosomal recessive neurodegenerative disease FRDA, characterized mainly by ataxia and degeneration of the spinocerebellar tract, is caused by a GAA expansion in the first intron of the FXN gene [2, 18, 77, 99]. In patients, an overall reduction of 70–85% in FXN transcript levels and 70–95% in frataxin protein levels are observed in the cerebellum [18]. FXN KO mouse models are embryonically lethal, whereas the inducible hemizygous FXN mice recapitulate hallmarks of FRDA (severe ataxia, mitochondrial and cardiac abnormalities) [19]. Given the recessive inheritance, significantly reduced protein levels in patients and FRDA-like phenotype in FXN hemizygous KO mice, it is believed that LOF is the primary mechanism of FRDA [77, 99].
Fragile X syndrome (FXS)
FMRP translational regulator 1 (FMR1) harbors a trinucleotide repeat (i.e. CGG) in its 5′UTR region [56]. When this repeat is expanded above 200 repeats, it induces the neurodevelopmental syndrome FXS characterized by learning disabilities and cognitive impairment. Shorter (60 < n < 200) repeat expansions cause the neurodegenerative Fragile X-associated tremor/ataxia syndrome (FXTAS), in which increased repeat-containing FMR1 transcripts are believed to cause a toxic RNA GOF [13, 111]. In FXS the long repeat expansion (n > 200) induces silencing of the FMR1 gene (through hypermethylation), leading to an obvious LOF [56, 86, 89, 112]. This is supported by the observation of an FMR1 KO mouse model recapitulating all hallmarks of FXS patients [52].
Huntington’s disease-like-2 (HDL-2)
HDL-2 resembles Huntington’s disease both clinically (movement disorder and cognitive decline) and pathologically (intranuclear inclusions) [76]. It is caused by a CAG repeat expansion (n = 50–60) in the alternatively spliced exon 2A of the junctophilin 3 (JPH3) gene [76]. This gene encodes a component of junctional complexes between the ER and plasma membrane. The mechanism underlying the autosomal dominant HDL-2 is mainly RNA and RAN toxicity consisting of toxic inclusions [76]. However, as transcript (~ 90%) and protein (~ 70–80%) decreases have been observed in the frontal cortex of patients [100], the contribution of a LOF mechanism is not excluded. Notably, a JPH3 KO mouse model exhibits a motor phenotype. However, neurodegeneration or intranuclear inclusions have not been observed [85, 100].
Myotonic dystrophy type 2 (DM2)
The autosomal dominant neuromuscular disorder DM2, characterized by progressive muscle wasting and weakness, is caused by a CCTG repeat expansion in the first intron of the Zinc Finger Protein 9 (ZNF9) gene [14]. RNA toxicity is the predominant mechanism and is mediated by the sequestration of MBNL leading to aberrant splicing of several genes [57, 97]. There is no consensus on altered ZNF9 transcript and protein levels [14, 91]. Apart from a partial KO of ZNF9 in mice reflecting some of the aspects of DM2, including heart conductivity abnormalities and myotonic deficits [20], there is little evidence for a LOF mechanism to be involved in DM2.
Overall, non-coding repeat expansion disorders with LOF as principal disease mechanism (i.e. FRDA and FXS) display a 70–100% reduction of the respective transcript and protein levels and knockout mouse models recapitulate clinical and pathological phenotypes of patients (Table 5). This is in clear contrast with C9 ALS/FTD, where protein reduction (25%) is rather limited and KO mouse models lack a motor phenotype and neurodegenerative hallmarks. More related to C9 ALS/FTD, in HDL-2 and DM2, multiple lines of evidence imply GOF (RNA toxicity, RAN toxicity) as a primary mechanism with a lesser contribution of LOF. Altogether, comparing non-coding repeat expansion disorders based on their inheritance pattern, transcript, and protein level in patients, location of the repeat region, and knockout mouse models suggests a predominant GOF mechanism in C9 ALS/FTD.
Conclusions
With the uprising evidence for GOF (i.e. RNA toxicity and DPR toxicity) mechanisms in C9 ALS/FTD, the contribution of a C9orf72 LOF is still debated. Inconclusive findings regarding transcript and protein levels, functionality, and the correlation with an ALS/FTD phenotype in patients and different LOF model systems complicate this assessment. Nevertheless, in vitro and in vivo models made clear that the extent of C9orf72 transcript and protein level reduction observed in patients seems to be insufficient to cause neurodegeneration in these models. Together with the findings inferred from other non-coding repeat expansion disorders, we conclude that C9orf72 LOF is unlikely to be the main culprit in C9 ALS/FTD. Nevertheless, a synergistic mechanism in which C9orf72 LOF might exacerbate the GOF mechanisms is not excluded and multiple recent studies even support such synergy. In the event of this multiple-hit mechanism, a therapeutic strategy to target both mechanisms will be even more challenging to develop. Nonetheless, targeting one of these mechanisms could be enough to prevent accumulative events causative for neurodegeneration. Either way, all these insights highlight the importance of studies taking into account, not one, but multiple C9 ALS/FTD mechanisms.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ASO:
-
Antisense oligonucleotide
- CNS:
-
Central nervous system
- DENN:
-
Differentially expressed in normal and neoplastic cells
- DPR:
-
Dipeptide repeat protein
- ER:
-
Endoplasmic reticulum
- FTD:
-
Frontotemporal dementia
- GEF:
-
Guanine exchange factor
- GOF:
-
Gain-of-function
- iPSC:
-
Induced pluripotent stem cell
- iPSN:
-
IPSC-derived neuron
- KD:
-
Knockdown
- KO:
-
Knockout
- LOF:
-
Loss-of-function
- MN:
-
Motor neurons
- RAN:
-
Repeat-associated non-ATG
- RBP:
-
RNA binding protein
- SMCR8:
-
Smith–Magenis chromosome region 8
- UTR:
-
Untranslated region
- WDR41:
-
WD repeat-containing protein 41
References
Abo-Rady M, Kalmbach N, Pal A, Schludi C, Janosch A, Richter T et al (2020) Knocking out C9ORF72 exacerbates axonal trafficking defects associated with hexanucleotide repeat expansion and reduces levels of heat shock proteins. Stem Cell Rep 14:390–405. https://doi.org/10.1016/j.stemcr.2020.01.010
Al-Mahdawi S, Ging H, Bayot A, Cavalcanti F, La Cognata V, Cavallaro S et al (2018) Large interruptions of GAA repeat expansion mutations in Friedreich ataxia are very rare. Front Cell Neurosci 12:443. https://doi.org/10.3389/fncel.2018.00443
Almeida S, Gascon E, Tran HH, Chou HJ, Gendron TF, Degroot S et al (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126:385–399. https://doi.org/10.1007/s00401-013-1149-y
Amick J, Ferguson SM (2017) C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic 4:267–276. https://doi.org/10.1016/S2214-109X(16)30265-0
Amick J, Roczniak-Ferguson A, Ferguson SM (2016) C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 27:3040–3051. https://doi.org/10.1091/mbc.E16-01-0003
Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL et al (2017) C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140:887–897. https://doi.org/10.1093/brain/awx024
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6:23204. https://doi.org/10.1038/srep23204
Belzil VV, Bauer PO, Gendron TF, Murray ME, Dickson DW, Petrucelli L (2014) Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res 1584:15–21. https://doi.org/10.1016/j.brainres.2014.02.015
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK et al (2013) Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 126:895–905. https://doi.org/10.1007/s00401-013-1199-1
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NCA, Brown PH et al (2015) Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol 130:863–876. https://doi.org/10.1007/s00401-015-1480-6
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A et al (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6:20877. https://doi.org/10.1038/srep20877
Boivin M, Pfister V, Gaucherot A, Ruffenach F, Negroni L, Sellier C et al (2020) Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J 39:e100574. https://doi.org/10.1016/j.ejmg.2017.11.001
Boivin M, Willemsen R, Hukema RK, Sellier C (2018) Potential pathogenic mechanisms underlying Fragile X Tremor Ataxia Syndrome: RAN translation and/or RNA gain-of-function? Eur J Med Genet 61:674–679. https://doi.org/10.1016/j.ejmg.2017.11.001
Botta A, Caldarola S, Vallo L, Bonifazi E, Fruci D, Gullotta F et al (2006) Effect of the [CCTG]n repeat expansion on ZNF9 expression in myotonic dystrophy type II (DM2). Biochim Biophys Acta Mol Basis Dis 1762:329–334. https://doi.org/10.1016/j.bbadis.2005.11.004
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172. https://doi.org/10.1056/NEJMra1603471
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH et al (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8:93. https://doi.org/10.1126/scitranslmed.aaf6038
Cali CP, Patino M, Tai YK, Ho WY, McLean CA, Morris CM et al (2019) C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol 138:795–811. https://doi.org/10.1007/s00401-019-02045-5
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427. https://doi.org/10.1126/science.271.5254.1423
Chandran V, Gao K, Swarup V, Versano R, Dong H, Jordan MC et al (2017) Inducible and reversible phenotypes in a novel mouse model of Friedreich’s ataxia. Elife 6:e30054. https://doi.org/10.7554/eLife.30054
Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP (2007) Haploinsuffciency for Znf9 in Znf9+/- mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol 368:8–17. https://doi.org/10.1016/j.jmb.2007.01.088
Chitiprolu M, Jagow C, Tremblay V, Bondy-Chorney E, Paris G, Savard A et al (2018) A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun 9:2794. https://doi.org/10.1038/s41467-018-05273-7
Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A et al (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol 74:180–187. https://doi.org/10.1002/ana.23946
Cooper-Knock J, Higginbottom A, Connor-Robson N, Bayatti N, Bury JJ, Kirby J et al (2013) C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology 81:1719–1721. https://doi.org/10.1212/01.wnl.0000435295.41974.2e
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB et al (2015) Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol 130:63–75. https://doi.org/10.1007/s00401-015-1429-9
Corrionero A, Horvitz HR (2018) A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr Biol 28:1522–1535.e5. https://doi.org/10.1016/j.cub.2018.03.063
Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM et al (2019) Ensembl 2019. Nucleic Acids Res 47:D745–D751. https://doi.org/10.1093/nar/gky1113
Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H et al (2016) C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34:2063–2078. https://doi.org/10.1002/stem.2388
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Donnelly CJ, Zhang P, Pham JT, Haeusler AR, Mistry NA, Vidensky S et al (2014) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428. https://doi.org/10.1016/j.neuron.2013.10.015
Douglas AGL (2018) Non-coding RNA in C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia: A perfect storm of dysfunction. Non-Coding RNA Res 3:178–187. https://doi.org/10.1016/j.ncrna.2018.09.001
Edbauer D, Haass C (2016) An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr Opin Neurobiol 36:99–106. https://doi.org/10.1016/j.conb.2015.10.009
Esanov R, Belle KC, van Blitterswijk M, Belzil VV, Rademakers R, Dickson DW et al (2016) C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp Neurol 277:171–177. https://doi.org/10.1016/j.expneurol.2015.12.022
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RAK, Levina V et al (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23:3579–3595. https://doi.org/10.1093/hmg/ddu068
Fomin V, Richard P, Hoque M, Li C, Gu Z, Fissore-O’Leary M et al (2018) The C9ORF72 gene, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, encodes a protein that functions in control of endothelin and glutamate signaling. Mol Cell Biol 38:e00155-18. https://doi.org/10.1128/MCB.00155-18
Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J et al (2013) Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 126:401–409. https://doi.org/10.1007/s00401-013-1147-0
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH et al (2015) GGGGCC repeat expansion in C9ORF72 compromises nucleocytoplasmic transport. Nature 525:129–133. https://doi.org/10.1038/nature14974
Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci 10:35. https://doi.org/10.3389/fnmol.2017.00035
Frick P, Sellier C, Mackenzie IRA, Cheng CY, Tahraoui-Bories J, Martinat C et al (2018) Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun 6:72. https://doi.org/10.1186/s40478-018-0579-0
Fujikake N, Shin M, Shimizu S (2018) Association between autophagy and neurodegenerative diseases. Front Neurosci 12:255. https://doi.org/10.3389/fnins.2018.00255
Fumagalli L, Young FL, Boeynaems S, De Decker M, Mehta A, Swijsen A et al (2019) C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. bioRxiv 835082. https://doi.org/10.1101/835082
Gagliardi S, Pandini C, Garofalo M, Bordoni M, Pansarasa O, Cereda C (2018) Long non coding RNAs and ALS: Still much to do. Non-Coding RNA Res 3:226–231. https://doi.org/10.1016/j.ncrna.2018.11.004
Gao FB, Almeida S, Lopez-Gonzalez R (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J 36:2931–2950. https://doi.org/10.15252/embj.201797568
Gendron TF, Petrucelli L (2018) Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb Perspect Med 8:a024224. https://doi.org/10.1101/cshperspect.a024224
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G et al (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. https://doi.org/10.1016/S1474-4422(11)70261-7
Gijselinck I, Van Mossevelde S, Van Der Zee J, Sieben A, Engelborghs S, De Bleecker J et al (2016) The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 21:1112–1124. https://doi.org/10.1038/mp.2015.159
Greenberg MVC, Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20:590–607. https://doi.org/10.1038/s41580-019-0159-6
Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw G, Kim M et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. https://doi.org/10.1038/nature13124.C9orf72
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. https://doi.org/10.1038/nature04724
Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P et al (2017) Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 34:2234.e13–2234.e19. https://doi.org/10.1016/j.neurobiolaging.2013.03.006
Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S (2003) Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol 23:6494–6506. https://doi.org/10.1128/mcb.23.18.6494-6506.2003
Hornburg D, Drepper C, Butter F, Meissner F, Sendtner M, Mann M (2014) Deep proteomic evaluation of primary and cell line motoneuron disease models delineates major differences in neuronal characteristics 13:3410–3420. https://doi.org/10.1074/mcp.M113.037291
Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99:7746–7750. https://doi.org/10.1073/pnas.122205699
Jackson JL, Finch NA, Baker MC, Kachergus JM, DeJesus-Hernandez M, Pereira K et al (2020) Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol Neurodegener 15:7. https://doi.org/10.1186/s13024-020-0359-8
Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164:487–498. https://doi.org/10.1016/j.cell.2015.12.038
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A et al (2016) Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90:535–550. https://doi.org/10.1016/j.neuron.2016.04.006
Jin P, Warren ST (2000) Understanding the molecular basis of fragile X syndrome. Hum Mol Genet 9:901–908. https://doi.org/10.1093/hmg/9.6.901
Jones K, Jin B, Iakova P, Huichalaf C, Sarkar P, Schneider-Gold C et al (2011) RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am J Pathol 179:2475–2489. https://doi.org/10.1016/j.ajpath.2011.07.013
Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070. https://doi.org/10.1126/science.1063852
Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB et al (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18:1226–1229. https://doi.org/10.1038/nn.4085
Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. https://doi.org/10.1038/nature04723
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CAC, Vieira De Sá R et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438. https://doi.org/10.1002/ana.24453
Laflamme C, McKeever P, Kumar R, Schwartz J, Chen C, You Z et al (2018) Implementation of an antibody characterization process: application to the major ALS/FTD disease gene C9ORF72. bioRxiv 499350. https://doi.org/10.1101/499350
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 110:4530–4539. https://doi.org/10.1073/pnas.1318835110
Lan Y, Sullivan PM, Hu F (2019) SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 15:871–885. https://doi.org/10.1080/15548627.2019.1569914
Langseth AJ, Kim J, Ugolino JE, Shah Y, Hwang HY, Wang J et al (2017) Cell-Type specific differences in promoter activity of the ALS-linked C9orf72 mouse ortholog. Sci Rep 7:5685. https://doi.org/10.1038/s41598-017-05864-2
Lee K-HH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167:774–788. https://doi.org/10.1016/j.cell.2016.10.002
Leskelä H, Rostalski N, Remes T, Hiltunen H (2019) C9orf72 proteins regulate autophagy and undergo autophagosomal or proteasomal degradation in a cell type-dependent manner. Cells 8:1233. https://doi.org/10.3390/cells8101233
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29:499–503. https://doi.org/10.1093/bioinformatics/bts725
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Liu EY, Russ J, Wu K, Neal D, Suh E, McNally AG et al (2014) C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 128:525–541. https://doi.org/10.1007/s00401-014-1286-y
Liu F, Liu Q, Lu CX, Cui B, Guo XN, Wang RR et al (2016) Identification of a novel loss-of-function C9orf72 splice site mutation in a patient with amyotrophic lateral sclerosis. Neurobiol Aging 47:219.e1–219.e5. https://doi.org/10.1016/j.neurobiolaging.2016.07.027
Liu Y, Beyer A, Aebersold R (2016) Leading edge review on the dependency of cellular protein levels on mRNA abundance. Cell 165:535–550. https://doi.org/10.1016/j.cell.2016.03.014
Longatti A, Lamb CA, Razi M, Yoshimura SI, Barr FA, Tooze SA (2012) TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol 197:659–675. https://doi.org/10.1083/jcb.201111079
Maharjan N, Künzli C, Buthey K, Saxena S (2017) C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol Neurobiol 54:3062–3077. https://doi.org/10.1007/s12035-016-9850-1
Marat AL, Dokainish H, McPherson PS (2011) DENN domain proteins: regulators of Rab GTPases. J Biol Chem 286:13791–13800. https://doi.org/10.1074/jbc.R110.217067
Margolis RL, Holmes SE (2003) Huntington’s disease-like 2: a clinical, pathological, and molecular comparison to Huntington’s disease. Clin Neurosci Res 3:187–196. https://doi.org/10.1016/S1566-2772(03)00061-6
Martelli A, Napierala M, Puccio H (2012) Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis Model Mech 5:165–176. https://doi.org/10.1242/dmm.008706
Martier R, Liefhebber JM, García-Osta A, Miniarikova J, Cuadrado-Tejedor M, Espelosin M et al (2019) Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther Nucleic Acids 16:26–37. https://doi.org/10.1016/j.omtn.2019.02.001
McAlpine W, Sun L, Wen WK, Liu A, Jain R, Miguel MS et al (2018) Excessive endosomal TLR signaling causes inflammatory disease in mice with defective SMCR8-WDR41-C9ORF72 complex function. Proc Natl Acad Sci USA 115:E11523–E11531. https://doi.org/10.1073/pnas.1814753115
McMillan CT, Russ J, Wood EM, Irwin DJ, Grossman M, McCluskey L et al (2015) C9orf72 promoter hypermethylation is neuroprotective. Neurology 84:1622–1630. https://doi.org/10.1212/WNL.0000000000001495
Miller ZA, Sturm VE, Camsari GB, Karydas A, Yokoyama JS, Grinberg LT et al (2016) Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts. Neurol Neuroimmunol Neuroinflamm 3:e301. https://doi.org/10.1212/NXI.0000000000000301
Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. https://doi.org/10.1007/s00401-013-1189-3
Mori K, Nihei Y, Arzberger T, Zhou Q, Mackenzie IR, Hermann A et al (2016) Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Rep 17:1314–1325. https://doi.org/10.15252/embr.201541724
Nassif M, Woehlbier U, Manque PA (2017) The enigmatic role of C9ORF72 in autophagy. Front Neurosci 11:442. https://doi.org/10.3389/fnins.2017.00442
Nishi M, Takeshima H, Hashimoto K, Kano M, Hashimoto K, Kano M et al (2002) Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292:318–324. https://doi.org/10.1006/bbrc.2002.6649
Oostra BA, Willemsen R (2003) A fragile balance: FMR1 expression levels. Hum Mol Genet 12:R249–R257. https://doi.org/10.1093/hmg/ddg298
O'rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AKMG et al (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351:1324–1329. https://doi.org/10.1126/science.aaf1064
Pal A, Kretner B, Abo-Rady M, Glaß H, Naumann M, Japtok J et al (2020) High content live profiling reveals concomitant gain and loss of function pathomechanisms in C9ORF72 amyotrophic lateral sclerosis. bioRxiv 2020.04.15.040394. https://doi.org/10.1101/2020.04.15.040394
Pieretti M, Zhang F, Fu YH, Warren ST, Oostra BA, Caskey CT et al (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66:817–822. https://doi.org/10.1016/0092-8674(91)90125-I
Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, Atkin JD (2019) Motor neuron susceptibility in ALS/FTD. Front Neurosci 13:532. https://doi.org/10.3389/fnins.2019.00532
Raheem O, Olufemi SE, Bachinski LL, Vihola A, Sirito M, Holmlund-Hampf J et al (2010) Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol 177:3025–3036. https://doi.org/10.2353/ajpath.2010.100179
Renton AE, Majounie E, Waite A, Simón-sánchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010.A
Rizzu P, Blauwendraat C, Heetveld S, Lynes EM, Castillo-Lizardo M, Dhingra A et al (2016) C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol Commun 4:37. https://doi.org/10.1186/s40478-016-0306-7
Russ J, Liu EY, Wu K, Neal D, Suh ER, Irwin DJ et al (2015) Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol 129:39–52. https://doi.org/10.1007/s00401-014-1365-0
Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D et al (2017) Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol 135:459–474. https://doi.org/10.1007/s00401-017-1793-8
Sakkas LI, Bogdanos DP, Kousvelari EE (2017) Loss of C9orf72 function leads to autoimmunity. Ann Transl Med 5:60. https://doi.org/10.21037/atm.2017.01.33
Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B et al (2009) Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol 175:748–762. https://doi.org/10.2353/ajpath.2009.090047
Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M et al (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5:208ra149. https://doi.org/10.1126/scitranslmed.3007529
Schmucker S, Puccio H (2010) Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum Mol Genet 19:R103–R110. https://doi.org/10.1093/hmg/ddq165
Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, Pruitt JL et al (2012) Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol 71:245–257. https://doi.org/10.1002/ana.22598
Sellier C, Campanari M, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M et al (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 35:1276–1297. https://doi.org/10.15252/embj.201593350
Selvaraj BT, Livesey MR, Zhao C, Gregory JM, James OT, Cleary EM et al (2018) C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat Commun 9:347. https://doi.org/10.1038/s41467-017-02729-0
Shao Q, Yang M, Liang C, Ma L, Zhang W, Jiang Z et al (2019) c9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy. https://doi.org/10.1080/15548627.2019.1703353
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST et al (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24:313–325. https://doi.org/10.1038/nm.4490
Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A et al (2016) C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci 19:1610–1618. https://doi.org/10.1038/nn.4407
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CAC et al (2016) Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 132:145–147. https://doi.org/10.1007/s00401-016-1581-x
Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB et al (2016) The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun 4:51. https://doi.org/10.1186/s40478-016-0324-5
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670. https://doi.org/10.1038/nrneurol.2014.184
Swinnen B, Robberecht W, Van Den Bosch L (2020) RNA toxicity in non-coding repeat expansion disorders. EMBO J 39:e101112. https://doi.org/10.15252/embj.2018101112
Tang BL (2016) C9orf72’s Interaction with Rab GTPases—Modulation of Membrane Traffic and Autophagy. Front Cell Neurosci 10:228. https://doi.org/10.3389/fncel.2016.00228
Tassone F, Adams J, Berry-Kravis EM, Cohen SS, Brusco A, Leehey MA et al (2007) CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet Part B Neuropsychiatr Genet 144:566–569. https://doi.org/10.1002/ajmg.b.30482
Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ (2000) Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet Semin Med Genet 97:195–203. https://doi.org/10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R
Therrien M, Rouleau GA, Dion PA, Parker JA (2013) Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 8:e83450. https://doi.org/10.1371/journal.pone.0083450
Turner MR, Al-Chalabi A, Chio A, Hardiman O, Kiernan MC, Rohrer JD et al (2017) Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry 88:1042–1044. https://doi.org/10.1136/jnnp-2017-315995
Vanneste J, Vercruysse T, Boeynaems S, Sicart A, Van Damme P, Daelemans D et al (2019) C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci Rep 9:15728. https://doi.org/10.1038/s41598-019-52035-6
Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O et al (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35:1779.e5–1779.e13. https://doi.org/10.1016/j.neurobiolaging.2014.01.016
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L et al (2016) The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 35:1656–1676. https://doi.org/10.15252/embj.201694401
Webster CP, Smith EF, Grierson AJ, De Vos KJ (2018) C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 9:399–408. https://doi.org/10.1080/21541248.2016.1240495
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y et al (2014) Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84:1213–1225. https://doi.org/10.1016/j.neuron.2014.12.010
Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811. https://doi.org/10.1038/nn.2575
Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P et al (2015) The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 129:715–727. https://doi.org/10.1007/s00401-015-1401-8
Xiao S, MacNair L, McGoldrick P, McKeever P, McLean JR, Zhang M et al (2015) Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78:568–583. https://doi.org/10.1002/ana.24469
Xiao S, MacNair L, McLean J, McGoldrick P, McKeever P, Soleimani S et al (2016) C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res 1647:43–49. https://doi.org/10.1016/j.brainres.2016.04.062
Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R et al (2016) A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2:e1601167. https://doi.org/10.1126/sciadv.1601167
Yeh TH, Liu HF, Li YW, Lu CS, Shih HY, Chiu CC et al (2018) C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp Neurol 304:114–124. https://doi.org/10.1016/j.expneurol.2018.03.002
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhang Y, Burberry A, Wang JY, Sandoe J, Ghosh S, Udeshi ND et al (2018) The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev 32:929–943. https://doi.org/10.1101/gad.313932.118
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A et al (2020) Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci 23:615–624. https://doi.org/10.1038/s41593-020-0619-5
Zoppino FCM, Militello RD, Slavin I, Álvarez C, Colombo MI (2010) Autophagosome formation depends on the small GTPase rab1 and functional ER exit sites. Traffic 11:1246–1261. https://doi.org/10.1111/j.1600-0854.2010.01086.x
Acknowledgements
Research of the authors is supported by VIB, KU Leuven (C1 and “Opening the Future” Fund), the “Fund for Scientific Research Flanders” (FWO-Vlaanderen, G0C1620N), the Thierry Latran Foundation, the “Association Belge contre les Maladies neuro-Musculaires-aide à la recherché ASBL” (ABMM), the Muscular Dystrophy Association (MDA), the ALS Liga België (A Cure for ALS), Target ALS and the ALS Association (ALSA). Elke Braems is a PhD fellow at FWO-Vlaanderen (1145619N).
Author information
Authors and Affiliations
Contributions
EB contributed to the original draft preparation and writing of the manuscript. BS contributed to the writing, reviewing and editing. LVDB reviewed and edited the manuscript before submission and supervised the writing process.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare to have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Braems, E., Swinnen, B. & Van Den Bosch, L. C9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD?. Acta Neuropathol 140, 625–643 (2020). https://doi.org/10.1007/s00401-020-02214-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-020-02214-x