
Abstract
Host–parasite interactions include effects on both proximate and ultimate levels: parasite infections affect individual’s fitness and play a significant role in shaping the life history of host species. Global environmental changes as well as significant shifts in abiotic factors might impact the dynamics of parasite–host interactions, especially in Arctic regions, where the climate is changing at an alarming rate. With global warming, parasites and their vectors are predicted to spread to polar latitudes, and it is crucial to follow the changes occurring in the ecosystems in the era of global changes. We studied blood parasites (Haemosporidae) of passerine birds (Passeriformes: Aves) in southwest Yamal (north-western Siberia) using genetic and morphological methods. We found an overall parasite prevalence of 76.3%, with highest values for Leucocytozoon (72.0%) and lower values for Parahaemoproteus and Plasmodium (8.9 and 8.2%, respectively). We determined 26 genetic lineages in total, five of them were novel. The most common parasite lineages were TRPIP2 (18%), BT1 (14%), novel ACAFLA06 (13%), BT2 (7%), novel ACAFLA07 (6%), BT4 (5%) for Leucocytozoon; SISKIN1 (4%) for Parahaemoproteus; TURDUS1 (4%) for Plasmodium. For redpoll (Acanthis flammea), brambling (Fringilla montifringilla), bluethroat (Luscinia svecica) and little bunting (Emberiza pusilla) overall prevalence exceeded 90%. We also found significant differences in prevalence and lineage composition between sample sites, species and sexes, and a positive correlation between number of infections and host body mass. Our study provides knowledge about haemosporidian infections in the region, which had been barely studied for blood parasites. Gathered information is essential for the future monitoring and assessing potential shifts due to global change.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global climate change and ensuing habitat alterations are expected to have a major influence on infectious disease dynamics. Such effects may include encountering novel hosts and/or vectors by parasites as well as occupation of biotopes inaccessible earlier (e.g. higher altitudes) and subsequent broadening of parasite geographical distribution (Loiseau et al. 2012; Caminade et al. 2019). Any predictions of effects are controversial, but it is crucial to follow the changes occurring in the ecosystem in the era of global changes.
Haemosporidia (Apicomplexa) is a group of parasitic organisms with a complex evolutionary history influenced by multiple transitions in life-history strategies and hosts (Galen et al. 2018a). Avian malaria parasites of the genus Plasmodium and the subgenera of Haemoproteus, H. (Haemoproteus) and H. (Parahaemoproteus) as well as other haemosporidian parasites (Leucocytozoon) infect a broad variety of avian host species and cause malaria-like conditions in birds (Valkiūnas 2005; Rivero and Gandon 2018; Fecchio et al. 2021), however, data on haemosporidian pathogenicity and influence on hosts remain contradictory (e.g. van Riper et al. 1986; Bensch et al. 2007). Those parasites display complex life cycles with sexual stages of reproduction proceeding in blood-feeding insects and asexual ones taking place in the vertebrate hosts (Santiago‐Alarcon et al. 2012). Each genus of parasites is transmitted mainly by one group of dipterans. Thus, Plasmodium is transmitted by mosquitoes (Culicidae), Haemoproteus by hippoboscid flies (Hippoboscidae), Parahaemoproteus by biting midges (Culicoides, Ceratopogonidae) and Leucocytozoon by black flies (Simuliidae) (Fecchio et al. 2020). Currently, the species diversity of Haemosporidia in birds seems to be as high as avian diversity, and each species contains numerous genetic lineages that may represent distinct evolutionary entities (Bensch et al. 2004).
Such vector-borne parasites with complex life cycles as haemosporidians require specific environmental and biological conditions to develop successfully and face many constraints shaping their geographical distribution (Valkiūnas 2005). Their abundance is shaped not only by the limitations for their own life cycles, but also by factors influencing their host and vector distributions. Moreover, prevalence and infection rates may also vary between sexes or host ages within one host species (Hasselquist 2007; Lachish et al. 2011).
One of the most important abiotic factors directly and indirectly affecting the abundance of parasites is temperature. Thermal constraints exist for several stages in the life cycles of haemosporidian parasites and their vectors, such as the parasite development in the mosquito, the larval abundance and vector development itself (Paaijmans et al. 2010; Gardner et al. 2012). Temperature limits and trade-offs exist during the life cycle stage in the avian host as well (Andreasson et al. 2020).
In Arctic regions, temperatures are increasing significantly and ecosystems are changing rapidly (MacDonald 2010; Huang et al. 2017). Due to climate change, plenty of diseases considered as “tropical” or “temperate” may spread to the higher latitudes and altitudes inaccessible earlier; certainly, avian malaria is not an exception (Garamszegi 2011; Rocklöv and Dubrow 2020).
Birds, as the organisms conducting long-distance migrations, are one of the few groups of vertebrates that may facilitate the expansion of haemosporidian parasites into arduous places including high altitudes or high latitudes. However, earlier studies conducted in the high arctic tundra of North America failed to find avian blood parasites, which was explained by the absence of vectors (Bennett et al. 1992). Nevertheless, more recent studies show the abundance of haemosporidians in Passeriformes captured in Arctic biotopes (MalAvi database; e.g. Krams et al. 2010; Marzal et al. 2011; Loiseau et al. 2012; Oakgrove et al. 2014 and others) and even demonstrate first evidences of avian Plasmodium transmission in northern latitudes. These confirmations of transmission include observations of resident avian species infected by malaria parasites in Norway (69.4°N, 30°E; Marzal et al. 2011) and Finland (66°N, 29°E; Krams et al. 2010) as well as resident species and juvenile birds in Alaska (61–64°N, 147–150°W; Loiseau et al. 2012).
Knowledge about parasite distributions in the Russian Arctic and north-western Siberia in particular is scarce and sparse (see, for instance, Clark et al. 2014). In 1984–1988, Leucocytozoon simondi was found in blood smears from ducks (Anatidae) abundant in several geographically distant plots, but no Haemoproteus or Plasmodium were found at the Arctic latitudes (66–68°N, 65–171°E; including one sampling site on the Yamal peninsula; Valkiūnas et al. 1990). A recent study revealed Plasmodium and Leucocytozoon in adult bluethroats Luscinia svecica on Kola Peninsula (68°N, 34°E; Svoboda et al. 2015).
The Yamal peninsula is a region of the north-western Siberia which extends roughly 700 km from Polar Ural Mountains to Kara Sea and contains all the biogeographical zones from forest–tundra and woodland habitats to shrub and Arctic tundra (Ryabitsev 2001). The region itself is currently experiencing significant environmental and anthropogenic pressure including general temperature raising, permafrost melting, as well as rapid development of the oil and gas industry and growth of reindeer herds, which may lead to environmental changes and significant impacts on the ecosystems in general and bird populations in particular (Sokolov et al. 2012). Yamal tundra is a breeding area for many avian species as well as for potential vectors—blood-sucking dipterans, especially black flies, reaching population densities of up to million larvae per m2 under favourable conditions (Currie and Adler 2008). Most Arctic birds leave their breeding sites and migrate to southern wintering quarters which may be located thousands of kilometres away (Ryabitsev 2001), which, as discussed earlier, increases the chance to be infected by parasites from a different geographical region.
The precise northern boundaries of haemosporidian distribution as well as ecological constraints shaping this frontier are unknown. However, with all the existing premises and impacts as well as under the set of additional favourable conditions the new foci of disease might appear in Arctic latitudes. Therefore, it is essential to gather knowledge about the present parasite distribution as well as their host range and lineage diversity. The general aim of this pilot study was to explore the prevalence and distribution patterns of Haemosporidae (genera Plasmodium, Haemoproteus and Leucocytozoon) in avian hosts occurring in the Yamal tundra, as well as assess potential differences between sexes and a relation between number of infections and body mass.
Materials and methods
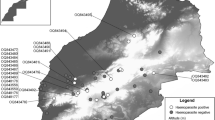
The sample collection was conducted during fieldwork at two locations roughly 210 km distant from each other in the southwest part of Yamal peninsula in June and July 2021. We had several sites of sampling (Fig. 1): lowland shrub tundra with willow thickets plots located in 2 km range to the Erkutayakha (Erkuta) river (68°13′N, 69°09′E), as well as heterogeneous woodland habitats in town Labytnangi (66°39′N, 66°24′E) and village Oktyabrsky (66°41′N, 66°34′E)—geographically located in a transition zone in between northern taiga and forest–tundra. Henceforth, Labytnangi refers to both town Labytnangi and village Oktyabrsky, unless otherwise explicitly explained.

The Yamal peninsula and passerine birds sampling locations: Erkuta and Labytnangi. Satellite images (source: google maps) displaying the sampling plots and their surroundings are given in black rectangle. Sampling sites in Erkuta region (68°13′N, 69°09′E): yellow—in the vicinity of the camp, green—by the turn of the river, grey—in the vicinity of the Henado cliff, blue—beside the Merzyampertyata lake, orange—beside the “trousers-like” lake. Sampling sites in Labytnangi: in the town (66°39′N, 66°24′E) and near the village° (66°41′N, 66°34′E)
In total, we caught and blood sampled 141 individuals from 15 avian species (redpoll Acanthis flammea, bluethroat Luscinia svecica, willow warbler Phylloscopus trochilus, little bunting Emberiza pusilla, chiffchaff Phylloscopus collybita, brambling Fringilla montifringilla, Arctic warbler Phylloscopus borealis, redstart Phoenicurus phoenicurus, redwing Turdus iliacus, sedge warbler Acrocephalus schoenobaenus, reed bunting Schoeniclus schoeniclus, meadow pipit Anthus pratensis, Siberian accentor Prunella montanella, common rosefinch Carpodacus erythrinus, red-flanked bluetail Tarsiger cyanurus; see Online Resource 1; all the taxonomic positions are given in the text as stated in MalAvi database). All the birds were captured by mist-netting and blood sampled by brachial venepuncture (using a heparinised capillary tube after puncturing the brachial vein with a sterile syringe needle); captures were carried out in accordance with the local legislation. All the individuals were ringed and weighed, and their age and sex (for species with detectable sexual dimorphism) were recorded.
Blood was stored on Whatman FTA classic cards (Whatman®, UK) for genetic analyses and on blood smears (two per individual) for microscopic examination. In most cases we collected enough blood (less than 50 µl) for both methods (n = 133), but for some individuals we obtained FTA (n = 2) or smears (n = 6) only.
Microscopic examination
The blood smears were fixed in 100% methanol for 1 min or 100% ethanol for 5 min in the field and later stained with Giemsa in a solution prepared with buffer pH 7.0 (ratio 1:5) for 30 min in the laboratory (Valkiūnas 2005).
Blood smears were examined at ×1000 magnification for at least 10,000 monolayered erythrocytes using a light microscope (PrimoStar Zeiss, Germany). The examined smear was just marked as containing parasitic gametocytes (“positive”) or not (“negative”) and served as an additional control for detecting successful infections.
Preparation of genetic samples
A 3 × 3 mm piece of each sample was cut out from the FTA cards for DNA isolation. DNA was extracted according to the ammonium-acetate protocol by Martínez et al. (2009) and purified with NZYGelpure columns (NZYTech, Portugal). The concentration of isolated DNA was measured by NanoDrop2000c UV–Vis Spectrophotometer (Thermo Fisher Scientific, Wilmington, USA).
Parasite detection
To determine the presence of haemosporidians we performed a nested polymerase chain reaction (PCR) targeting a 479 bp region of the mitochondrial cytochrome b gene (cyt b, Bensch et al. 2000) and followed the PCR protocol suggested by Hellgren et al. (2004). For the initial step we applied the primer pair HaemNFI/HaemNR3. A 4 μl aliquot of this PCR product was subsequently used as template DNA for the second PCR reactions with specific primer pairs. We used HaemF/HaemR2 for Haemoproteus and Plasmodium; HaemFL/HaemR2L for Leucocytozoon detection. We included additional positive (samples with known infection status) and negative controls (deionised water) for each PCR run. Cycling conditions followed the protocol given by Hellgren et al. (2004) and included an incubation step at 94 °C for 3 min, a final extension at 72 °C for 10 min and a thermal profile of 30 s at 94 °C, 30 s at 50 °C and 45 s at 72 °C for 20 cycles in the initial PCR and for 35 cycles in the parasite-specific PCR. PCR protocols were carried out on a Biometra TOne Cycler (Analytik Jena, Germany).
PCR products that displayed clear bands during gel electrophoresis (QIAxcel Advanced, Qiagen, Switzerland) were bidirectionally Sanger sequenced in Microsynth-Seqlab (Sequence Laboratories Goettingen GmbH, Germany). Forward and reverse sequences were assembled and trimmed in CLC Main WorkBench (Qiagen, Switzerland).
During the process of assembly, forward/reverse or both sequences in the consensus alignment sometimes displayed conflicts—missing nucleotides or double peaks. In the case of missing nucleotides, we used the second sequence (reverse or forward, respectively) to fill the gap. If the observed double peaks were equal or one peak was slightly lower, we considered such sequences as containing mixed homogeneric infections (Online Resource 2a–b). If the lineage associated with the lower peak never appeared in our results as a distinct infection, we did not consider the case as a mixed infection and analysed the lineage associated with higher peak only (Online Resource 2d). Some of our sequences contained several conflicts and following all the plausible coinfections seemed barely possible (however, we managed to do that for TRPIP2/BT1 and TRPIP2/BT4 coinfections). We repeated sequencing for all the files, containing more than 20 mismatches that could not be resolved, and unless the sequences were significantly improved, we considered them further as “unknown lineages”.
Phylogenetic and statistical analysis
In order to identify genetic lineages, we aligned the quality-checked sequences with the references deposited in the MalAvi database via BLAST algorithm (BLASTN 2.3.0+ ; Zhang et al. 2000). All the identified sequences were then aligned and trimmed in BioEdit (Hall 1999). To construct lineage networks, we applied median-joining network methods with PopART 1.7 (Leigh and Bryant 2015).
We ran additional Bayesian inference analysis for our Haemoproteus and Plasmodium and Leucocytozoon lineages with one Theileria annulata (GenBank: ON706267) as an outgroup in order to reconstruct the phylogeny as well as clarify if our lineages belong to Haemoproteus (Haemoproteus) or Haemoproteus (Parahaemoproteus). In this study by “Haemoproteus” we will mostly refer to the genus, unless otherwise stated. For that we included in our analysis fifteen (eight H. Haemoproteus and seven Parahaemoproteus) additional 479 bp cyt b sequences from the GenBank with known subgenera status (e.g. described in Dimitrov et al. 2016; Schumm et al. 2021 and other works). For the phylogenetic reconstruction we used transitional model including variation among sites (TIM2+ G; Posada 2008), suggested by jModelTest 2.1.7 software (Darriba et al. 2012) as the best-fit model according to Bayesian Information Criterion (BIC) and decision-theoretic performance-based approach (DT). Bayesian phylogenetic reconstruction was performed by BEAST 1.8.4. (Drummond et al. 2012), using strict clock and Yule speciation process (Yule 1925; Gernhard 2008) as tree priors selected in BEAUTi 1.8.4. Markov chain Monte Carlo (MCMC) simulations were run with 25 000 000 chain length, sampling every 1000 generations with 10% of the samples later discarded as burn-in in TreeAnnotator (BEAST package). The results of the analyses were validated in Tracer 1.6 (Drummond and Rambaut 2007) and the final tree was built in FigTree 1.4.4 (Rambaut 2007).
We calculated the general prevalence according to both genetic and morphological methods, while the prevalence per species and per sites were assessed based on genetics data only. We also compared lineage prevalence between females and males for bird species with clear sexual dimorphism. The association between number of infections and body mass per individual was also assessed.
For the statistical analyses, we used Chi-squared tests, two-factor ANOVAs and Spearman correlations conducted in R (R core team 2022). For the multiple testing we used Bonferroni correction. We applied statistical comparisons to species with sample size exceeding five individuals (redpoll, bluethroat, willow warbler, little bunting, chiffchaff, brambling, Arctic warbler, redstart; see Online Resource 1). To create the graphs outlining the findings of our research, we used the ggplot2 package in R (Wickham 2016).
Results
Parasite prevalence
Screening the DNA from 135 birds revealed 103 (76.3%, Online Resource 3) individuals infected by at least one parasite genus; the remaining 32 displayed no detectable infection. The most common parasite was Leucocytozoon, which was found in 97 individuals (72%) from 13 avian species. They were infected by 16 genetic lineages belonging to two clades (Table 1; Fig. 2). Beyond identified lineages of this genus, our data contained nine Leucocytozoon sequences that displayed plenty of conflicts in assembling and therefore could not be identified to lineage level.

Phylogeny of mitochondrial cytochrome b gene lineages (479 bp fragment) of avian haemosporidian parasites (two clades of Leucocytozoon, subgenera Parahaemoproteus and Haemoproteus, as well as Plasmodium with Theileria as an outgroup) with lineages shown on the left and genera/bigger clades only on the right. Scale is given for the image on the left (and the long branch of Theileria is not shown), branch lengths are arbitrary for the image on the right. All the lineage names are given according to the MalAvi database; additional GenBank accession numbers are included for the lineages that were not found in our study and were used as “control” sequences for two subgenera of Haemoproteus. Nodal support values indicate posterior clade probability. Asterisks mark novel lineages
All the lineages pertaining to Haemoproteus sensu lato were proved to belong to H. (Parahaemoproteus). We did not find any H. (Haemoproteus) infection in our dataset (Fig. 2).
Parahaemoproteus and Plasmodium appeared to be far less common than Leucocytozoon, with 12 individuals (8.9%) from four avian species infected by Parahaemoproteus and another 11 individuals (8.2%) from six avian species infected by Plasmodium. We found five distinct genetic lineages for each of these parasite genera (Table 1).
Blood parasite prevalence differed significantly among species (χ27 = 120.6, p < 0.0001; Fig. 3). For all the examined species the most frequently abundant parasite was Leucocytozoon, except for the one sampled rosefinch infected by Parahaemoproteus only. All the Parahaemoproteus and Plasmodium infections occurred in adult birds, while several Leucocytozoon infections were found in juvenile birds.

Overall prevalence of blood parasites of passerine bird species sampled on the Yamal peninsula. Colours mark combinations of infections: L—Leucocytozoon, H—Parahaemoproteus, P—Plasmodium, single infections; LL, LH, LP—mixed infections, respectively; u—unknown Leucocytozoon infections, uP—mixed infections with unknown Leucocytozoon and Plasmodium. Total sample size per each species (including non-infected individuals) are given in brackets
Mixed infections
Of 103 infected birds, single infections were found genetically in 63 individuals with one lineage of Leucocytozoon (L), two individuals with Parahaemoproteus (shortly referenced as H) and four individuals with Plasmodium (P); 25 individuals (18.5% from all the sampled birds) contained confirmed multiple mixed infections (LL—11 samples, LH—10, LP—4). Additionally, nine unidentified sequences most likely contained mixed Leucocytozoon infections. Moreover, three of them displayed Plasmodium infection, and these Plasmodium sequences were perfectly assembled and identified. Therefore, we suggested six of these sequences presented at least LL infection and three were most likely LLP (Fig. 3).
Each species with heterogeneric infections contained either Leucocytozoon and Parahaemoproteus (redpoll and willow warbler) or Leucocytozoon and Plasmodium (bluethroat, little bunting, Arctic warbler, redstart; Fig. 3). Only one species was infected by all three genera of parasites—brambling.
Lineage diversity
We found 26 genetic lineages of parasites in our sample, and five of them were not described earlier. We gave names to novel lineages and registered them in MalAvi; four of them (ACAFLA06, ACAFLA07, TURILI01, EMBPUS01) belonged to Leucocytozoon and one (PHYBOR05) belonged to Plasmodium. We also uploaded novel lineages into the GenBank under the accession numbers OP763386-90.
We considered eight lineages as common, appearing in five or more individuals; others were considered as rare (Table 1). Common Leucocytozoon lineages (Figs. 4, 5) were TRPIP2 (18%), as well as BT1 (14%), novel ACAFLA06 (13%), BT2 (7%), novel ACAFLA07 (6%), BT4 (5%). The most common Haemoproteus lineage was SISKIN1 (4%) and the most common Plasmodium lineage was TURDUS1 (4%).

Median-joining network of mitochondrial cytochrome b sequences (479 bp) of Leucocytozoon lineages belonging to passerine birds sampled on the Yamal peninsula (number of infections = 99, total number of infected birds = 103). Circles represent lineages. Circle sizes are proportional to the frequency of the lineage in the sample set; avian families are represented by different colours. One hatch mark represents one mutation. Asterisks mark novel lineages

Proportion of common (ACAFLA06, ACAFLA07, BT1, BT2, BT4, TRPIP2, SISKIN1, TURDUS1), rare and unknown blood parasite lineages per each avian species sampled on the Yamal peninsula (proportions are given from total number of infections per each species). The genus particular lineage belongs to is given in brackets (L—Leucocytozoon, H—Parahaemoproteus, P—Plasmodium), “rare” category combines lineages belonging to all three genera. Asterisks mark novel lineages
The highest lineage diversity was found in redpoll, little bunting and brambling (7 lineages each species), followed by willow warbler (6) and bluethroat (5) (Table 1, Fig. 5).
We also found a correlation between sample sizes and numbers of detected lineages per species (rs = 0.74, n = 14, p = 0.0025) as well as per sites (rs = 0.95, n = 7, p = 0.0008).
Sampling sites
General parasite prevalence according to genetic methods was 83% at Erkuta and 57% in Labytnangi. We found significant differences in general prevalence between all seven sampled plots (χ26 = 19, p = 0.0040), albeit distinct comparisons among five sites of Erkuta or two sites of Labytnangi revealed no significant differences (Online Resource 4; χ24 = 0.22, p = 0.99 and χ21 = 2.76, p = 0.1, respectively).
We found more lineages in Erkuta (11 unique, 18 in total) than in Labytnangi (8 unique, 15 in total), seven of them abundant on both sites (TRPIP2, BT1, ACAFLA06, BT2, CARFLA03 belonging to Leucocytozoon, SISKIN1 and TURDUS1 belonging to Parahaemoproteus and Plasmodium, respectively). Also, most of the lineages from both Erkuta and Labytnangi belonged to Leucocytozoon, each site contained three Plasmodium lineages, and Haemoproteus diversity was higher in Labytnangi compared to Erkuta (four and two lineages, respectively). One of our novel lineages (ACAFLA06) was common at both sampling sites, but other three novel Leucocytozoon lineages (ACAFLA07, EMBPUS01, TURILI01) were found in Erkuta only, and novel Plasmodium lineage (PHYBOR05) was found in Labytnangi only.
The highest lineage diversity was found at Erkuta at the site in the vicinity of the camp (Fig. 1) and in the town Labytnangi (13 lineages each site, Online Resource 4). The rare lineages (14; except NEVE01, ROFI6, WW1, BT7) and all the novel lineages (3; except broadly distributed ACAFLA06 and ACAFLA07) were found in these two sampling sites only.
Transmission
Several infections occurred in first-year birds, which had never migrated and therefore prove a local transmission of the parasites (Leucocytozoon only in our case). Thus, we found juvenile bramblings infected by TRPIP2 and BRAM3 as well as redstart infected by PARUS16 (all of aforementioned birds were caught in Labytnangi) and little bunting infected by BT1 (the only one infected juvenile bird from Erkuta). Therefore, at least two broadly distributed common lineages (TRPIP2 and BT1) are transmitted locally.
Body mass
Infected birds tended to weigh slightly more than individuals without parasites, and there was a positive correlation between body mass and total number of infections (by lineage) per individual (rs = 0.49, n = 110, p < 0.0001; Fig. 6). A two-factor ANOVA revealed significant effect of species (F7 = 144.2, p < 0.0001) and number of infections (F1 = 9.2, p = 0.0031) on the body mass, and no interaction between these two factors (F7 = 0.79, p = 0.5943).

Body mass of the passerine birds (log scale) sampled on the Yamal peninsula, in relation to the number of detected blood parasite infections for the same individuals
Sex
We compared lineage prevalence between females and males for bird species with clear sexual dimorphism and found significant differences in redpolls (χ26 = 58.1, p < 0.0001), bramblings (χ212 = 308.3, p < 0.0001) and bluethroats (χ24 = 68.4, p < 0.0001); all the p values are given according to Bonferroni correction. Particular lineages tended to be more frequently found in one of the sexes (Online Resource 5). For example, the most common lineage found in female redpolls (44%) was TRPIP2, while just 12% of males were infected by the same lineage. Most redpoll males (53%) displayed ACAFLA06 infection; and just 17% of females had that lineage. The majority of Parahaemoproteus (SISKIN1) infections occurred in redpoll males compared to females (18 and 6%, respectively).
Blood smears
Microscopic examination revealed 16 individuals out of 139 (11.5%), whose blood contained parasitic gametocytes. The prevalence of parasite genera differed significantly from results obtained by genetic methods (χ22 = 28.5, p < 0.0001). Moreover, according to blood smear examination, the most common parasite was Parahaemoproteus (11 infected individuals compared to three containing Plasmodium and two containing Leucocytozoon). We found no heterogeneric mixed infections in blood smears. The highest prevalence was found in brambling (54.6%) and little bunting (15.4%), while all other species had less than 12% blood parasite prevalence detected by blood smear examination (Online Resource 3).
Successful infections
Blood smear examination revealed gametocytes (sexual stages of life cycle occurring in avian blood cells) and, therefore, successful infections; thus, we could connect morphological data with certain genetic lineages. We found gametocytes of the subsequent lineages in the blood smears: TRPIP2 in one little bunting and BT1 or BT4 (genetic analysis showed mixed infection) in one bluethroat; SGS1 in one redstart, BT7 in one bluethroat, SW2 in one little bunting; SISKIN1 in four redpolls, CCF3 in two bramblings, BRAM1 in two bramblings, ROFI3 in one rosefinch. Images of infected erythrocytes are available in Online Resource 6.
SISKIN1 (Parahaemoproteus) infection in all the four infected redpolls was acute; we observed more infected blood cells than normally during the smear screening (Online Resource 6). Moreover, it is the only case of consensus between infections detected by genetic and morphological methods; in other words, genetic screening revealed four infected individuals and Parahaemoproteus gametocytes were found in blood smears from the same four individuals. In all the other cases, no consensus was obtained.
Discussion
Parasite prevalence
We screened 135 birds from the north-western Siberia genetically, and detected an overall blood parasite prevalence of 76.3%, which was higher than previously reported for other northern regions (e.g. Alaska, 53%; Oakgrove et al. 2014). In particular, a prevalence of 90% and more for certain species was rather an unusual result compared to other studies for particular species abundant in northern areas (80%, Krams et al. 2010; 68.5%, Svoboda et al. 2015; 69%, Van Hemert et al. 2019). The majority of detected infections refer to Leucocytozoon, while Parahaemoproteus and Plasmodium infections were less abundant and mostly appeared as a double infection together with Leucocytozoon, which is, most likely, common for the northern latitudes (Valkiūnas 1989; Valkiūnas et al. 1990; Valkiūnas 2005; Oakgrove et al. 2014).
Infection rates
Infection rate for several species was very high: 94% for redpoll, 92% for little bunting, 91% for bluethroat and 90% for brambling. We could not associate such a high prevalence with any known parameter, e.g. shared wintering area, breeding biotope or phylogenetic relationships. However, such pattern might be a feature of northern populations. For example, a previous study showed that the northern population of bluethroats contained the highest prevalence of blood parasites compared to more southern populations (Svoboda et al. 2015).
The most surprising high value of prevalence was the one obtained for the redpoll, the only resident species in our dataset. Normally, parasite prevalence in juveniles and resident birds is lower compared to the adult long-distance migrants (Oakgrove et al. 2014; de Angeli Dutra et al. 2021). However, in our study (adult) redpolls displayed the highest prevalence value (Fig. 3).
We also had species with relatively low infection rate (all the Phylloscopus birds: willow and Arctic warblers, chiffchaff). Redstarts might also be considered as species with rather low infection rate, but about 80% of sampled individuals were juveniles, and such comparison to other species might be irrelevant. Another remarkable result was 9% blood parasite prevalence in chiffchaff, where only one individual was infected by Leucocytozoon. Unusually low prevalence had already been reported for this species, however, it referred to Parahaemoproteus only, while Leucocytozoon prevalence was higher (40–70%, Bensch et al. 2012).
Sampling sites
Generally, there are twice as much passerine species breeding in northern taiga and forest–tundra zone at 66° on Yamal peninsula (Labytnangi) than in shrub tundra at 68° (Erkuta) (Ryzhanovskiy 2012), and we could have expected higher diversity of avian species caught as well as consequent higher diversity of parasite lineages in Labytnangi. However, we caught 11 avian species in Erkuta and 10 in Labytnangi, which is almost equal, but the sample size (number of individuals) from Erkuta was approximately thrice as much as Labytnangi sample size. Moreover, the most common and numerous species in our sample from Erkuta were redpoll, bluethroat and little bunting (all three rather not common, but still abundant in Labytnangi), while the main dominant species from Labytnangi was brambling, totally absent in Erkuta and in tundra in general due to niche incompatibility.
Differences in sample sizes, sampled avian species, as well as other methodological aspects such at timing (sampling in Labytnangi occurred in July only and much more juvenile birds were caught, 70% of them uninfected) might partly explain the differences in prevalence and lineage composition between two sites. Most likely these sites have their own unique composition of lineages, partly shaped by differences in avian biodiversity between shrub tundra and woodland habitat.
Our findings of broadly distributed novel ACAFLA06 (Leucocytozoon) lineage in these northern habitats as well as less common novel lineages (ACAFLA07, EMBPUS01, TURILI01, found in Erkuta only) might suggest the evolution of Leucocytozoon lineages above the polar circle, especially in tundra zones.
Lineage diversity: generalists and specialists
Although common lineages infected a relatively broad range of sampled hosts, some specific tendencies might be observed. Thus, closely related BT1 and BT4 never infected Fringillidae (except Emberiza and Schoeniclus), and ACAFLA06/07, TRPIP2 never infected Turdidae (bluethroats mostly) (Fig. 4).
Most of the identified Leucocytozoon lineages seem to be generalists capable of infecting more than one Passeriformes genera, however, displaying certain patterns (MalAvi database). For example, according to MalAvi, TRPIP2 infects Fringillidae, Motacillidae (Anthus) and Paridae birds while BT1—Turdidae (Luscinia), Sylviidae, Muscicapidae (Phoenicurus), Paridae. Our findings support these patterns as well (Table 1, Fig. 4). Our research presents novel hosts for PARUS16, BT2, BT4 and BT5. NEVE01 was previously found in blackbird only, and in our study just the redwings were infected with this lineage, thus, it is the only one Leucocytozoon candidate for being a specialised (to genus Turdus) lineage.
In general, Plasmodium is known to be a generalist, while Haemoproteus—as more host-specific parasite (Fecchio et al. 2020; Doussang et al. 2021). All the identified Plasmodium lineages (TURDUS1, BT7, SGS1, SW2; except the novel PHYBOR05) are indeed broadly distributed and infect a broad range of hosts. Knowledge about Haemoproteus lineages is scarcer (which could be due to general bias towards Plasmodium studies; Valkiūnas 2005). Nevertheless, according to MalAvi, SISKIN1 and CCF3 infect mostly Fringillidae, WW1—mostly Sylvidae (the most frequent host is willow warbler; but this particular lineage was also reported from Anseriformes and Bucerotiformes), which totally corresponds to our results. BRAM1 and ROFI3 are only known from brambling and rosefinch, respectively, and in our dataset, they are represented in these particular species only.
However, all the specialist–generalist suggestions are quite vague. All the identified entities are based on one gene, cyt b, which is involved oxidative phosphorylation energy metabolism in vector, so, most likely, cyt b genetic lineages are bound to vectors rather than to hosts (Hall et al. 2005; Fecchio et al. 2020). Therefore, observed differences in host ranges might be severely dependent from exposure to compatible vectors. Moreover, several lineages might be considered as one species, as it was recently shown with Leucocytozoon lineages (Galen et al. 2018b).
Transmission
Life of a bird usually includes seasonal migrations, giving plenty of opportunities for manifold infectious agents: assuming abundance of compatible vectors, transmission of blood parasites can occur on the breeding sites, along migration routes and in wintering areas (Hubálek 2004; Valkiūnas 2005). Furthermore, if the infection occurs successfully, it remains in bird hosts either for many years or for life (Garnham 1966), but demonstrates certain seasonal dynamics (Valkiūnas 2005). In other words, abundance of those parasites does not necessarily mean transmission in polar latitudes.
Nonetheless, Yamal breeding habitats have enough potential to be the foci for the diseases caused by haemosporidians. Northern taiga, forest–tundra and tundra zones are perfect breeding habitats for vectors—blood-sucking insects, such as black flies (genera Simulium, Odagmia, Schoenbaueria), mosquitoes (Ochlerotatus) and biting midges (Culicoides). Some species were observed recently for the first time suggesting a potential shift in geographical distribution (Fyodorova et al. 2019; full list of blood-sucking insects of Yamalo–Nenets Autonomous Region is available in the paper by Fiodorova et al. 2019).
However, distribution patterns and periods of mass imago abundance differ between these insects. Numbers of Yamal black fly species increase from northern taiga to tundra with intermediate value in forest–tundra, and the opposite trend is observed for biting midges; mosquitoes are also more diverse in tundra (Fyodorova et al. 2019). Moreover, abundance of (imago) mosquitoes reaches its peak in June and July, black flies and biting midges—in July and August with biting midges being the latest (Fyodorova et al. 2019). Possibly, Parahaemoproteus infections in juvenile birds (sampled mostly in early–middle July) were not detectable in our study, because the vectors—biting midges—are abundant and active later during the summer.
All the lineages found in juvenile birds (TRPIP2, BRAM3, PARUS16, BT1) belong to Leucocytozoon, and, interestingly, to the clade 1, suggesting that successful local transmission might be bound to compatibility between the certain features of parasite metabolism and local vectors’ physiology (because cyt b of parasite is involved in metabolism in vector). Therefore, Leucocytozoon clade 1 might be more favourable for transmission in northern latitudes.
Moreover, it is known that redpolls are non-philopatric species performing no transcontinental migrations, but committing short-distance seasonal relocations (Ryzhanovskiy and Ryabitsev 2021). Therefore, most likely they were still infected (by Leucocytozoon: ACAFLA06, ACAFLA07, BT2, ROFI6, CARFLA03 from clade 2 and again TRPIP2 from clade 1; as well as one Parahaemoproteus lineage SISKIN1) in rather northern latitudes (but not necessarily locally or above the polar circle). We did not find any proof for Plasmodium transmission in our study either with juveniles or resident species.
Body mass
A positive correlation between body mass and number of infections, obtained in our study, led us directly onto the question of pathogenicity of haemosporidian parasites and their effect on hosts. In general, studies obtained rather contradictory results on pathogenicity: from minimal effect on individuals (Bensch et al. 2007), their behaviour (Knowles et al. 2010; Dunn et al. 2011) and fitness (Merino et al. 2000; Marzal et al. 2005) to huge impact on the whole population (Warner 1968; van Riper et al. 1986). The same applies to body mass. It is known that severe parasitemia leads to the decrease of body mass (e.g. Valkiūnas 2005; Valkiūnas et al. 2006). However, there are surveys showing no clear effect on weight (Smith and Cox 1972; Bennett et al. 1988) or a positive correlation between host body mass and infection rate as obtained in our study (e.g. Scheuerlein and Ricklefs 2004; González et al. 2014; Fecchio et al. 2021).
Such a visible contradiction in observed physiological effects might occur due to different intensities of infection. In a study assessing an association between haemosporidian infection and another physiological parameter, fatness level of migratory birds, many haemosporidian hosts with medium and low intensity of infection had quite high level of fatness, while birds with intense parasitemia mostly displayed insufficient levels of fatness (Valkiūnas 1983). Moreover, host nutrition has a strong effect on infection dynamics and parasite virulence (Cornet et al. 2014). If the body mass is a proxy for the general condition of the bird, then individuals in a better condition could be more exposed to vectors and parasites than individuals in poor condition.
Thus, our correlation might reflect these two processes: hosts in a better condition “attract” more vectors and parasites, but generally low intensity of infection (supported by results of blood smear examination) does not affect the body mass dramatically. However, to follow-up the reasons and the precise mechanism of association between body mass and number of infections, further research is essential.
Sex
Observed differences in prevalence might be due to various biotopes chosen by certain groups of birds during the migrations or spatial movements and subsequent exposure to various vectors (Svoboda et al. 2015). However, to find out if these differences are real, a larger sample size is needed (especially for bluethroats and bramblings).
Restrictions of the methods
We obtained very different results from screening blood smears and applying genetic methods (Online Resource 3). Blood smear examination revealed successful acute infections (via detection of gametocytes), while infections in certain seasonal periods of avian life cycle and mild infections could be missed. However, they had to be detected by genetic methods together with unsuccessful infections, when the parasite does not complete its entire life cycle and infect the host (Valkiūnas 2005; Moens et al. 2016). Most likely, blood smear examination tends to underestimate blood parasite prevalence, while genetic methods might overestimate the same value (Valkiūnas 2005; Bensch et al. 2021). Both methods are thus imperfect and could lead to contradictory results. For example, smear from one brambling contained Parahaemoproteus plenty of gametocytes, albeit electrophoresis showed a peak in Leucocytozoon PCR products, but not in Haemoproteus/Plasmodium. We repeated the PCRs for this individual and obtained the same result.
Double Leucocytozoon coinfections appeared to happen between certain lineages. Thus, the most common combinations occurred between couples of closely related lineages: BT1 and BT4 (differing in 2 nucleotides in 479 bp cyt b fragment; 5 individuals) as well as ACAFLA06 and ACAFLA07 (differing in 1 nucleotide; 4 individuals). In several sequences with mixed BT1/BT4 infection from bluethroats there were signs of additional RS2 infection (secondary lower peak; another closely related lineage). This lineage never occurred in our data separately, so we did not count these cases as triple coinfection. When coinfections of more distant lineages or between several lineages occur, it is much more difficult to detect which signal belongs to which lineages, and, therefore, lineage identification is not a trivial task. We managed to obtain such results from TRPIP2/BT1 and TRPIP2/BT4 mixed infections, but nine sequences remained unresolved.
We also found no individuals displaying triple infection LHP. However, we might simply miss them, because nested PCR for Haemoproteus and Plasmodium favours the amplification of the most abundant parasite in the sample or the parasite for which the primers are a better match (Ciloglu et al. 2019).
We found correlations between sample sizes and number of lineages. Also, some of avian species and sample sites from our study were clearly underrepresented, so more lineages might be revealed in the research with bigger sample sites for these underrepresented sets of species or sites.
Conclusions
Our pilot study revealed avian blood parasites from three genera (Leucocytozoon, H. (Parahaemoproteus), Plasmodium) infecting Passeriformes breeding on the Yamal peninsula. The prevalence of haemosporidians was relatively high, especially for certain avian species. We found novel genetic lineages as well as novel host species for lineages described earlier. Our results suggest the potential expansion of blood parasites above the polar circle. Further avian haemosporidian research in the Yamal tundra, especially with larger sample sizes, could shed light on some of the discussed questions, including further climate-change effects, sex differences in infection rates and health impacts on birds.
Data availability
All raw data as well as additional supplementary figures presented in this study are available in the supplemental online materials (Online Resource 7). All the relevant details about lineages are also submitted to the “MalAvi” database. Novel lineages are deposited in GenBank under accession numbers OP763386-90.
References
Andreasson F, Nilsson JÅ, Nord A (2020) Avian reproduction in a warming world. Front Ecol Evol. https://doi.org/10.3389/fevo.2020.576331
ASAB Ethical Committee/ABS Animal Care Committee (2023) Guidelines for the ethical treatment of nonhuman animals in behavioural research and teaching. Anim Behav 195:1–11. https://doi.org/10.1016/j.anbehav.2022.09.006
Bennett GF, Caines JR, Bishop MA (1988) Influence of blood parasites on the body mass of passeriform birds. J Wildl Dis 24:339–343. https://doi.org/10.7589/0090-3558-24.2.339
Bennett GF, Montgomerie R, Seutin G (1992) Scarcity of haematozoa in birds breeding on the arctic tundra of North America. Condor 94:289–292
Bensch S, Stjernman M, Hasselquist D, Hannson B, Westerdahl H, Pinheiro RT (2000) Host specificity in avian blood parasites: a study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc R Soc B 267:1583–1589. https://doi.org/10.1098/rspb.2000.1181
Bensch S, Péarez-Tris J, Waldenströum J, Hellgren O (2004) Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: multiple cases of cryptic speciation? Evolution 58:1617–1621. https://doi.org/10.1111/j.0014-3820.2004.tb01742.x
Bensch S, Waldenström J, Jonzán N, Westerdahl H, Hansson B, Sejberg D, Hasselquist D (2007) Temporal dynamics and diversity of avian malaria parasites in a single host species. J Anim Ecol. https://doi.org/10.1111/j.1365-2656.2006.01176.x
Bensch S, Jönsson J, Copete JL (2012) Low prevalence of Haemoproteus infections in Chiffchaffs. Parasitology 139:302–309. https://doi.org/10.1017/S0031182011002009
Bensch S, Inumaru M, Sato Y, Lee Cruz L, Cunningham AA, Goodman SJ, Levin II, Parker PG, Casanueva P, Hernández MA, Moreno-Rueda G, Rojo MA (2021) Contaminations contaminate common databases. Mol Ecol Resour 21:355–362. https://doi.org/10.1111/1755-0998.13272
Caminade C, McIntyre KM, Jones AE (2019) Impact of recent and future climate change on vector-borne diseases. Ann NY Acad Sci 1436:157–173. https://doi.org/10.1111/nyas.13950
Ciloglu A, Ellis VA, Bernotienė R, Valkiūnas G, Bensch S (2019) A new one-step multiplex PCR assay for simultaneous detection and identification of avian haemosporidian parasites. Parasitol Res 118:191–201. https://doi.org/10.1007/s00436-018-6153-7
Clark NJ, Clegg SM, Lima MR (2014) A review of global diversity in avian haemosporidians (Plasmodium and Haemoproteus: Haemosporida): new insights from molecular data. Int J Parasitol 44:329–338. https://doi.org/10.1016/j.ijpara.2014.01.004
Cornet S, Bichet C, Larcombe S, Faivre B, Sorci G (2014) Impact of host nutritional status on infection dynamics and parasite virulence in a bird-malaria system. J Anim Ecol 83:256–265. https://doi.org/10.1111/1365-2656.12113
Currie DC, Adler PH (2008) Global diversity of black flies (Diptera: Simuliidae) in freshwater. Hydrobiol 595:469–475. https://doi.org/10.1007/s10750-007-9114-1
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. https://doi.org/10.1038/nmeth.2109
de Angeli DD, Fecchio A, Braga ÉM, Poulin R (2021) Migratory birds have higher prevalence and richness of avian haemosporidian parasites than residents. Int J Parasitol 51:877–882. https://doi.org/10.1016/j.ijpara.2021.03.001
Dimitrov D, Iezhova TA, Zehtindjiev P, Bobeva A, Ilieva M, Kirilova M, Bedev K, Sjöholm C, Valkiūnas G (2016) Molecular characterisation of three avian haemoproteids (Haemosporida, Haemoproteidae), with the description of Haemoproteus (Parahaemoproteus) palloris n. sp. Systematic Parasitol 93:431–449. https://doi.org/10.1007/s11230-016-9638-8
Doussang D, Sallaberry-Pincheira N, Cabanne GS, Lijtmaer DA, González-Acuña D, Vianna JA (2021) Specialist versus generalist parasites: the interactions between host diversity, environment and geographic barriers in avian malaria. Int J Parasitol 51:899–911. https://doi.org/10.1016/j.ijpara.2021.04.003
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. https://doi.org/10.1093/molbev/mss075
Dunn JC, Cole EF, Quinn JL (2011) Personality and parasites: sex-dependent associations between avian malaria infection and multiple behavioural traits. Behav Ecol Sociobiol 65:1459–1471. https://doi.org/10.1007/s00265-011-1156-8
Fecchio A, Chagas CR, Bell JA, Kirchgatter K (2020) Evolutionary ecology, taxonomy, and systematics of avian malaria and related parasites. Acta Trop 204:105364. https://doi.org/10.1016/j.actatropica.2020.105364
Fecchio A, Clark NJ, Bell JA, Skeen HR, Lutz HL, De La Torre GM, Vaughan JA, Tkach VV, Schunck F, Ferreira FC, Braga ÉM (2021) Global drivers of avian haemosporidian infections vary across zoogeographical regions. Glob Ecol Biogeogr 30:2393–2406. https://doi.org/10.1111/geb.13390
Fiodorova OA, Khlyzova TA, Siben AN, Domatsky VN, Beletskaya NI (2019) Current fauna of parasitic flies of Yamalo-Nenets autonomous region. Ukrainian J Ecol 9:448–458
Fyodorova OA, Siben AN, Khlyzova TA (2019) Distribution and flight dates of bloodsucking dipterans and gadflies on the territory of the Yamalo-Nenets autonomous district. Vet Kubani. https://doi.org/10.33861/2071-8020-2019-1-21-24
Galen SC, Borner J, Martinsen ES, Schaer J, Austin CC, West CJ, Perkins SL (2018a) The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R Soc Open Sci 5:171780. https://doi.org/10.1098/rsos.171780
Galen SC, Nunes R, Sweet PR, Perkins SL (2018b) Integrating coalescent species delimitation with analysis of host specificity reveals extensive cryptic diversity despite minimal mitochondrial divergence in the malaria parasite genus Leucocytozoon. BMC Evol Biol 18:1–15. https://doi.org/10.1186/s12862-018-1242-x
Garamszegi LZ (2011) Climate change increases the risk of malaria in birds. Glob Change Biol 17:1751–1759. https://doi.org/10.1111/j.1365-2486.2010.02346.x
Gardner AM, Hamer GL, Hines AM, Newman CM, Walker ED, Ruiz MO (2012) Weather variability affects abundance of larval Culex (Diptera: Culicidae) in storm water catch basins in suburban Chicago. J Med Entomol 49:270–276. https://doi.org/10.1603/ME11073
Garnham PCC (1966) Malaria parasites and other Haemosporidia. Blackwell Scientific, Oxford
Gernhard T (2008) Yule process. J Theor Biol 253:769–778
González AD, Matta NE, Ellis VA, Miller ET, Ricklefs RE, Gutierrez HR (2014) Mixed species flock, nest height, and elevation partially explain avian haemoparasite prevalence in Colombia. PLoS ONE 9:e100695. https://doi.org/10.1371/journal.pone.0100695
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hall N, Karras M, Raine JD, Carlton JM, Kooij TWA, Berriman M, Florens L, Janssen CJ, Pain A, Christophides GK, James K, Rutherford K, Harris B, Harris D, Churcher C, Quail MA, Ormond D, Doggett J, Trueman HE, Mendoza J, Bidwell SL, Rajandream MA, Carucci DJ, Yates JR III, Kafatos FC, Janse CJ, Barrell B, Turner CMR, Waters AP, Sinden RE (2005) A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307:82–86. https://doi.org/10.1126/science.1103717
Hasselquist D (2007) Comparative immunoecology in birds: hypotheses and tests. J Ornithol 148:571–582. https://doi.org/10.1007/s10336-007-0201-x
Hellgren O, Waldenström J, Bensch S (2004) A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol 90:797–802. https://doi.org/10.1645/GE-184R1
Huang J, Zhang X, Zhang Q, Lin Y, Hao M, Luo Y, Zhao Z, Yao Y, Chen X, Wang L, Nie S (2017) Recently amplified arctic warming has contributed to a continual global warming trend. Nat Clim Change 7:875–879. https://doi.org/10.1038/s41558-017-0009-5
Hubálek Z (2004) An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wildl Dis 40:639–659. https://doi.org/10.7589/0090-3558-40.4.639
Knowles SCL, Palinauskas V, Sheldon BC (2010) Chronic malaria infections increase family inequalities and reduce parental fitness: experimental evidence from a wild bird population. J Evol Biol 23:557–569. https://doi.org/10.1111/j.1420-9101.2009.01920.x
Krams I, Cīrule D, Krama T, Hukkanen M, Rytkönen S, Orell M, Iezhova TA, Rantala MJ, Tummeleht L (2010) Effects of forest management on haematological parameters, blood parasites, and reproductive success of the Siberian tit (Poecile cinctus) in northern Finland. Ann Zool Fenn 47:335–346. https://doi.org/10.5735/086.047.0504
Lachish S, Knowles SC, Alves R, Wood MJ, Sheldon BC (2011) Infection dynamics of endemic malaria in a wild bird population: parasite species-dependent drivers of spatial and temporal variation in transmission rates. J Anim Ecol 80:1207–1216. https://doi.org/10.1111/j.1365-2656.2011.01893.x
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Loiseau C, Harrigan RJ, Cornel AJ, Guers SL, Dodge M, Marzec T, Carlson JS, Seppi B, Sehgal RNM (2012) First evidence and predictions of Plasmodium transmission in Alaskan bird populations. PLoS ONE 7:e44729. https://doi.org/10.1371/journal.pone.0044729
MacDonald GM (2010) Global warming and the Arctic: a new world beyond the reach of the Grinnellian niche? J Exp Biol 213:855–861. https://doi.org/10.1242/jeb.039511
MalAvi (2022) A database for avian haemosporidian parasites. Table: Great lineage summary. http://130.235.244.92/Malavi/index.html
Martínez J, Martínez de la Puente J, Herrero J, Cerro Gómez SD, Lobato E, Rivero de Aguilar J, Vásquez RA, Merino S (2009) A restriction site to differentiate Plasmodium and Haemoproteus infections in birds: on the inefficiency of general primers for detection of mixed infections. Parasitology 136:713–722. https://doi.org/10.1017/S0031182009006118
Marzal A, Lope FD, Navarro C, Møller AP (2005) Malarial parasites decrease reproductive success: an experimental study in a passerine bird. Oecologia 142:541–545. https://doi.org/10.1007/s00442-004-1757-2
Marzal A, Ricklefs RE, Valkiūnas G, Albayrak T, Arriero E, Bonneaud C, Czirják GA, Ewen J, Hellgren O, Hořáková D, Iezhova TA, Jensen H, Križanauskienė A, Lima MR, de Lope F, Magnussen E, Martin LB, Møller AP, Palinauskas V, Pap PL, Pérez-Tris J, Sehgal RNM, Soler M, Szöllősi E, Westerdahl H, Zetindjiev P, Bensch S (2011) Diversity, loss, and gain of malaria parasites in a globally invasive bird. PLoS ONE 6:e21905. https://doi.org/10.1371/journal.pone.0021905
Merino S, Moreno J, José Sanz J, Arriero E (2000) Are avian blood parasites pathogenic in the wild? A medication experiment in blue tits (Parus caeruleus). Proc R Soc B-Biol Sci 267:2507–2510. https://doi.org/10.1098/rspb.2000.1312
Moens MAJ, Valkiūnas G, Paca A, Bonaccorso E, Aguirre N, Pérez-Tris J (2016) Parasite specialization in a unique habitat: hummingbirds as reservoirs of generalist blood parasites of Andean birds. J Anim Ecol 85:1234–1245. https://doi.org/10.1111/1365-2656.12550
Oakgrove KS, Harrigan RJ, Loiseau C, Guers S, Seppi B, Sehgal RN (2014) Distribution, diversity and drivers of blood-borne parasite co-infections in Alaskan bird populations. Int J Parasitol 44:717–727. https://doi.org/10.1016/j.ijpara.2014.04.011
Paaijmans KP, Blanford S, Bell AS, Blanford JI, Read AF, Thomas MB (2010) Influence of climate on malaria transmission depends on daily temperature variation. Proc Natl Acad Sci U S A 107:15135–15139. https://doi.org/10.1073/pnas.1006422107
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. https://doi.org/10.1093/molbev/msn083
R Core Team (2022) R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. https://www.R-project.org/
Rambaut A (2007) FigTree. https://tree.bio.ed.ac.uk/software/figtree/
Rivero A, Gandon S (2018) Evolutionary ecology of avian malaria: past to present. Trends Parasitol 34:712–726. https://doi.org/10.1016/j.pt.2018.06.002
Rocklöv J, Dubrow R (2020) Climate change: an enduring challenge for vector-borne disease prevention and control. Nature Immunol 21:479–483. https://doi.org/10.1038/s41590-020-0648-y
Ryabitsev VK (2001) Birds of the Ural, Ural foothills and western Siberia: a guide. Ural University Press, Ekaterinburg
Ryzhanovskiy VN (2012) Ecological factors determining the northern boundaries of the passerine birds ranges in north-western Siberia. Russian J Ornithol 21:769–780
Ryzhanovskiy VN, Ryabitsev VK (2021) Biology and ecology of the redpoll (Acanthis flammea sensu lato, Passeriformes, Fringillidae) on Yamal Peninsula and in the near-Ob forested tundra. Biol Bull 48:1347–1357. https://doi.org/10.1134/S1062359021080240
Santiago-Alarcon D, Palinauskas V, Schaefer HM (2012) Diptera vectors of avian Haemosporidian parasites: untangling parasite life cycles and their taxonomy. Biol Reviews 87:928–964. https://doi.org/10.1111/j.1469-185X.2012.00234.x
Scheuerlein A, Ricklefs RE (2004) Prevalence of blood parasites in European passeriform birds. Proc R Soc B-Biol Sci 271:1363–1370. https://doi.org/10.1098/rspb.2004.2726
Schumm YR, Bakaloudis D, Barboutis C, Cecere JG, Eraud C, Fischer D, Hering J, Hillerich K, Lormée H, Mader V, Masello JF (2021) Prevalence and genetic diversity of avian haemosporidian parasites in wild bird species of the order Columbiformes. Parasitol Res 120:1405–1420. https://doi.org/10.1007/s00436-021-07053-7
Smith VW, Cox FEG (1972) Blood parasites and the weights of Palaearctic migrants in central Nigeria. Ibis 114:105–106. https://doi.org/10.1111/j.1474-919X.1972.tb02595.x
Sokolov V, Ehrich D, Yoccoz NG, Sokolov A, Lecomte N (2012) Bird communities of the Arctic shrub tundra of Yamal: habitat specialists and generalists. PLoS ONE 7:e50335. https://doi.org/10.1371/journal.pone.0050335
Svoboda A, Marthinsen G, Pavel V, Chutný B, Turčoková L, Lifjeld JT, Johnsen A (2015) Blood parasite prevalence in the Bluethroat is associated with subspecies and breeding habitat. J Ornithol 156:371–380. https://doi.org/10.1007/s10336-014-1134-9
Valkiūnas GA (1983) On the pathogenicity of Haemosporidia (Sporozoa, Haemosporidia) in birds. Parazitologiya 17:375–381
Valkiūnas G (2005) Avian malaria parasites and other Haemosporidia. CRC Press, New York
Valkiūnas G, Zickus T, Shapoval AP, Iezhova TA (2006) Effect of Haemoproteus belopolskyi (Haemosporida: Haemoproteidae) on body mass of the black-cap Sylvia atricapilla. J Parasitol 92:1123–1125. https://doi.org/10.1645/GE-3564-RN.1
Valkiūnas GA (1989) On the role of seasonal migrations of palaearctic migrants in the spread of haemosporidia of birds (Sporozoa, Haemosporidia). Parazitologiya 23:208–215
Valkiūnas GA, Sruoga AA, Paulauskas AP (1990) Geographical distribution of Leucocytozoon simondi (Haemosporidia, Leucocytozoidae). Parazitologiya 24:400–407
Van Hemert C, Meixell BW, Smith MM, Handel CM (2019) Prevalence and diversity of avian blood parasites in a resident northern passerine. Parasit Vectors 12:1–16. https://doi.org/10.1186/s13071-019-3545-1
Ripervan C III, Van Riper SG, Goff ML, Laird M (1986) The epizootiology and ecological significance of malaria in Hawaiian land birds. Ecol Monogr 56:327–344. https://doi.org/10.2307/1942550
Warner RE (1968) The role of introduced diseases in the extinction of the endemic Hawaiian avifauna. Condor 70:101–120. https://doi.org/10.2307/1365954
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Yule GU (1925) Yule process. Philos Trans Proc R Soc B-Biol Sci 213:21–87
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214. https://doi.org/10.1089/10665270050081478
Acknowledgements
We would like to thank Dr. Noémie Becker for helping us to settle the project officially and giving remarks on the data analyses. We are very grateful to Anvar Kerimov, Tatiana Golubeva and Vyacheslav Red’kin for crucial lessons in taking blood samples and handling birds, to Olga Pokrovskaya for the support with research and communication issues, to Vyacheslav Ryzhanovskiy for the help in definition of birds’ age by moulting stages, as well as to Julia Loschagina and Svetlana Artemieva for the assistance in acquiring the necessary equipment. We would also thank Victor Shtro and other members of Labytnangi Arctic Research Station as well as Erkuta field team (Marcus Spiegel, Dorothee Ehrich, Alexandra Terekhina, Alexander Volkovitskiy, Natalia, Nikita and Ilya Sokolov, Pavel Orekhov) for the technical support during the fieldwork. We also thank Wiebke Schäfer, Sabine Wagner, Lucie Michel, Naemi Lederer-Ponzer and other members of the Behavioural Ecology and Ecophysiology research group for the technical laboratory support as well as Staffan Bensch for helping with parasite DNA sequences. Remarks given by two anonymous reviewers and Dieter Piepenburg, Journal’s editor in chief, significantly improved the article.
Funding
Open Access funding enabled and organized by Projekt DEAL. No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
PQ and AAS conceived and designed the research as well as contributed reagents and materials. YRS assisted with analysis. DAY conducted the field and laboratory work as well as analysed the data and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare.
Ethical approval
ASAB Ethical Committee recommendations for the care and use of animals were followed (ASAB Ethical Committee/ABS Animal Care Committee; 2023
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yusupova, D.A., Schumm, Y.R., Sokolov, A.A. et al. Haemosporidian blood parasites of passerine birds in north-western Siberia. Polar Biol 46, 497–511 (2023). https://doi.org/10.1007/s00300-023-03130-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-023-03130-y