
Abstract
Purpose
Adavosertib may alter exposure to substrates of the cytochrome P450 (CYP) family of enzymes. This study assessed its effect on the pharmacokinetics of a cocktail of probe substrates for CYP3A (midazolam), CYP2C19 (omeprazole), and CYP1A2 (caffeine).
Methods
Period 1: patients with locally advanced or metastatic solid tumors received ‘cocktail’: caffeine 200 mg, omeprazole 20 mg, and midazolam 2 mg (single dose); period 2: after 7- to 14-day washout, patients received adavosertib 225 mg twice daily on days 1–3 (five doses), with cocktail on day 3. After cocktail alone or in combination with adavosertib administration, 24-h pharmacokinetic sampling occurred for probe substrates and their respective metabolites paraxanthine, 5-hydroxyomeprazole (5-HO), and 1′-hydroxymidazolam (1′-HM). Safety was assessed throughout.
Results
Of 33 patients (median age 60.0 years, range 41–83) receiving cocktail, 30 received adavosertib. Adavosertib co-administration increased caffeine, omeprazole, and midazolam exposure by 49%, 80%, and 55% (AUC0–12), respectively; AUC0–t increased by 61%, 98%, and 55%. Maximum plasma drug concentration (Cmax) increased by 4%, 46%, and 39%. Adavosertib co-administration increased 5-HO and 1′-HM exposure by 43% and 54% (AUC0–12) and 49% and 58% (AUC0–t), respectively; paraxanthine exposure was unchanged. Adavosertib co-administration decreased Cmax for paraxanthine and 5–HO by 19% and 7%; Cmax increased by 33% for 1′-HM. After receiving adavosertib, 19 (63%) patients had treatment-related adverse events (six [20%] grade ≥ 3).
Conclusion
Adavosertib (225 mg bid) is a weak inhibitor of CYP1A2, CYP2C19, and CYP3A.
ClinicalTrials.gov
NCT03333824
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclin-dependent kinase 1 (CDK1; also known as cell division cycle 2 protein [CDC2]) drives a cell from the G2 phase of the cell cycle into mitosis. In response to DNA damage, the nuclear tyrosine kinase Wee1 inhibits CDK1 to prevent the cell from dividing until the damaged DNA is repaired (G2 checkpoint arrest) [1]. CDK2 drives a cell into, and through, the S phase of the cell cycle, during which the genome is duplicated in preparation for cell division; inhibition of Wee1 is expected to cause aberrantly high CDK2 activity in S-phase cells that, in turn, leads to unstable DNA replication structures and, ultimately, DNA damage [2].
Fully functional CDK1- and CDK2-mediated checkpoints are necessary for the DNA damage response (DDR) to minimize replication stress for proliferating cells [3,4,5,6]. Adavosertib (AZD1775) is a highly selective ATP-competitive small-molecule inhibitor of Wee1, with a half-maximal inhibitory concentration (IC50) of 5.2 nmol/L in in vitro kinase assays [7, 8]. Inhibition of Wee1 releases tumor cells from DNA-damage-induced arrest at the G2/M boundary, so that unrepaired DNA damage may be taken into mitosis (M phase); as cancer cells show higher levels of endogenous damage than normal cells, as well as exhibiting loss of one or more DDR capabilities, this is predicted to preferentially enhance cancer cell death through mitotic catastrophe compared with normal cells [2, 9].
Adavosertib has been evaluated as mono- and combination therapy with chemotherapy, olaparib, and durvalumab in numerous phase I and II studies in patients with a wide variety of solid tumors [10,11,12,13,14,15]. Preclinical (in vitro) studies indicate that adavosertib is metabolized by, and is both a substrate for and an inhibitor of, certain enzymes of the cytochrome P450 (CYP) family [16]. Metabolism of adavosertib is predominantly by CYP3A, with a flavin-containing monooxygenase (FMO) 3 and/or FMO5 component (AstraZeneca, data on file, 2022). Adavosertib is also a weak reversible inhibitor (IC50 14 μM) and a time-dependent (irreversible) inhibitor of CYP3A (maximal inactivation [kinact] 0.061/min, concentration at 50% of kinact 6.04 μM); modeling data predicted an eight- to tenfold increase in the exposure of sensitive CYP3A substrates when administered with adavosertib (250 mg twice daily [bid] for five doses; AstraZeneca, data on file, 2020) [16]. Adavosertib is a weak inducer of CYP1A2 (39% increase in activity of positive control; AstraZeneca, data on file, 2022) [16].
The primary objective of this prospective, two-period, open-label, drug–drug interaction (DDI) study (NCT03333824) was to determine whether there was a clinically significant increase of exposure to substrates for CYP1A2 (caffeine), CYP2C19 (omeprazole), and CYP3A (midazolam) in the presence of adavosertib, and if co-administration of the probe CYP substrates had a clinically meaningful effect on adavosertib pharmacokinetics (PKs) [17]. The doses of the probe drugs (midazolam: 1 mL of 2 mg/mL syrup; omeprazole: 20 mg capsules; caffeine: 200 mg tablet) included in the cocktail were previously validated in this cocktail [18].
Methods
Objectives
The primary study objective was to assess the effect of adavosertib on the PKs of probe substrates for CYP3A (midazolam), CYP2C19 (omeprazole), and CYP1A2 (caffeine). The secondary study objectives were to: describe the PKs of midazolam, omeprazole, and caffeine and their respective metabolites (1ʹ-hydroxymidazolam [1ʹ-HM], 5-hydroxyomeprazole [5-HO], and paraxanthine) in the absence and presence of adavosertib; describe the PKs of adavosertib; and observe the clinical and laboratory safety and tolerability of adavosertib.
Study design
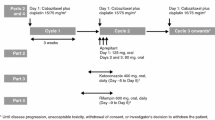
This manuscript focuses on PK data (part A, periods 1 and 2) from NCT03333824, which was conducted at seven clinical sites in the USA. Part B of NCT03333824 was an investigation of the effect of adavosertib on the electrocardiogram QT interval, the results of which are reported separately. The study was of a prospective, open-label, non-randomized, sequential, two-period design (Fig. 1).

a Study design and b patient disposition flow chart. This manuscript focuses on pharmacokinetic data from part A of NCT03333824; part B of NCT03333824 is an investigation of the effect of adavosertib on the QT interval, results of which are reported separately. *Informed consent received; †A number of patients enrolled more than once; there were 49 unique enrollments; ‡Study treatment refers to treatment with either cocktail or adavosertib; §One each as a result of death (pancreatic cancer), study termination by the sponsor, and withdrawal by the patient. bid twice daily
Treatment started with single-dose administration of the probe cocktail of caffeine (200 mg tablet), omeprazole (20 mg capsule), and midazolam (1 mL of 2 mg/mL syrup formulation) on day − 8, followed by PK sampling for 24 h (period 1) and a washout period of 7–14 days. In period 2, adavosertib (225 mg; 3 × 75 mg capsules) was administered bid until steady state for 2.5 days (total of five doses; maximum tolerated/recommended phase II dose for combination therapy [19]), and the final dose was administered in combination with single doses of the probe cocktail drugs on the morning of day 3, followed by PK sampling for 24 h. Venous blood samples were taken immediately pre-dose, as well as post-dose at 15, 30, and 45 min and 1, 2, 3, 4, 6, 8, 10, 12, and 24 h following a single dose of the cocktail alone on day − 8 or the cocktail with adavosertib 225 mg on day 3. Patients were screened 28 days before the first adavosertib dose and within 14–21 days before the first cocktail administration. Patients were required to fast from 2 h before until 2 h after the probe cocktail drug administration, as well as each administration of adavosertib alone or in combination with the cocktail.
Patients received granisetron 1 mg orally as anti-emetic prophylaxis 30 min prior to administration of the cocktail drugs or adavosertib capsules. Water intake was prohibited from 1 h before until 1 h after administration of the probe cocktail alone or with adavosertib (days − 8 and 3, respectively), excluding the 240 mL of water used for administration of adavosertib and the cocktail. Dexamethasone was prohibited until after the last PK sample was drawn; prochlorperazine, promethazine, and/or lorazepam could be used as required to treat nausea/vomiting.
The study was performed in accordance with the ethical principles of the Declaration of Helsinki and is consistent with the International Council for Harmonisation’s Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on bioethics [20].
Patients
Patients were eligible for the study if they met the following inclusion criteria: presence of a histologically or cytologically confirmed locally advanced or metastatic solid tumor, excluding lymphoma, for which standard therapy did not exist or had proven ineffective or intolerable; any prior palliative radiation must have been completed at least 7 days prior to the start of study treatment, and patients must have recovered from any acute adverse effects prior to the start of study treatment; Eastern Cooperative Oncology Group performance status of 0 or 1; no abnormalities in laboratory values within 7 days of initiation of study treatment; ≥ 18 years of age. Key exclusion criteria included: patients who were suffering from conditions that were likely to adversely affect gastrointestinal motility and/or transit or drug absorption; patients taking prescription medicines or foods known to interfere with the PKs of adavosertib or the probe cocktail drugs (see Supplementary Methods for restricted medications and additional exclusion criteria).
Pharmacokinetic assessment
After cocktail alone (day − 8) and with adavosertib co-administration (period 2 day 3), 24-h venous blood sampling took place for PK measurements of caffeine, omeprazole, midazolam, and their respective metabolites paraxanthine, 5-HO, and 1ʹ-HM. This sampling period was selected based on the half-lives of the probe substrates. The PK parameters measured included maximum plasma drug concentration (Cmax), area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC0–∞), AUC from time zero to time of the last quantifiable concentration (AUC0–t), time to reach maximum plasma concentration (tmax), plasma terminal half-life (t½), elimination rate constant (λz), apparent clearance (CL/F), and apparent volume of distribution (Vz/F) for the cocktail parent compounds (midazolam, omeprazole, and caffeine).
AUC from time zero to 12 h (AUC0–12), AUC0–t, tmax, Cmax, t½, and λz were determined for the cocktail metabolites (1ʹ-HM, 5–HO, and paraxanthine), and AUC and Cmax ratios were determined in relation to the parent compounds. Following adavosertib administration on period 2 day 3, PK parameters for adavosertib measured included AUC0–12, tmax, Cmax, minimum plasma drug concentration (Cmin), apparent average plasma concentration over a dosing interval (Cavg), apparent clearance at steady state (CLss/F), and fluctuation index over a dosing interval (FI).
Plasma concentrations for caffeine, paraxanthine, omeprazole, 5-HO, midazolam, and 1ʹHM were all determined by high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) at Labcorp Bioanalytical Laboratories (Madison, WI, USA).
Plasma concentrations of caffeine and paraxanthine collected with K2EDTA as anticoagulant were determined by HPLC–MS/MS detection simultaneously. The standard curves of caffeine and paraxanthine both ranged from 25.0 to 20,000 ng/mL. Precision (percentage coefficient of variance [%CV]) and accuracy (percentage bias) for the quality control (QC) samples were ≤ 7.0% and ≤ 7.5% CV and within − 3.1% to − 1.8% and − 4.9% to − 1.8% bias for caffeine and paraxanthine, respectively. The lower limit of quantification (LLOQ) was 25.0 ng/mL for caffeine and paraxanthine.
Plasma concentrations of omeprazole and 5-HO were collected with lithium heparin as anticoagulant and were determined by HPLC–MS/MS detection simultaneously. The standard curves of omeprazole and 5-HO both ranged from 20.0 to 20,000 nmol/L. Precision (%CV) and accuracy (percentage bias) for the QC samples were ≤ 3.9% and ≤ 4.4% CV and within 0.0 − 9.0% and − 1.9% to 2.2% bias for omeprazole and 5-HO, respectively. The LLOQ was 20.0 nM for omeprazole and 5-HO.
Plasma concentrations of midazolam and 1ʹ-HM were collected with K2EDTA as anticoagulant and were determined by HPLC–MS/MS detection simultaneously. The standard curves of midazolam and 1ʹ-HM both ranged from 0.100 to 100 ng/mL. Precision (%CV) and accuracy (percentage bias) for the QC samples were ≤ 6.4% and ≤ 5.1% CV and within − 0.8% to 1.0% and − 1.0% to 0.0% bias for midazolam and 1ʹ-HM, respectively. The LLOQ was 0.1 ng/mL for midazolam and 1ʹ-HM.
The concentration of adavosertib in human plasma was determined by Labcorp Bioanalytical Laboratories using the same validated method described previously [21]. The assay had a linear dynamic range of 2–1000 ng/mL, with an LLOQ of 2 ng/mL [22].
All plasma samples were stored at − 70 °C (or below) prior to analysis and analyzed within the validated time frame.
Safety and tolerability assessments
Safety was appraised throughout the study by the assessment of clinical and laboratory adverse events (AEs; graded by Common Terminology Criteria for Adverse Events [CTCAE], version 4.03), physical examination, and evaluation of vital signs and laboratory data (clinical chemistry and hematology).
Statistical analyses
No formal sample size estimation was conducted. Based on an estimate of within-patient standard deviation of 0.294 and assuming a true interaction effect of 100%, it was estimated that 20 evaluable patients would provide 80% power to show that the 90% confidence interval (CI) for the adavosertib effect lies entirely below 2.67, i.e. it would rule out a 167% increase of exposure in the presence of adavosertib. The number of patients was based on careful clinical consideration to gain adequate information on the primary endpoints while exposing as few patients as possible to study procedures. Enrollment of approximately 30 patients, with a target of 20 evaluable patients, was considered adequate and sufficient to meet the objectives of this study.
The safety analysis set included all patients who received at least one dose of study treatment (adavosertib or probe cocktail drugs). Patients were evaluated according to the treatment received. A treatment-emergent AE was defined as an AE with its start date and time on or after the first dose of probe cocktail on day − 8 up to and including 30 days after the last dose date of adavosertib, or, for pre-existing pre-treatment AEs, the date on which they worsened in severity after the first dose of study treatment. The PK analysis set included all dosed patients who had at least one quantifiable plasma concentration for any of the cocktail drugs (or metabolites) or adavosertib collected post-dose without protocol deviations or events that could affect the PK analysis.
PK parameters were derived using non-compartmental methods with Phoenix® WinNonlin® version 6.4 (Certara, LP, Princeton, NJ, USA). All descriptive and inferential statistical computations were performed using SAS® version 9.1 (SAS Institute, Cary, NC, USA). Estimates of the mean difference between treatments (adavosertib + probe cocktail substrate compared with probe cocktail substrate alone) and corresponding 90% CIs were calculated using a linear mixed-effects model, with a fixed effect for treatment and a random effect for patient. The natural-log-transformed PK parameters (Cmax, AUC0-∞, and AUC0–t) of the cocktail parent compounds and metabolites were used in the mixed-effects models as dependent variables. The mean differences and CIs were back transformed to the original scale to give estimates of the ratios and the associated 90% CIs; additionally, back-transformed geometric means, together with 95% CIs, were estimated and are presented by treatment.
Results for AUC0–12 and Cmax obtained following adavosertib administration on period 2 day 3 were compared with those obtained on period 2 day 3 following the same adavosertib dosage schedule but without cocktail (AstraZeneca, data on file, 2022) to probe the potential effects of the cocktail drugs and to obtain an estimate of intra-patient adavosertib variability.
Results
Patients
Of 57 patients enrolled (from 49 unique patients, as several patients enrolled more than once because of rescreening), 33 were assigned to treatment (see Fig. 1 for patient disposition flow chart). Patient baseline characteristics are shown in Table 1; the median age of patients assigned to treatment was 60 years (range 41–83), and the gender distribution was balanced. Six patients used disallowed concomitant medications, including three patients who were taking a weak CYP3A inhibitor that could have affected the PK results for midazolam and its metabolite 1ʹ-HM (two patients took amlodipine, one patient took alprazolam); these patients had their midazolam and 1ʹ-HM PK results excluded. Details of the number of patients with evaluable data for each drug/period are provided in Supplementary Table S1.
Pharmacokinetic assessments
Pharmacokinetic parameter estimates for substrates of CYP1A2 (caffeine), CYP2C19 (omeprazole), and CYP3A (midazolam) and their metabolites (paraxanthine, 5-HO, and 1ʹ-HM, respectively) in the presence and absence of adavosertib are shown in Table 2.
Co-administration of adavosertib increased caffeine total exposure by 49% (AUC0–12) to 61% (AUC0–t; Table 2). Caffeine AUC was only reliably characterized in eight patients in period 2. With the exception of one patient, caffeine AUC0–12 increased following co-administration of adavosertib in all patients with paired data for both periods; hence, the results obtained for AUC0–12 from this inferential analysis were considered to be representative of the data.
Paraxanthine AUC0–12 was only reliably characterized in 10 patients in period 1 and one patient in period 2. Thus, the treatment comparison for AUC0–12 was not considered meaningful.
Co-administration of adavosertib increased omeprazole total exposure by 80% (AUC0–12) to 98% (AUC0–t) and increased Cmax by 46% (Table 2).
Co-administration of adavosertib increased midazolam total exposure by 55% (AUC0–12 and AUC0–t; Table 2).
The individual and geometric mean AUC0–12 and Cmax of adavosertib plus cocktail (part A) versus adavosertib alone (part B), and the individual and geometric mean AUC0–t and Cmax of caffeine, omeprazole, and midazolam alone versus each cocktail drug plus adavosertib, are shown in Fig. 2.

Individual and geometric mean AUC0–t and Cmax of a caffeine, b omeprazole, and c midazolam alone versus each cocktail drug plus adavosertib, and d individual and geometric mean of adavosertib plus cocktail (part A) versus adavosertib alone (part B). *Individual patient and geometric mean AUC0–t and Cmax in the presence and absence of adavosertib 225 mg bid for caffeine (n = 25), omeprazole (n = 27), and midazolam (n = 23); †Adavosertib + cocktail: caffeine (200 mg tablet), omeprazole (20 mg capsule), and midazolam (1 mL of 2 mg/mL syrup formulation) on day 3 and adavosertib 225 mg (3 × 75 mg capsules) on day 3 (part A). Adavosertib: adavosertib 225 mg (3 × 75 mg capsules) bid on days 1 and 2 and once on day 3 (part B/pharmacodynamic study). AUC0–12 area under the plasma concentration–time curve from time zero to 12 h, AUC0–t area under the plasma concentration–time curve from time zero to time of the last quantifiable concentration, Cmax maximum plasma drug concentration
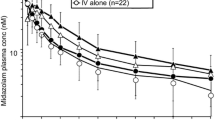
The geometric mean plasma concentrations of caffeine, omeprazole, and midazolam versus time, with and without adavosertib, are shown in Fig. 3. In the presence of adavosertib, the rate of paraxanthine formation appeared to be decreased, resulting in lower mean concentrations and a lower rate of elimination over the sampling period (Supplementary Figure S1).

Geometric mean (± SD) plasma concentration of a caffeine, b omeprazole, and c midazolam ± adavosertib versus time. Exponential of (mean of log concentration ± SD of log concentration). Probe cocktail: caffeine (200 mg tablet), omeprazole (20 mg capsule), and midazolam (1 mL of 2 mg/mL syrup formulation) on day − 8. Adavosertib + cocktail: cocktail and adavosertib 225 mg (3 × 75 mg capsules) on day 3. SD standard deviation
The PK parameters of adavosertib following co-administration of cocktail are shown in Table 3. Point estimates of the AUC0–12 and Cmax geometric least-squares (LS) mean ratios were 108% and 97%, respectively. Accumulation over 2.5 days of bid dosing with adavosertib was approximately 2.3 and 2.1 for AUC0–12 and Cmax, respectively. The adavosertib concentration in human plasma ranged from 4 to 2000 nM. Precision (%CV) was ≤ 4.2% and accuracy (percentage bias) was within 0.7–1.5% for the QC samples. Reproducibility was confirmed by re-analysis of 87 study samples (with adavosertib concentrations above the LLOQ) selected at random; 98.9% of the results obtained following the initial and repeat analyses were within 20.0% of the mean of the two values, thus meeting the acceptance criteria [23].
Safety
Causally related AEs (stratified by any grade and grade ≥ 3) are shown in Supplementary Table 2 for patients who received only adavosertib and patients who received both adavosertib and the probe cocktail. Four of 33 (12.1%) patients experienced AEs with cocktail alone, compared with 16/26 (61.5%) patients who received probe cocktail plus adavosertib and 16/30 (53.3%) patients after receiving adavosertib alone on days 1 and 2. Treatment-related AEs were reported by 12/26 (46.2%) patients receiving probe cocktail plus adavosertib and 11/30 (36.7%) patients after receiving adavosertib alone on days 1 and 2. The most common treatment-related AEs in patients receiving probe cocktail plus adavosertib (n = 26 patients) were diarrhea and nausea (both in four [15.4%] patients) and dizziness (in two [7.7%] patients). The most common treatment-related AEs in patients receiving adavosertib alone (n = 30 patients) were diarrhea (in 10 [33.3%] patients), vomiting (in six [20.0%] patients), and nausea (in three [10.0%] patients).
Adverse events of CTCAE grade ≥ 3 were observed in: 4/26 (15.4%) patients receiving probe cocktail plus adavosertib (anemia, neutropenia, pulmonary embolism, and portal vein thrombosis in one [3.8%] patient each); 5/30 (16.7%) patients receiving adavosertib alone on days 1 and 2 (diarrhea in three [10.0%] patients, necrotizing soft-tissue infection, neutropenia, thrombocytopenia, dehydration, hypokalemia, hypoxia, nausea, pancreatitis, small-intestinal obstruction, vomiting, and acute kidney injury in one [3.3%] patient each [multiple events were experienced by the same five (16.7%) patients]); and 0/33 patients receiving cocktail alone.
One patient receiving adavosertib alone on days 1 and 2 reported two serious AEs (SAEs) considered related to adavosertib (acute kidney injury, which resolved after 2 days, and pancreatitis, which did not resolve during the study but improved to grade 2 after 2 days) that led to discontinuation; four patients reported five SAEs that were not considered related to treatment (grade 2 bacterial pneumonia and deep vein thrombosis, which did not resolve; grade 3 necrotizing soft-tissue infection, which resolved; grade 3 small-bowel obstruction, which did not resolve; and grade 4 pulmonary embolism, which resolved).
Two patients discontinued treatment because of AEs: one as a result of grade 3 diarrhea considered related to adavosertib, which resolved, and the other for grade 3 small-bowel obstruction, which was not considered related to adavosertib and did not resolve.
No AEs with an outcome of death were observed; one patient died as a result of disease progression (pancreatic cancer) following receipt of the probe cocktail but prior to receiving adavosertib.
Discussion
In this study, adavosertib exposure, when given alone or co-administered with the cocktail drugs, was similar to that observed in previous studies when given alone [5]. Therefore, concomitant administration of drugs metabolized by CYP3A, CYP2C19, and CYP1A2 is unlikely to have a significant clinical effect on adavosertib PKs. Adavosertib, when given bid at a dose of 225 mg for 2.5 days, exhibited low intra-patient variability (eg < 10% for AUC0–12 and Cmax), and adavosertib accumulation over 2.5 days of bid dosing was approximately twofold. The detailed PK data from this study reveal that the point estimates of the AUC0–12 and AUC0–t geometric LS mean ratios of all three probe cocktail drugs in the presence of adavosertib (225 mg bid) compared with probe cocktail drugs administered alone showed a 1.25- to < twofold increase [17]. Adavosertib therefore meets the US Food and Drug Administration (FDA) guidance for definition of a weak inhibitor of CYP1A2, CYP2C19, and CYP3A [17].
A physiologically based PK (PBPK) model (based on in vitro studies of absorption, distribution, metabolism, and excretion, clinical adavosertib PK data for doses of 175–300 mg once daily [qd] or bid, and multiple dosing schedules [3 days on/4 days off and 5 days on/9 days off]) to assess multifaceted CYP modulation predicted that adavosertib is mainly metabolized by CYP3A with an FMO3 and/or FMO5 component and is a time-dependent (irreversible) inhibitor of CYP3A and a weak reversible (direct) inhibitor of CYP2C8, CYP2C9, and CYP2C19 [16, 24]. Model predictions of adavosertib being a weak reversible inhibitor of CYP2C19 were confirmed by this study. Concentration–time curves for midazolam, the substrate of CYP3A, and its metabolite 1ʹ-HM show both midazolam and 1ʹ-HM plasma concentrations increasing then returning towards baseline/elimination over the sampling period. The PBPK model predictions of adavosertib being a weak reversible inhibitor of CYP2C8 and CYP2C9 [16, 24] remain clinically unvalidated but are in line with observed effects of adavosertib being a weak inhibitor of CYP1A2, CYP2C19, and CYP3A.
The observed safety profile of adavosertib in this study was concordant with that found in previous clinical studies [7, 15]. The most prevalent AEs were gastrointestinal toxicities (diarrhea, vomiting, and nausea). No new or unexpected adavosertib-related safety concerns were identified.
Although the study was terminated early because of the difficulty in enrolling patients (i.e. screening/rescreening failures) and evaluable data not being available for all enrolled patients (as a result of protocol deviations/events), this had minimal impact on the interpretation of the PK results. Overall, the data from evaluable patients were adequate and representative of the primary objective of assessing the DDI potential of adavosertib. Instances of pre-dose concentrations > 5% of Cmax were especially prevalent for caffeine and paraxanthine, despite the study design including washout periods and restriction of caffeine consumption. A potential explanation for this finding is that caffeine/paraxanthine elimination may be slower in this patient population than in participants in previous DDI studies; also, and perhaps the most likely explanation, patients may not have been strictly compliant with the caffeine restrictions in the protocol. The true exposure increase when caffeine/paraxanthine were co-administered with adavosertib may therefore be lower than reported here. Compared with the recommended phase II dose for monotherapy (300 mg qd, 5 days on/2 days off for 2 of 3 weeks) and the maximum tolerated dose of monotherapy (125 mg bid, 5 days on/9 days off for 2 of 3 weeks), both of which were determined after the present study commenced, the adavosertib dose in this study (225 mg bid) was higher and the dosing duration (2.5 days) sufficient to reach steady state; therefore, DDIs at the recommended phase II dose for monotherapy are not expected to be worse than observed during this study [25]. The relatively small study sample size facilitates the balance of needing to gain adequate PK data against exposing as few patients as possible to study procedures.
Overall, the results from this study suggest that adavosertib is a weak inhibitor of CYP1A2, CYP2C19, and CYP3A according to the US FDA guidance for definition of a weak inhibitor [17]. Although the limited impact on probe drug exposures suggests that it is unlikely that dose adjustments of drugs metabolized by CYP1A2, CYP2C19, and CYP3A will be needed when co-administered with adavosertib, adequate safety monitoring and dose modification may be necessary, particularly when co-administration occurs with substrates with a narrow therapeutic index.
Conclusions
Adavosertib, when administered at a dose of 225 mg bid, is a weak inhibitor of CYP1A2, CYP2C19, and CYP3A and therefore has a low risk of causing clinically significant DDIs with drugs metabolized by these enzymes.
Data availability
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Data for studies directly listed on Vivli can be requested through Vivli at www.vivli.org. Data for studies not listed on Vivli could be requested through Vivli at https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. AstraZeneca Vivli member page is also available outlining further details: https://vivli.org/ourmember/astrazeneca/.
References
Russell P, Nurse P (1987) Negative regulation of mitosis by Wee1+, a gene encoding a protein kinase homolog. Cell 49:559–567. https://doi.org/10.1016/0092-8674(87)90458-2
Do K, Doroshow JH, Kummar S (2013) Wee1 kinase as a target for cancer therapy. Cell Cycle 12:3159–3164. https://doi.org/10.4161/cc.26062
Coleman TR, Dunphy WG (1994) CDC2 regulatory factors. Curr Opin Cell Biol 6:877–882. https://doi.org/10.1016/0955-0674(94)90060-4
Parker LL, Piwnica-Worms H (1992) Inactivation of the P34CDC2-cyclin B complex by the human Wee1 tyrosine kinase. Science 257:1955–1957. https://doi.org/10.1126/science.1384126
Elbæk CR, Petrosius V, Sørensen CS (2020) Wee1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat Res 819–820:111694. https://doi.org/10.1016/j.mrfmmm.2020.111694
Macheret M, Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10:425–448. https://doi.org/10.1146/annurev-pathol-012414-040424
Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S (2015) Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 33:3409–3415. https://doi.org/10.1200/jco.2014.60.4009
Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, Sakai T, Arakawa H, Sakamoto T, Sagara T, Yoshizumi T, Mizuarai S, Kotani H (2009) Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 8:2992–3000. https://doi.org/10.1158/1535-7163.mct-09-0463
Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC (2012) Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of Wee1. Cancer Discov 2:524–539. https://doi.org/10.1158/2159-8290.cd-11-0320
Bauer TM, Moore K, Rader JS, Simpkins F, Mita A, Beck JT, Hart L, Chu Q, Oza A, Tinker AV, So K, Imedio ER, Kumar S, Mugundu GM, Jenkins S, Chmielecki J, Jones S, Spigel DR, Fu S (2019) Open-label, multicenter, phase Ib study to assess safety, tolerability and efficacy of adavosertib monotherapy in patients with advanced solid tumors: expansion cohorts. Cancer Res 79(13 Suppl):CT012. https://doi.org/10.1158/1538-7445.am2019-ct012. (abstract)
Hamilton E, Falchook GS, Wang JS, Fu S, Oza A, Karen S, Imedio ER, Kumar S, Ottesen L, Mugundu GM, Chmielecki J, Jones S, Spigel DR, Li BT (2019) Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: dose escalation. Cancer Res 79(13 Suppl):CT025. https://doi.org/10.1158/1538-7445.am2019-ct025. (abstract)
Moore KN, Chambers SK, Hamilton EP, Chen LM, Oza AM, Ghamande SA, Konecny GE, Plaxe SC, Spitz DL, Geenen JJJ, Troso-Sandoval TA, Cragun JM, Rodrigo Imedio E, Kumar S, Mugundu GM, Lai Z, Chmielecki J, Jones SF, Spigel DR, Cadoo KA (2022) Adavosertib with chemotherapy in patients with primary platinum-resistant ovarian, fallopian tube, or peritoneal cancer: an open-label, four-arm, phase II study. Clin Cancer Res 28:36–44. https://doi.org/10.1158/1078-0432.Ccr-21-0158
Patel MR, Falchook GS, Wang JS-Z, Imedio ER, Kumar S, Motlagh P, Miah K, Mugundu GM, Jones SF, Spigel DR, Hamilton EP (2019) Open-label, multicenter, phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors. J Clin Oncol 37(15 Suppl):2562. https://doi.org/10.1200/JCO.2019.37.15_suppl.2562. (abstract)
Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, Olawaiye AB, Ellard SL, Weberpals JI, Wahner Hendrickson AE, Fleming GF, Welch S, Dhani NC, Stockley T, Rath P, Karakasis K, Jones GN, Jenkins S, Rodriguez-Canales J, Tracy M, Tan Q, Bowering V, Udagani S, Wang L, Kunos CA, Chen E, Pugh TJ, Oza AM (2021) Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397:281–292. https://doi.org/10.1016/s0140-6736(20)32554-x
Liu JF, Xiong N, Campos SM, Wright AA, Krasner C, Schumer S, Horowitz N, Veneris J, Tayob N, Morrissey S, West G, Quinn R, Matulonis UA, Konstantinopoulos PA (2021) Phase II study of the Wee1 inhibitor adavosertib in recurrent uterine serous carcinoma. J Clin Oncol 39:1531–1539. https://doi.org/10.1200/jco.20.03167
Moorthy G, Nagard M, Johnson M, Ah-See M, Fretland L, Ottesen G, Mugundu GM (2020) A physiologically based pharmacokinetic (PBPK) model for adavosertib to assess multifaceted CYP modulation: verification using cocktail DDI study. Clin Pharmacol Ther 107(S1):E-011. https://doi.org/10.1002/cpt.1732. (abstract)
FDA (2020) Clinical drug interaction studies – cytochrome P450 enzyme- and transporter-mediated drug interactions: guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed October 2022
Turpault S, Brian W, Van Horn R, Santoni A, Poitiers F, Donazzolo Y, Boulenc X (2009) Pharmacokinetic assessment of a five-probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol 68:928–935. https://doi.org/10.1111/j.1365-2125.2009.03548.x
Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, Freshwater T, Shumway S, Liang LW, Oza AM, Schellens JH, Shapiro GI (2016) Phase I study evaluating Wee1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol 34:4371–4380. https://doi.org/10.1200/jco.2016.67.5991
AstraZeneca (2015) Global policy: bioethics. https://www.astrazeneca.com/content/dam/az/PDF/2019/Bioethics%20Policy%20final.pdf. Accessed Oct 2022
Någård M, Ah-See ML, So K, Vermunt M, Thistlethwaite F, Labots M, Roxburgh P, Ravaud A, Campone M, Valkenburg-van Iersel L, Ottesen L, Li Y, Mugundu G (2020) Effect of food on the pharmacokinetics of the WEE1 inhibitor adavosertib (AZD1775) in patients with advanced solid tumors. Cancer Chemother Pharmacol 86:97–108. https://doi.org/10.1007/s00280-020-04101-4
Xu Y, Fang W, Zeng W, Leijen S, Woolf EJ (2012) Evaluation of dried blood spot (DBS) technology versus plasma analysis for the determination of MK-1775 by HILIC-MS/MS in support of clinical studies. Anal Bioanal Chem 404:3037–3048. https://doi.org/10.1007/s00216-012-6440-6
FDA (2018) Bioanalytical method validation guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry. Accessed October 2022
Miah K, Imedio E, Kumar S, Ottesen L, Mugundu G (2020) Clinical pharmacokinetics of adavosertib in the presence or absence of PD-L1 inhibitor durvalumab in patients with refractory solid tumors. ASCPT Vision 107(Suppl 1):S49 PII-051 (abstact)
Falchook GS, Sachdev J, Imedio ER, Kumar S, Mugundu G, Chmielecki J, Jones S, Spigel DR, Johnson M (2019) A phase Ib study of Wee1 inhibitor adavosertib in patients with advanced solid tumors. Cancer Res 79(13 Suppl):CT022. https://doi.org/10.1158/1538-7445.am2019-ct022. (abstract)
Acknowledgements
The authors wish to thank all the patients, their families, and the clinical teams who worked on this study. Medical writing assistance was provided by CL Attwell PhD, from AMICULUM Ltd, funded by AstraZeneca. LDL acknowledges the role of the Dartmouth Cancer Center Clinical Pharmacology Shared Resource, which is supported in part by NCI P30CA023108, in participating in this study.
Funding
This study was funded by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LDL is a consultant to G1 Therapeutics and 7 Hills Pharma LLC. He also received, through an award to his institution, clinical trial funding for this study from AstraZeneca and other similar clinical trial funding support from the following sponsors: Bristol Myers Squibb, Bayer Pharmaceuticals, and AbbVie. WJE is a consultant for Chimerix Corp. MA and MN are employees and shareholders of AstraZeneca. LHO was an employee of AstraZeneca when this study was designed, executed, and reported. LHO is also a shareholder of AstraZeneca. All other authors have declared no conflicts of interest.
Ethical approval
This study was performed in accordance with the ethical principles of the Declaration of Helsinki and is consistent with the International Council for Harmonisation’s Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on bioethics [20].
Consent to participate
The institutional review boards or independent ethics committees of all investigational sites approved the protocol, and all patients provided written, informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Någård, M., Ah-See, ML., Strauss, J. et al. Phase I study to assess the effect of adavosertib (AZD1775) on the pharmacokinetics of substrates of CYP1A2, CYP2C19, and CYP3A in patients with advanced solid tumors. Cancer Chemother Pharmacol 92, 193–203 (2023). https://doi.org/10.1007/s00280-023-04554-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-023-04554-3